

1 Introduction
In 1971, it was published [1] that cyanide anions were able to reduce electron acceptor molecules such as fluorenone, anthraquinone or phenanthrenequinone through the formation of the corresponding quinone anion radical intermediates under aprotic polar solvents such as DMF, DMSO or HMPA. The formation of these paramagnetic anion radicals was proven by recording the ESR and visible spectra of their strongly coloured solutions. More recently, Roginsky et al. [2] described the similar one-electron reduction of Ubiquinone-0 by ascorbate under aerobic conditions. Nevertheless, the synthetic-scale way has not yet been explored regarding these processes.
On the other hand, many imides, such as naphthalimides, have been employed in the treatment of a host with a cellular proliferative disease [3] together with an antiproliferative agent, but also 1H-azepine-2,7-diones have demonstrated hypolipidemic and good hypotriglyceridemic activity [4].
The cathodic reduction of o-quinones and of other 1,2-dicarbonyl compounds in aprotic organic solvents has been studied in depth in our research group, both in the absence and in the presence of arene diazonium salts. In the absence of diazonium salts, o-quinones can be transformed in good yield and only one pot to 1,3-dioxoles when electrolysis is performed in dichloromethane as the solvent. Other 1,2-dicarbonyl derivatives, such as benzil or 1,2-naphtoquinone, have been respectively converted into methylenediesters [5] or lactones [6] by cathodic reduction in the presence of oxygen. When diazonium salts are present in the catholyte, concomitant reduction of the latter and of 1,2-quinones produces coupling products with radicals generated from the solvent. In this case, aryl radicals, obtained in the diazonium salt discharge, led to cyanomethylated, alkylated or dimethylaminocarbonylated coupling products with the anion radicals of different quinones, by hydrogen atom abstraction from acetonitrile, dichloroethane or DMF, respectively used as the solvent. This reduction of 1,2-quinones in the presence of benzenediazonium tetrafluorborate was extended to other solvents such as dichloromethane, chloroform, ethyl acetate or chloroacetonitrile. A very special case in this topic was the electrosynthesis of 1,3,4-oxadiazol-2(3H)-ones and dibenzo[c,e]azepines when N-methylformamide was used as the solvent [7].
In the present paper, we describe the reduction of 1,2-dicarbonyl compounds and their imine derivatives in acetonitrile, but not only at the electrode: a comparison between the electrochemical and a conventional electron transfer process from cyanide anion to the quinone is made. The subsequent formation, when oxygen is present in solution, of superoxide anion provides an interesting entry to 1H-azepine 2,7-diones.
2 Results and discussion
When 9,10-phenanthrenequinone (1a), 10-(phenylimino)phenanthren-9(10H)-one (1b), acenaphthenequinone (1c), 2-(phenylimino)acenaphthylen-1(2H)-one (1d), benzil (1e) or 1,2-diphenyl-2-(phenylimino)ethanone (1f) were stirred for 24 h in the presence of a large excess of sodium cyanide, a redox process was observed between them, allowing one to get results similar to those obtained in the corresponding preparative-scale electrochemical reduction of 1.
The expected cyanhydrins were obtained as side products by direct attack of the nucleophile on the carbonyl group.
The treatment of 1b with cyanide afforded 1-phenyl-1H-azepine-2,7-dione (2b) as the main product (70% yield) after an electron transfer reaction in solution from cyanide to 1b, as indicated in Scheme 1. 9,10-Dihydro-9-hydroxy-10-(phenylimino) phenanthrene-9-carbonitrile (3b) was obtained in 18% yield.
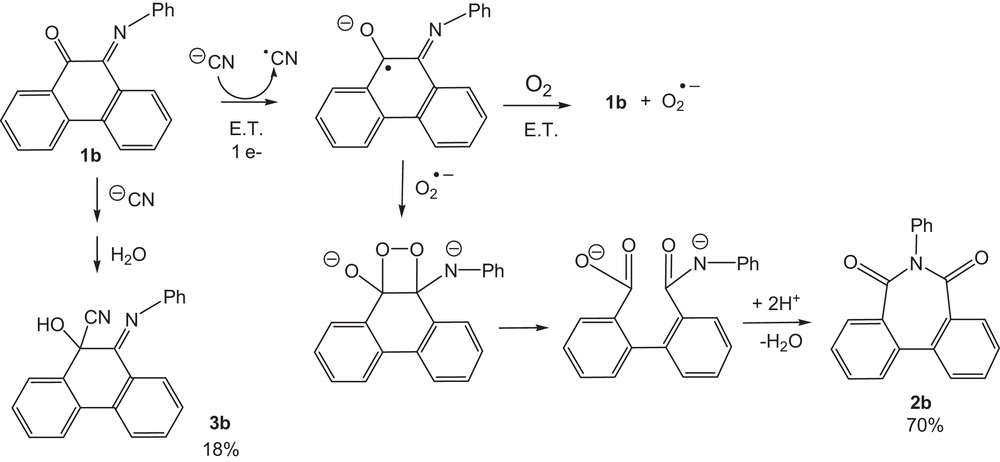
Proposed pathway for the reduction of 1b by the cyanide anion.
