

1 Introduction
In the last decades, the drastically increasing environmental consciousness urged scientists to search for more efficient methods to reduce pollutants. Volatile Organic Compounds (VOCs), which are emitted by various industrial processes and automotive exhausts, are the main class of air pollutants [1] because of their harmful effects on man’s health and environment [1,2]. Among the various technologies available for VOCs abatement, especially for low pollutant concentrations (<1%), catalytic oxidation is cheaper and very efficient [2,3]. It consists of a complete oxidizing of VOCs into carbon dioxide and water. By shifting the temperature required for conventional thermal incineration to lower values, this technology saves energy as well as it minimizes the formation of harmful by-products [1,2]. Therefore, highly active catalysts which work at lower temperatures are required.
Two groups of catalysts were widely used for the oxidation of VOCs: noble metals (Pt, Pd, Rh, and Au) [4,5] and transition metals (Mn, Co, Cu, Fe, and Ni) [6–10]. In general, the catalytic activity of noble metals is higher than that of transition metals [11–14]. Out of all noble metals, Pd-based catalysts have been studied and showed high activity, at relatively low temperatures, and selectivity towards CO2 and H2O in the oxidation of volatile organic compounds (VOCs) with a high tolerance to moisture [15–20]. Therefore, they are promising catalysts for practical applications. After the study done by Haruta [21], supported gold catalysts have attracted much attention. Many studies reported high activity of Au-supported catalysts in VOCs oxidation [22–27] even in the presence of moisture [27]. However, it is found that the support nature [6–8, 28–30] and the preparation method [23, 30–32] play an important role in the improvement in the catalyst’s efficiency, particularly in oxidation reaction.
Hydroxyapatite (Hap) based materials have attracted much interest to be used in a variety of applications because they are safe, non-toxic [33], inexpensive, and readily available in the natural environment. Furthermore, these compounds present a high chemical and thermal stability and a weak solubility in water. In addition to this, they have a structural flexibility; all the elements can be exchanged if the charge balance is maintained, and they also have base and acid sites at the same time [34–36]. These properties make them good candidates to be used as catalysts [37,38] or catalytic supports. Stoichiometric Hap (Hap-S) has the chemical formula Ca10(PO4)6(OH)2 where the ratio Ca/P is of 1.67. Calcium deficient Hap (Hap-D) has a Ca/P ratio less than 1.67 (Ca10−x(HPO4)x(PO4)6−x(OH)2−x, 0 < x ≤ 1), and carbonate-rich hydroxyapatite (Hap-E) (Ca10−yNay[(PO4)6−y(CO3)y][(OH)2−2x(CO3)x]) has a Ca/P ratio higher than 1.67.
To date, few studies [24,30,39–42] investigated Hap as a catalyst or catalyst support for total VOCs oxidation. Aellach et al. [30] assessed the effect of the stoichiometry of apatite support (Hap-S and Hap-D), and that of the synthesis method (coprecipitation and impregnation) on Co-Hap materials performances towards the total oxidation of methanol. Wet impregnation of cobalt on deficient Hap support (Hap-D) leads to better catalytic results as a consequence of the better reducibility of Co3O4 species. Conversely, Nishikawa et al. [39] found that stoichiometric Hap is more active than the deficient one in toluene, ethyl acetate and iso-propanol oxidative decomposition because of the higher quantity of electrons trapped in vacancies, proved by Electron Paramagnetic Resonance (EPR), which could be responsible for the oxygen activation that oxidizes VOC. Xu et al. [40] reported the adsorption and activation of formaldehyde by hydroxyl groups bonded with the Ca2+ of the hydroxyapatites. Sun et al. [41] confirmed that a higher hydroxyl group content in Hap leads to better catalytic conversions in formaldehyde oxidation since interaction between Hap and formaldehyde was increased. Wang et al. [24] showed that gold supported on hydroxyapatites are highly active and stable catalysts for benzene and formaldehyde oxidation and that hydroxyapatites stabilized nano-gold particles via interaction with phosphate (at T < 400 °C) and hydroxyl groups (at T < 600 °C). Qu et al. [42] investigated the oxidation of formaldehyde on Hap modified by copper. They concluded that small dispersed Cu(II) clusters, formed from the substitution of Ca2+ on the surface, mainly catalyzed formaldehyde oxidation.
To the best of our knowledge, no reports in the literature deal with toluene catalytic oxidation over noble metals supported on Hap. Thus, the aim of the present study is to investigate the effect of (1) the support’s nature (different Hap supports and alumina), (2) the noble metal content, (3) the preparation method (wet impregnation or nanoparticle deposition), and (4) the noble metal nature (Pd or Au), on total oxidation of toluene. Toluene was used as a VOC model molecule because it is an aromatic molecule typically found in emissions of several processes such as printing, pressing, and petrochemical industries [43,44], and also in Diesel exhaust gases [45]. Moreover, toluene has an important Photochemical Ozone Creation Potential (POCP = 67) [46], and like benzene, it has a high chemical lifetime [44,47]. Thus, it is relevant to decrease its emissions in the atmosphere.
2 Experimental
2.1 Catalysts preparation
2.1.1 Preparation of the support material
Hydroxyapatite (Hap) was used as a support and was prepared by a coprecipitation technique according to the following equation:
| 6 (NH4)H2PO4 + 10 Ca(NO3)2 + 14 NH4OH → Ca10(PO4)6(OH)2 (Hap) + 20 NH4NO3 + 12 H2O |
A 0.1 M (NH4)H2PO4 (Sigma–Aldrich; 98%) solution was added to 0.167 M Ca(NO3)2·4H2O (Sigma–Aldrich; 99%) solution at 80 °C, under magnetic stirring, and the pH was adjusted to 10 by adding ammonia solution (25%; Verbièse). A white suspension was obtained. The suspension was further stirred for 1 h, then filtered and washed with hot deionized water. The resultant solid was dried at 80 °C for 20 h and then calcined for 4 h at 400 °C under a flow of purified air. The as-prepared solid is considered as the stoichiometric hydroxyapatite and is designated by HapS.
A deficient hydroxyapatite HapD was prepared by the same way with appropriate concentrations of (NH4)H2PO4 and Ca(NO3)2·4H2O solutions in order to obtain Ca/P = 0.9.
A sodium containing carbonate-rich apatite, denoted as HapE, was also similarly synthesized where a suitable amount of NaNO3 (PROLABO; 99%) was added to Ca(NO3)2·4H2O solution in order to get a (Ca + Na)/P = 2.2 where Ca/P = 1.8. Indeed, introducing excess of positive charges into hydroxyapatite structure leads to the incorporation of carbonate ions, from atmospheric CO2 [34]. It is worth noting that carbonate ions can substitute either the hydroxyl ions (type A) or the phosphate ions (type B) [48,49].
2.1.2 Introduction of noble metals
Two different methods were used to introduce noble metals: the conventional wet impregnation and the wet impregnation of metal (0) nanoclusters.
2.1.2.1 Conventional wet impregnation
The required amount of Pd(NO3)2·2H2O (FLUKA), or HAuCl4.3H2O (ACROS) was dissolved in excess water to give the desired Pd or the Au loading before being added to the suitable amount of Hap support. The resulting mixture was stirred at 60 °C, in a rotary evaporator, until the sample was dried and then kept for about 20 h in an oven at 80 °C, in order to have a complete evaporation of the water. The prepared catalysts were calcined at 400 °C for 4 h under air. They are designated as: Pdx/HapY or Aux/HapY, where x represents the weight percentage of noble metal, x = 0.25 and 1, and Y the type of hydroxyapatite support (S, D or E).
Pd species was also impregnated on γ-Al2O3 support for comparison purposes, with x = 0.4. The reference alumina support was supplied by Alfa Aesar (purity: 99.97%; surface area = 72 m2 g−1), and also used as received.
2.1.2.2 Hydroxyapatite-supported noble metal (0) nanoclusters
2.1.2.2.1 Preparation of Pd@HEA16Cl suspension
106.4 mg (0.304 mmol, 2 equiv) of N,N-dimethyl-N-cetyl-N-(2-hydroxyethyl) ammonium chloride salt (HEA16Cl) were dissolved in 30 mL of water (solution A). HEA16Cl is a surfactant composed of a lipophilic chain of 16 carbons with chloride as counter-ion. 14.8 mg (0.391 mmol, 2.6 equiv) of sodium borohydride, NaBH4, were added to solution A to prepare a solution called B. 44.7 mg (0.152 mmol) of Na2PdCl4 were dissolved in 10 mL of water (solution C). Solution B was added, under vigorous stirring, to solution C. The reduction of palladium occurred immediately, and the resulting suspension, Pd@HEA16Cl, was kept under stirring during 24 h.
2.1.2.2.2 Wet impregnation of the nanoparticles
1.6 g of hydroxyapatite are stirred in 10 mL of water during 2 h. Then, a given volume of Pd@HEA16Cl suspension is added to the latter mixture to yield the desired metal content. The resulting mixture is stirred for 24 h. Then, the obtained solid is filtered, washed three times with 5 mL of water, dried in an oven at 80 °C, and finally calcined under air at 300 °C.
The same procedure was applied for gold, but only on the HapD support and with a gold content of 0.25 wt%. In this case, Na2PdCl4 was replaced by the suitable amount of HAuCl4.3H2O.
The as-prepared catalysts will be designated hereafter by Pdx@HapY (x = 0.25 and 0.5) or Au0.25@HapD.
2.2 Characterization techniques
Elemental analysis of noble metal loading and bulk Ca/P ratio was performed, on the calcined solids, using inductively coupled plasma – optical emission spectroscopy (ICP-OES) on an Agilent Technologies 700 Series spectrometer. Analysis was carried out at the REALCAT platform (Lille 1 University). Prior to analysis, the solids were treated by a mixture of concentrated hydrochloric and nitric acids (70%–30%).
Structural analyses were carried out by X-ray diffraction (XRD), at ambient temperature, of the calcined solids. Powder X-ray diffraction was recorded with Cu Kα (λ = 1.5418 Å) radiation on a D8 Advance Bruker diffractometer. The crystalline phases were determined by the comparison of the registered patterns with the ICDD-JCPDS powder diffraction files processed on EVA software. The diffraction patterns were recorded in the 2θ range 10–80° with a step of 0.02° and a count time of 1.5 s. The Fullprof Suite program was used for Rietveld refinement [50]. The Thompson–Cox–Hastings pseudo-Voigt function was chosen for describing the peak profiles. LaB6 was used as a standard to derive the instrument resolution [51]. Here, an isotropic size-broadening model based on linear combination of spherical harmonics was used to simulate the size broadening, and 6 additional parameters were refined. Out of this refinement, the individual size was derived for each crystallographic plane.
The textural properties were studied by N2 adsorption–desorption measurements at liquid nitrogen temperature. The experiences were performed in a Micromeritics TriStar-II Surface Areas and Porosity equipment. Before starting the analysis, the samples were degassed at 150 °C in vacuum. The sample weight was about 500 mg.
X-ray photoelectron spectra (XPS) were recorded at ambient temperature with a residual pressure of 10−9 mbar on two spectrometers: VG ESCALAB 220XL and KRATOS, AXIS Ultra with monochromated Al Kα anode (1486.6 eV) and a hemispherical analyzer with constant ΔE/E. The source power was kept at 300 W for ESCALAB and at 150 W for KRATOS.
All binding energies were calibrated by using contaminant carbon as an internal standard (C1s = 285 eV). Peak fitting was processed with CasaXPS. The fitting procedure allowed to determine the peak position, height, and width.