In the reaction of 1a with cyanide, the higher stability of the anion radical and of the dianion of 1a, related to that of 1b, is responsible for the lower conversion obtained in this case, even after 48 h of stirring. Together with the formation of the dicarboxylic acid and its anhydride (2a) [8] as main products, lactone 3a was formed as a minor product (12%) (see Scheme 2). A mechanism for the formation of 3a has already been proposed [6] through a Baeyer–Villiger/Dakin reaction involving oxygen.

Proposed pathway for the reduction of 1a by the cyanide anion.
Preparative-scaled electrolysis of 10-(phenylimino)phenanthren-9(10H)-one (1b) in dry CH3CN (AN)/lithium perchlorate at a mercury pool as the cathode was carried out applying a reduction potential of –1.3 V (vs. Ag/AgCl) (see Fig. 1). As indicated in Scheme 3, 1-phenyl-1H-azepine-2,7-dione (2b) was obtained as the major product, together with the expected cyanhydrin: 9,10-dihydro-9-hydroxy-10-(phenylimino)phenanthrene-9-carbonitrile (3b).
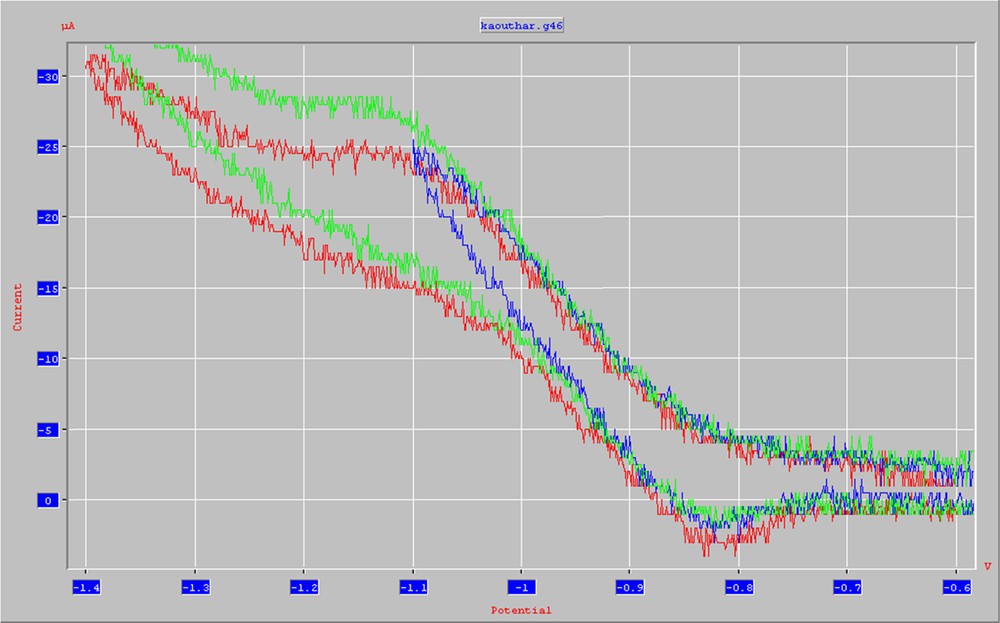
(Colour online.) Cyclic voltammogram of 1b in dry acetonitrile/LiClO4 at a platinum cathode. Reference electrode: Ag/AgCl. Scan rate: 100 mV/s.

Electrochemical reduction of 1b. Proposed pathway to explain the formation of the imide 2b.