2.3 Catalysts evaluation
Catalytic oxidation runs for abatement of toluene were performed in a continuous flow fixed bed Pyrex micro-reactor at atmospheric pressure. About 0.2 g of catalyst was placed in the reactor for each run. To obtain accurate and stable gas flow rates, the mass flow controllers were used. The concentration of toluene was 800 ppm, which was controlled by the temperature of a home-made saturator and the additional air stream. The flow rate of the gas mixture through the reactor was 100 mL min−1, which gave a Gas Hourly Space Velocity (GHSV) of 15,000 h−1. All the lines were sufficiently heated at 120 °C to prevent the adsorption and condensation of toluene and water in the tubes.
The micro-reactor is placed in an electrical furnace which provides the required temperature for catalytic reaction. Catalysts were evaluated in a temperature range of 300 °C–room temperature, with a rate decrease of 0.5 °C min−1. Before each run, the samples were pretreated for 4 h at 300 °C under an air flow of 75 mL min−1.
For the stability tests, the catalyst was fed with the reactant mixture at 165 °C, and the evolution of toluene conversion was followed for the next 45 h at the same temperature.
The concentrations of the inlet and outlet gas stream were analyzed online by Gas Chromatography (7860A Agilent Gas Chromatograph) equipped with a Thermal Conductivity Detector (TCD) and Flame Ionization Detector (FID) and with two columns: Restek Shin Carbon ST/Silco HP NOC 80/100 micropacked, to separate permanent gases (Air, CO and CO2) and a capillary column Cp-Wax 52 CB25 m, Ø = 0.25 mm, to separate hydrocarbons and aromatic compounds.
Toluene conversion (Ct) was calculated as follows:
The selectivity towards CO2 (SCO2) was determined by:
The specific rate r was taken as:
3 Results and discussion
3.1 Characterization of the supports
Chemical analysis results, BET surface areas and pore volumes of the supports are summarized in Table 1.
Chemical analysis of HapY supports and their textural properties.
| Catalyst | Theoretical Ca/P | Experimental Ca/P | Surface area (m2 g−1) | Pore volume (cm3 g−1) |
| HapS | 1.67 | 1.64 | 109 | 0.68 |
| HapD | 0.9 | 1.56 | 118 | 0.76 |
| HapE | 1.8 | 1.71 | 107 | 0.59 |
The ICP results show that HapS is nearly stoichiometric since its Ca/P ratio is close to 1.67. HapD is slightly deficient with Ca/P of 1.56 but not as lower as the targeted value of 0.9. HapE showed Ca/P greater than the stoichiometric value. Aellach et al. [30], Silvester et al. [36] and Lamonier et al. [37] obtained similar results. It is worth noting that even if we obtained deficient and rich hydroxyapatites, a difference exists between the experimental ratios Ca/P and the targeted ones. Diallo-Garcia [52] suggested that whatever the initial ratio of the reactants is, there is a trend towards the formation of the most stable phase, i.e., the stoichiometric one. Hence, by a simple adjustment of the initial reactants proportions, as in our work, it is hard to obtain hydroxyapatites with important deficiency or important excess of carbonates [52]. Some studies proved that parameters such as the pH [35,53] and the temperature of the synthesis [53] play an important role on the Ca/P ratio. Silvester et al. [36] also reported that the increase in the Ca/P ratio was accompanied by a linear increase in the carbon content (not analyzed in our samples), which could originate from the substitution of phosphate ions, located in the apatite structure, by carbonate ones.
Specific surface areas of the hydroxyapatite supports range between 110 and 120 m2 g−1. The value does not significantly differ between the different supports. However, HapD shows a trend to have a slightly higher surface area. It is to be noted that the values we got are higher than those found by some researchers who prepared and calcined apatites at 400 °C [30,37,54,55]. They are rather close to those of Silvester et al. [36].
The volumes of the pores of the supports are in the range of 0.6–0.8 cm3 g−1, where HapD shows a slightly higher value than that of the other supports.
The X-ray diffractograms of the different HapY supports are depicted in Fig. 1. All of the pure hydroxyapatites present diffraction lines due to crystalline hydroxyapatite phase Ca5(PO4)3OH (JCPDS n° 01-086-0740) [56]. The three prepared HapY crystallize in a hexagonal system with P63/m space group. All of the diffractograms are similar and no crystalline phase other than hydroxyapatite was detected.
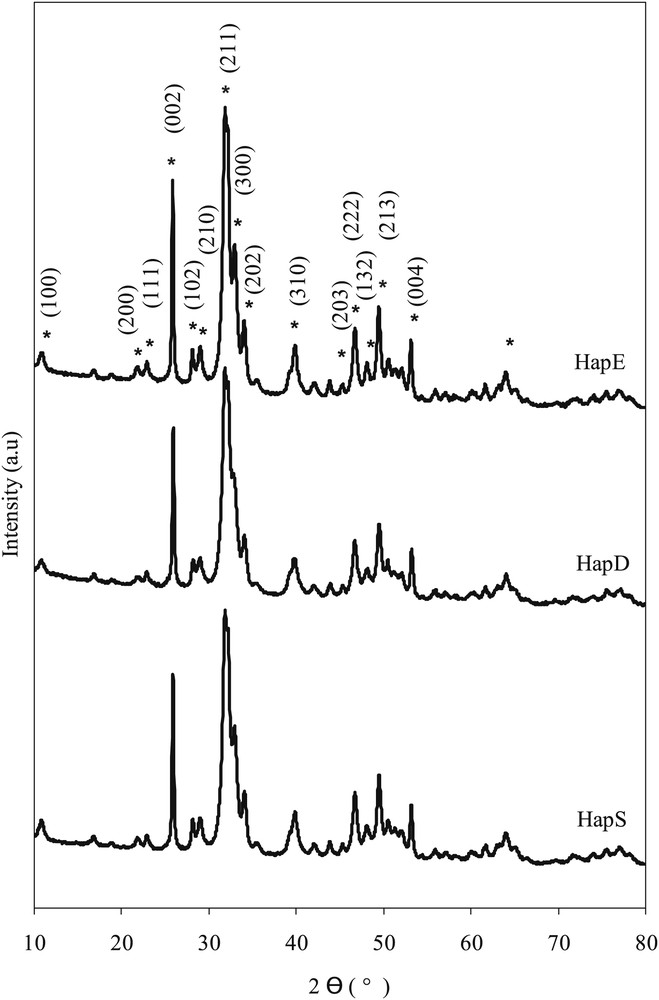
X-ray diffractograms of the HapS, HapD, and HapE materials.
By using the structure model proposed by Hughes et al. [56], the apatite structure was refined in the P63/m space group. A small difference was noticed between the value observed and the theoretical one. The cell parameters, as well as the crystallite sizes, were deduced from the Rietveld refinement and listed in Table 2. The cell parameters were deduced from the diffraction lines of (300) crystallographic plane at 32.9° and of (004) crystallographic plane at 53.1°. The crystallite sizes were deduced from (001) and (100) crystallographic planes. The information on (001) plane was deduced from (002) one at 25.8° (Fig. 1). The plane (100) is at 10.9° (Fig. 1).
Cell parameters and crystallite size of HapY supports determined using Rietveld refinement.
| Sample | Cell parametersa | Crystallite size (nm) corresponding to the crystallographic plane | Length/thickness ratio | ||
| a (±0.002 Å) | c (±0.002 Å) | (001) | (100) | (001)/(100) | |
| HapD | 9.4293 | 6.8830 | 28 | 8 | 3.5 |
| HapS | 9.4163 | 6.8873 | 31 | 9 | 3.4 |
| HapE | 9.4101 | 6.8915 | 33 | 10 | 3.3 |
a a calculated from the (300) line and c calculated from the (004) line.
When the Ca/P ratio increases, the amount of carbonates increases – as stated in our introduction and in reference [36] – and then the cell parameter a decreases. This observation could be explained by the fact that carbonate ions, which are smaller than phosphate ones, partially substitute these latter in the apatitic structure (type B substitution) [36,37,57].
In addition, it is to be noted here that the cell parameter c is slightly higher for the sodium containing apatite, HapE, than for the other apatites. Such a result which reveals an expansion in cell volume was already reported [36,37,58,59] for sodium containing apatites.
Cheng et al. [60] demonstrated that the substitution-type has different impacts on the Hap structure: type-A substitution leads to an elongation according to the a-axis and a contraction according to the c-axis, whereas type-B substitution leads to the reverse effect. The B substitution will lower the crystallinity of the Hap.
Moreover, as it is shown in Table 2, the crystallite size increases with the ratio Ca/P whereas the length/thickness ratio follows the reverse order (3.5 for HapD, 3.4 for HapS and 3.3 for HapE). This suggests that the anisotropic elongated shape of HapY crystallites became more spherical with the increase of Ca/P ratio. Our results are in good agreement with those of Silvester et al. [36] who showed an increase of the crystallite size with an increase of Ca/P ratio or carbonate content. Moreover, these researchers proved the elongated anisotropic shape of the HapY crystallites and found it to become more spherical for carbonate-rich apatites. It is to be reminded that in our case, HapE is supposed to be the support the most rich in carbonates and it has the highest Ca/P.
3.2 Effect of support nature
In our previous work [61], the different supports HapY were tested in toluene total oxidation and showed no toluene conversion between 100 and 250 °C. No oxidation occurs below 250 °C for HapE and below 290 °C for HapS and HapD [61]. It is also noted that without a catalyst, no reaction occurs at all at a temperature that ranges between 50 and 400 °C [61].
Light-off curves of Pd0.25/HapY and of palladium impregnated on a classical alumina support are plotted in Fig. 2. T10 and T90, the temperatures at which respectively 10% and 90% of toluene is converted, are listed in Table 3.
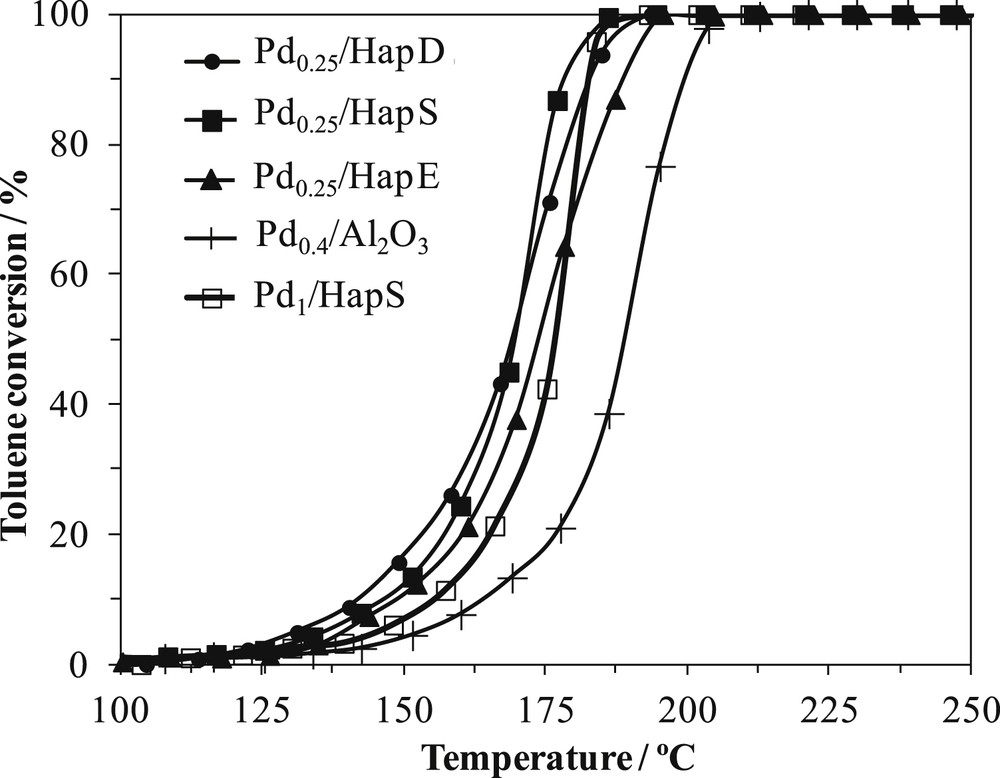
Toluene conversion versus reaction temperature on: Pd0.25/HapY, Pd1/HapS, and a classical catalyst Pd0.4/Al2O3.