The reaction of acenaphthenequinone (1c) with sodium cyanide produced, under the same experimental conditions as those specified above, the expected dicarboxylic acid and its anhydride (2c) as main products (75%) after 24 h of stirring (Scheme 4). The corresponding cyanhydrin: 1-hydroxy-2-oxo-acenaphthylene-1-carbonitrile (3c) was formed as a minor product.
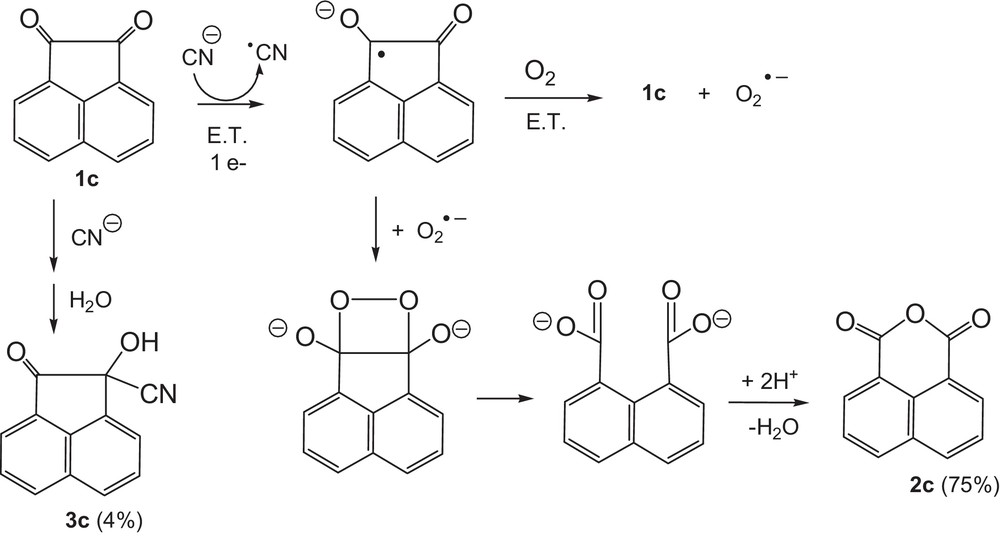
Proposed pathway for the reduction of 1c by the cyanide anion.
In this case, the azepine 4c was obtained as a trace product. The formation of 4c has to be explained by the presence of ammonia in the medium. The only source of ammonia involves, however, a simultaneous oxidation of the cyanide anion, as it is summarized in Scheme 5.

Proposed mechanism for the obtention of 4c from 1c.
The reduction by cyanide of 2-(phenylimino)acenaphthylen-1(2H)-one (1d) afforded, almost quantitatively, the N-phenylimide of naphthalene-1,8-dicarboxylic acid (2d), through a pathway similar to that described in Scheme 3.
Finally, the electron-transfer reaction from cyanide anion to benzil (1e) was carried out, producing benzoic acid and the expected cyanhydrin: 2-hydroxy-3-oxo-2,3-diphenylpropanenitrile (2e) in 50% yield.
The mechanism proposed to explain the formation of benzoic acid from cyanide and 1e is summarized in Scheme 6. It has been discussed previously in a paper reporting the electrochemical reduction of benzil in the presence of oxygen [5].

Already published mechanism proposed for the cathodic reduction of benzil (1e).
The monoimine 1f afforded 2-phenyl-2-(phenylimino)acetonitrile (2f) as the major product (72% yield) together with the low yielded (10%) 2-hydroxy-2,3-diphenyl-3-(phenylimino)propanenitrile (cyanhydrin) (3f). The formation of 2f could be rationalized through the logical pathway proposed in Scheme 7, where a double nucleophilic addition attack from cyanide to 1f can produce a dianion intermediate that, similarly to benzoin condensation, could evolve to 2f.

Double cyanide nucleophilic addition to 1f explaining the formation of 2f.
3 Conclusion
The reactions here described involve an unexpected electron transfer reaction in solution from the cyanide anion to conjugated 1,2-dicarbonyl derivatives to give interesting organic imides, such as 1H-azepine-2,7-diones. The expected nucleophilic behaviour of the cyanide anion to give the corresponding cyanhydrin has been minimized under our employed experimental conditions.
4 Experimental
4.1 General procedure
The electron transfer reaction from the cyanide anion to 1,2-dicarbonyl compounds and their monoimine derivatives 1a–f was performed under stirring conditions in dry acetonitrile (40 mL) solutions where the substrate (0,25 mmol) was maintained for 24 h (48 h for 1a and 1e) at room temperature and usual laboratory illumination conditions, in the presence of sodium cyanide (10 mmol) over an open-air vessel. The reaction was followed by TLC until complete disappearance of the starting compound.