Temperatures at 10% and 90% toluene conversions, respectively T10 and T90 (°C), and the specific rates r (mol toluene. h−1) at 150 °C for the different studied catalysts.
| Sample | T10 (°C) | T90 (°C) | r at 150 °C (mol toluene h−1) |
| Pd0.25/HapS | 146 | 178 | 0.05 |
| Pd0.25/HapD | 141 | 182 | 0.06 |
| Pd0.25/HapE | 150 | 190 | 0.04 |
| Pd1/HapS | 154 | 181 | 0.007 |
| Pd0.4/Al2O3 | 163 | 198 | 0.01 |
| Pd0.25@HapE | 147 | 180 | 0.04 |
First, Pd addition, even with a low content of 0.25 wt%, enhanced the toluene conversion of the supports; toluene is not oxidized below 250 °C on the HapY supports [61], whereas, T90 is in the range of 178–190 °C, when 0.25 wt% of Pd is present. Many scientists got such results [5,62–66]. Second, Pd0.25/HapY are more active than Pd0.4/Al2O3. Therefore, our supports with low palladium content are more performant than classical alumina one with a two-fold higher palladium content. Third, it could be said that Pd0.25/HapD and Pd0.25/HapS have similar toluene conversions with a trend for Pd0.25/HapD to be more active than Pd0.25/HapS at lower reaction temperatures, where toluene conversions are low, and to be slightly less active than it at higher reaction temperatures. Pd0.25/HapE is rather less active than both other apatites supporting palladium. It is worth mentioning that the toluene conversion difference between the different HapY supports is slight and could be linked to the slight difference in their Ca/P ratio which will result in a slight difference in their chemical properties. The comparison of the specific rates (r) obtained on the different studied systems at 150 °C (Table 3) confirmed the aforementioned trends: r increased by four to six times when the reaction was performed on Pd0.25/HapY instead of Pd0.4/Al2O3.
In addition, for low conversions, toluene was mainly oxidized into CO2. Secondary products obtained are benzene and some traces of non-identified hydrocarbons. No CO was detected consistent with the ability of supported Pd catalysts to catalyse the CO oxidation [67–70]. When toluene conversion reaches 100%, selectivity towards CO2 was 100%, in all the studied cases. Rooke et al. [64] obtained, in the presence of noble metals, some CO and benzene as major by-products during toluene total oxidation.
Okumura et al. [28] investigated the metal–support interaction between palladium and the different oxide supports (MgO, Al2O3, SiO2, SnO2, ZrO2, Nb2O5, WO3) with different acid-base properties. They reported that the combustion activity over Pd loaded on the strongest acidic (WO3) or basic (MgO) support was lower than when it was over Pd on weak acid-base ones (SnO2, Al2O3, SiO2 and Nb2O5). Therefore, it was considered that the combustion activity of Pd was controlled through the acid-base property of the support and through the electronic interaction between support and Pd which is linked to palladium dispersion on the support.
Furthermore, PdO formation on the hydroxyapatite surface was evidenced, in all our samples, hereafter, by XPS. No Pd(0) was detected. Hence, it could be postulated that PdO interaction with acidic supports leads to a slight enhancement of toluene oxidation. Indeed Popova et al. [8] and Aellach et al. [30] emphasized the role of the acidic centers of the catalysts in the toluene oxidation. The activity in total toluene oxidation is related to the interaction of the aromatic electrons of toluene with these acidic centers, increasing the possibility of an electrophilic attack of adsorbed oxygen and combustion of toluene molecules, the electrons trapped in vacancies of apatites are supposed to be responsible for oxygen activation [39].
Moreover, Tidahy et al. [29] reported that the activity of Pd should be controlled by the reducibility of PdO by the hydrocarbon and then through the electronic interaction between the support and palladium. Since the reduction step is an important factor for catalytic activity in total oxidation reactions, these researchers concluded that the pre-treatment of the used support could influence the electronic interaction between palladium particles and the corresponding support.
3.3 Effect of palladium content
Fig. 2 also compares toluene conversion data over two different palladium contents loaded on the same support, HapS. As already mentioned, adding Pd improved significantly support ability to oxidize toluene, since toluene conversion was of 100% at 200 °C. However, increasing palladium loading from 0.25 wt% to 1 wt% does not lead to a better enhancement in the catalytic oxidation of toluene. The same trend was observed whatever the support nature or the preparation method used was (results not shown). The specific rate on Pd0.25/HapS decreased about seven times (Table 3) when palladium content was increased to 1 wt%, Pd1/HapS. For 100% toluene conversion, toluene was selectively converted into CO2 and H2O.
0.5 wt% Pd/LaMnO3 was slightly less active than 0.1 wt% Pd/LaMnO3 versus toluene oxidation according to the work of Musialik-Piotrowska et al. [71]. However, Hosseini et al. [63] obtained a higher activity for 1.5 wt% Pd over nanostructured mesoporous TiO2–ZrO2 (20 wt%–80 wt%) than for 0.5 wt% Pd one. Besides, Okumura et al. reported that when Pd dispersion decreased, the catalytic activity in the toluene oxidation decreased [28]. A comparison of the specific surface areas of the reference [71] – range 15–20 m2 g−1 – with those of the reference [63] – about 372 m2 g−1 – , leads us to conclude that even when the palladium content is increased, the palladium species are still well dispersed on TiO2–ZrO2, whereas when more palladium is loaded on LaMnO3, palladium was no more dispersed and thus some palladium species were probably not accessible to the reactants and consequently no enhancement in toluene conversion could be observed. Then higher palladium loading on hydroxyapatite led certainly some palladium sites to be not accessible to reactants.
Li et al. [72] investigated catalytic oxidation of toluene over Pd-based FeCrAl wire mesh monolithic catalysts prepared by electrodes plating method, with palladium loadings of 0, 0.1, 0.2, 0.3, 0.4, and 0.5 wt%. For a catalyst without palladium, no toluene conversion was denoted. For 0.1 wt% of palladium, T50 was about 215 °C. Increasing palladium loading to 0.2 wt% leads to a shift of T50 to 206 °C. A further increase to 0.5 wt% does not lead to significant improvement. Thus, these researchers regarded 0.3–0.4 wt% as a suitable palladium loading.
3.4 Effects of preparation method and nature of noble metal
The two preparation methods we used were compared in terms of toluene conversion versus reaction temperature in Fig. 3. Light-off temperatures are slightly shifted to lower values for nanoparticle deposition method when toluene conversions become higher than 20%. For lower toluene conversions, both catalysts exhibit similar behaviors. Selectivity towards CO2 and other products are comparable between the two preparation methods.
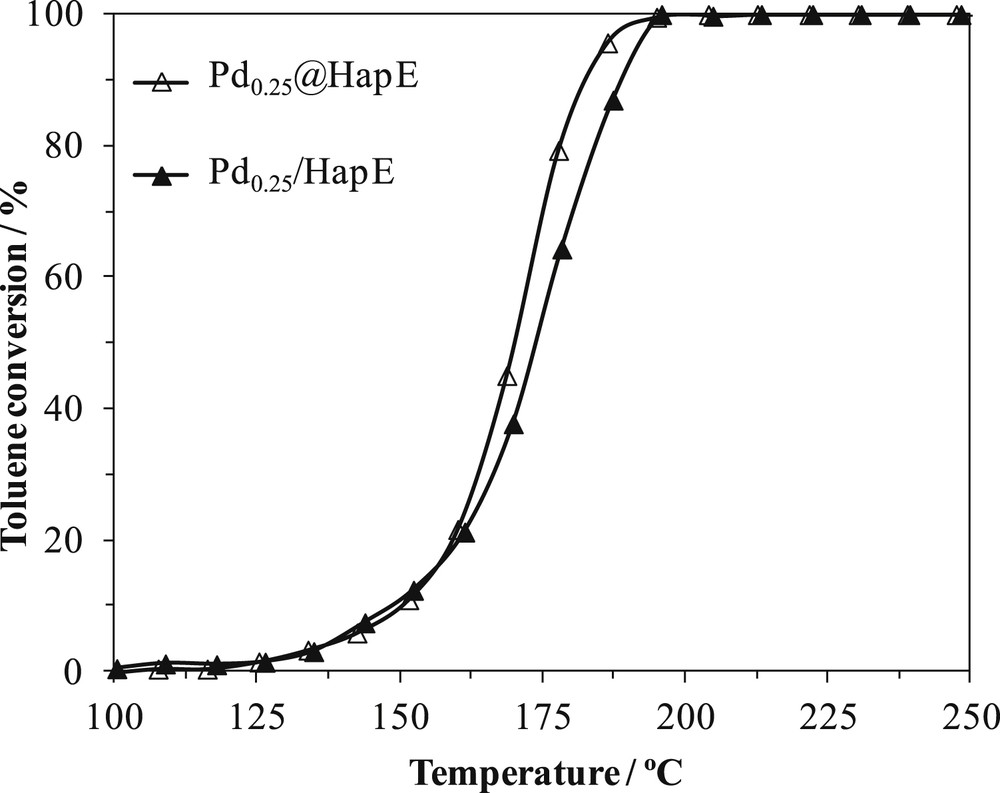
Toluene conversion versus reaction temperature on Pd0.25/HapE and Pd0.25@HapE.
Since a catalytic result similar to that of conventional impregnation was obtained, a palladium colloidal suspension can be successfully used to deposit Pd nanoparticles on a hydroxyapatite support. To the best of our knowledge, this type of impregnation on hydroxyapatite support has never been used, and further investigations are needed to improve this methodology approach. Indeed, for higher Pd loading (0.5 wt%), it has been difficult to deposit the targeted Pd content (see the characterization part).
The toluene conversion versus reaction temperature is shown in Fig. 4 for catalysts loaded with Pd and Au. It is clearly seen that Au0.25@HapD is far less active than Pd0.25@HapD. Toluene oxidation starts at 250 °C on Au0.25@HapD and the conversion is only at about 10% at 300 °C whereas at 200 °C it is at 100% on Pd0.25@HapD.
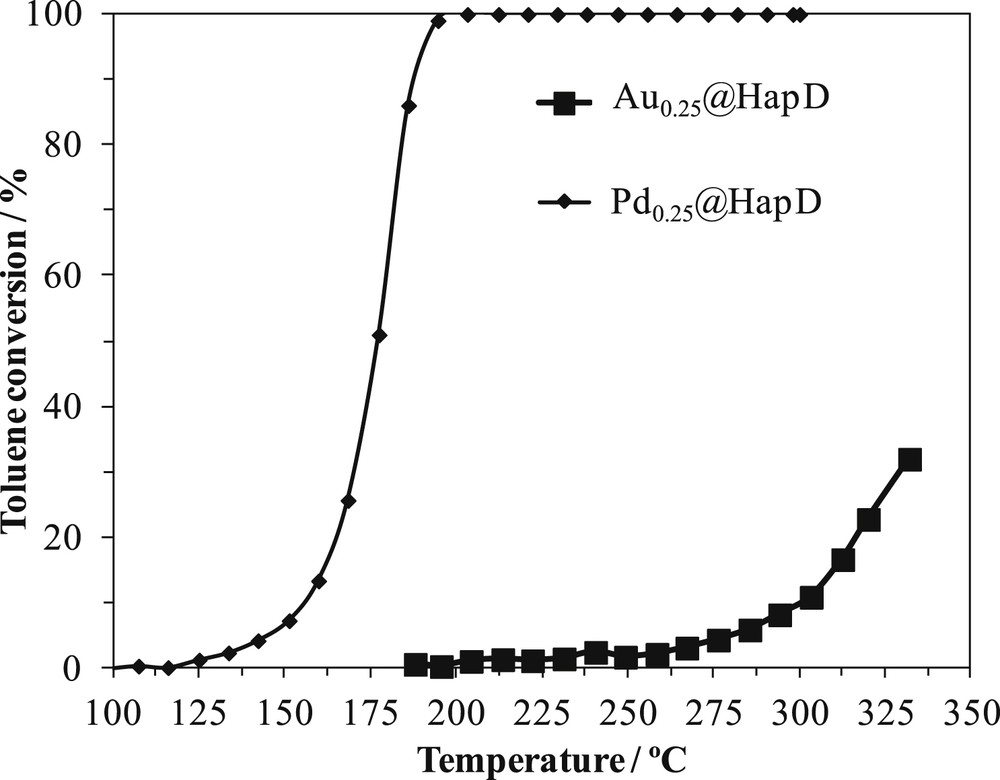
Toluene conversion versus reaction temperature on Au0.25@HapD and Pd0.25@HapD.