4.2 General electrochemical procedure
The electrochemical reduction of 1b was performed under potentiostatic conditions in a concentric cell with two compartments separated by a porous (D4) glass frit diaphragm and equipped with a magnetic stirrer. A mercury pool was used as the cathode (18 cm2), a platinum plate as the anode (9 cm2) and an Ag/AgCl electrode as the reference. The SSE was dry acetonitrile (AN) containing 0.1 M lithium perchlorate.
A solution of the electroactive substrate (1.0 mmol in 60 mL of SSE) was electrolyzed at a constant potential of –1.3 V (vs. Ag/AgCl), corresponding to the only reduction peak observed for this compound (see Fig. 1).
Once the reduction was finished, the solvent in the reaction solution was removed under reduced pressure. The residue was extracted with ether/water and the organic phase dried over Na2SO4 and concentrated by evaporation. The resulting solid was chromatographed on silica gel (22 × 3 cm) column, using PE/EtOAc (4:1) as an eluent. The spectroscopic description of the obtained compounds is given below. The aqueous phase was acidified with HCl (5%) and further extracted with ether, dried over Na2SO4 and concentrated by evaporation. From the aqueous solutions were isolated, when formed, carboxylic acids.
1-Phenyl-1H-azepine-2,7-dione (2b): (70% yield). Mp 195–197 °C. [Lit.[9] 197–198 °C]. MS m/e (relative intensity) EI: 299 (M+, 26), 254 (9), 206 (12), 180 (100), 152 (31), 76 (4).
9,10-Dihydro-9-hydroxy-10-(phenylimino)phenanthrene-9-carbonitrile (3b): (18% yield). MS m/e (relative intensity) EI: 311 (M+ + 1, 24), 310 (M+, 100), 309 (28), 281 (42), 267 (18), 254 (12), 190 (93), 180 (33), 165 (31), 152 (32), 77 (16).
1-Hydroxy-2-oxo-1-acenaphthylene-1-carbonitrile (3c): (4% yield). MS m/e (relative intensity) EI: 209 (M+, 57), 181 (52), 164 (16), 155 (100), 138 (9), 127 (68).
1,8-Naphthalenedicarboximide (4c): Mp 298 °C. [Lit.[10] 300–302 °C]. MS m/e (relative intensity) EI: 197 (M+, 100), 153 (78), 140 (6), 126 (40), 87 (9), 74 (9).
N-Phenyl 1,8-naphthalenedicarboximide (2d): (84% yield). Mp 205–206 °C. [Lit.[11] 207–209 °C]. MS m/e (relative intensity) EI: 273 (M+, 73), 272 (M+–1,100), 228 (26), 201 (7), 180 (20), 152 (11), 126 (18), 74 (4).
2-Hydroxy-3-oxo-2,3-diphenylpropanenitrile (2e): (50% yield). 1H NMR (300 MHz, CDCl3) δ: 6.7 (bs, 1H), 7.42–7.48 (m, 5H), 8.04 (d, 2H, J = 7.8 Hz). 13C NMR (75.4 MHz, CDCl3) δ: 63.5, 116.4, 127.9, 128.7, 129.3, 130.1, 130.4, 131.8, 134.2, 164.7. MS m/e (relative intensity) EI: 237 (M+, 9), 211 (17), 165 (3), 116 (14), 105 (100), 77 (12).
2-Phenyl-2-(phenylimino)acetonitrile (2f): (72% yield) Mp 73–75 °C. [Lit.[12] 76 °C]. MS m/e (relative intensity) EI: 206 (M+, 100), 205 (M+–1, 56), 180 (51), 129 (2), 103 (1), 77 (15).
2-Hydroxy-2,3-diphenyl-3-(phenylimino)propanenitrile (3f): (10% yield) MS m/e (relative intensity) EI: 312 (M+, 48), 286 (5), 268 (4), 207 (11), 106 (100), 77 (73).
Acknowledgements
The authors gratefully acknowledge the financial support of the University of Alcala – project No. CCG2014/EXP-010. K. Hamrouni thanks the Tunisian Ministry of Science that provided her a grant “bourse d’alternance” as financial support of her stay in Spain.