Many researchers obtained better catalytic activities for palladium-supported catalysts than gold ones in total oxidation of toluene, independently of the preparation method [5,62,63,65,66,73]. They explained the as-obtained catalytic trend by the oxidation mechanism. They suggested that VOC oxidation over supported noble metal catalysts usually involves the dissociative adsorption of oxygen [12,62,63]. They found, accordingly, that oxygen chemisorption energy is lower for gold catalysts than for other studied noble metals, including palladium, and that lower chemisorption energy was linked to a lower intrinsic activity [62]. Furthermore, some other researchers [74] proved by 18O/16O isotopic exchange study that gold did not promote the dissociation of dioxygen.
Furthermore, it is now well-known that the catalytic activity and selectivity of gold-based catalysts strongly depend on particles size, which in turn, depends mainly on the preparation method. Gold is catalytically active only under the form of ultrafine highly dispersed particles on metallic oxide supports [62–64]. Conventional preparation methods, such as wet impregnation, one of the methods used in our case, do not lead to the formation of such active particles [75]. Sintering of gold nanoparticles is one of the most important limitations to their catalytic performances. Our results show that even incorporating gold by colloidal suspension does not lead to good catalytic results. It could be then suggested that the latter preparation method does not give rise to the formation of highly dispersed gold nanoparticles. Of course, more deep characterization of gold catalysts should be carried out in order to check this hypothesis and also to verify the eventual influence of other parameters (oxidation state, support–metal interaction, reducibility…) on gold catalytic performances in our case.
Another explanation that could be postulated to explain the lower toluene conversion on gold impregnated supports compared to palladium homologous ones is the presence of chloride which is known to cause sinterization of gold particles, thus turning them less active or even inactive [55,58].
3.5 Characterization of noble metal based catalysts
Since gold-based catalyst showed lower reactivities than palladium ones towards toluene oxidation, characterization has been focused on palladium-based materials. The results of chemical analysis of palladium-based catalysts are summarized in Table 4.
Chemical analysis and textural properties of Pd-based hydroxyapatites.
| Catalyst | Experimental content of noble metal (%) ICP | Surface area (m2 g−1) | Pore volume (cm3 g−1) |
| Pd0.25/HapS | 0.24 | 93 | 0.59 |
| Pd0.25/HapD | 0.26 | 95 | 0.72 |
| Pd0.25/HapE | 0.30 | 99 | 0.52 |
| Pd1/HapS | 0.81 | 91 | 0.57 |
| Pd0.25@HapD | 0.22 | 92 | 0.55 |
| Pd0.25@HapE | 0.23 | 96 | 0.53 |
| Pd0.5@HapE | 0.34 | 92 | 0.55 |
Chemical analyses show that the amount of Pd found in the low Pd loading samples is generally in agreement with the quantities introduced in solutions. However, for high Pd content (Pd1/HapS and Pd0.5@HapE), palladium loading is somehow lower than the nominal content.
X-ray diffractograms of the different noble metal based catalysts are the same as those of the supports (figures not shown). After impregnation of noble metals, no crystalline phase other than Ca5(PO4)3OH was detected by XRD and hence the crystalline structure of the hydroxyapatite did not change as a consequence of noble metal addition. No diffraction line due to PdO or Pd(0) species was observed. Similar results after noble metal impregnation on hydroxyapatite have been obtained by different research groups [76–78]. They reported that impregnation of noble metals did not change the support structure, and that no XRD line ascribable to noble metal or oxide phase was observed because, probably, of the relatively low noble metal amount and/or the too small size of the diffracting domains.
Specific surface areas and the volumes of the pores are also listed in Table 4.
Surface areas of the supports decreased upon introduction of noble metals, whatever the preparation method was. When the metal content increases, in the range we studied, it is not accompanied by a further decrease in surface area since Pd0.25/HapS and Pd1/HapS show similar surface areas, also Pd0.25@HapE and Pd0.5@HapE. Pore volumes of the different HapY slightly decreased after noble metal introduction. All these results are consistent with the results obtained by Cheikhi et al. [55] who studied different palladium loadings on Hap. In a previous study [61], when we impregnate 2.5 wt% copper on different HapY, we obtain specific surface areas slightly lower than those of the corresponding supports and pore volumes similar to those of them. This observation was explained by the high dispersion of copper as suggested by XRD study [61]. A similar explanation could be stated for our present catalysts.
Fig. 5 gives examples of the XPS spectra obtained for our samples. All our palladium-based samples exhibited the same XPS spectra (not all shown in Fig. 5). A peak centered at 336.9 eV was obtained and assigned to Pd3d5/2 core level. This binding energy value is similar to those reported for Pd3d5/2 of PdO in the literature [28, 79–81]. No peak due to other palladium oxidation states was detected. The Full Width at Half Maximum (FWHM) of the XPS peak was nearly the same in all the studied cases. Thus, our two synthesis methods lead to PdO and not to Pd(0) formation. It seems that Pd(0) anchored on hydroxyapatites by the metal nanoparticle deposition method was oxidized, at least, for the species present on the surface, into PdO, after calcination under air. It is also noted that no chloride species was evidenced on the surface of our Pdx@HapY samples. It is then concluded that all chloride ions, whose origin is the precursor used, were removed by filtration and subsequent washings. XPS study of Pd introduced by different methods on mixed cobalt-aluminum oxide prepared by hydrotalcite route: coprecipitation, wet impregnation, ion exchange, and thermal combustion, also revealed the presence of PdO as the only surface palladium species [81].
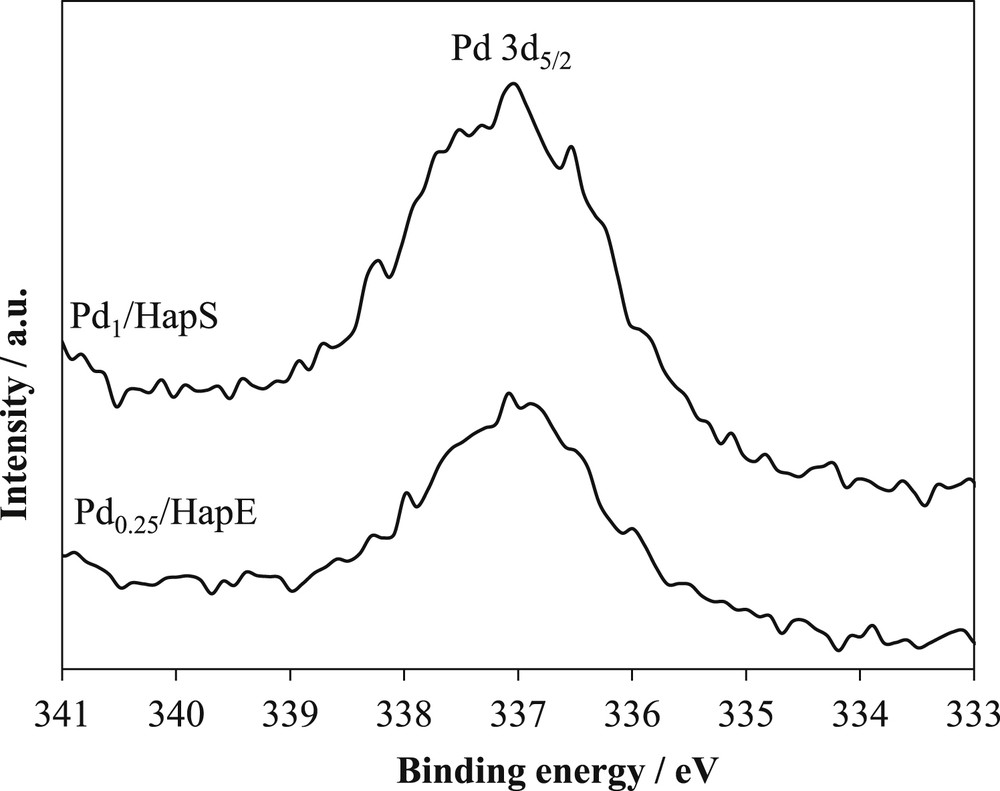
Pd3d5/2 XPS spectra of Pd1/HapS and Pd0.25/HapE materials.
Table 5 summarizes binding energies and atomic surface ratios given by XPS.
Binding energies, atomic surface ratios, and Full Width at Half Maximum (FWHM) data from XPS.
| Sample | Binding energy (eV) | Atomic ratios | |||||||
| Ca 2p | P2p | O1s | N1s | Pd3d5/2 (FWHM/eV) | Ca/P XPS (ICP) | Pd/(Ca + P) XPS (ICP) | Pd/Ca XPS (ICP) | Pd/P XPS (ICP) | |
| Pd1/HapS | 347.7 | 134.0 | 531.4 | 399.4 | 336.9 (1.9) | 1.46 (1.64) | 0.010 (0.007) | 0.017 (0.010) | 0.024 (0.017) |
| Pd0.25/HapS | 347.6 | 133.6 | 531.9 | 399.8 | 337.1 (2.5) | 1.38 (1.64) | 0.005 (0.002) | 0.008 (0.003) | 0.012 (0.005) |
| Pd0.25/HapE | 347.4 | 133.5 | 531.4 | 399.9 | 337.1 (1.7) | 1.50 (1.71) | 0.008 (0.002) | 0.013 (0.003) | 0.019 (0.005) |
| Pd0.25@HapE | 347.2 | 133.1 | 531.2 | – | 336.6 (2.2) | 1.46 (1.71) | 0.001 (0.002) | 0.002 (0.004) | 0.003 (0.002) |
A presence of N species is detected on the surface of the impregnated samples. N could be due to some residual nitrate coming from Pd(NO3)2.2H2O, the palladium precursor used in this method, not fully decomposed by calcination.
The Ca/P ratio obtained from XPS is lower than that obtained from ICP. Similar results were obtained by Diallo-Garcia [52] and Silvester et al. [36] and were explained by the fact that Ca species are less exposed to the surface than phosphates, at least within the 10 nm analysis depth limit of the XPS technique. Conversely, Pd/(Ca + P), Pd/Ca, and Pd/P XPS ratios are higher than those of ICP ones, indicating that both Ca and P of the surface were covered by Pd species. However, Pd/(Ca + P) and Pd/Ca obtained by XPS for Pd0.25@HapE are lower than those of ICP. It is thought that some diffusion (of palladium species towards the bulk, and of calcium species towards the surface) could occur. This hypothesis should be later verified.
3.6 Stability tests
The stability of the catalysts is crucial for a practical application. Fig. 6 shows, for Pd0.25/HapE and Pd0.25@HapE, the evolution of toluene conversion versus reaction time at 165 °C for 45 h. Pd0.25@HapE exhibits a decrease of toluene conversion from 30% to 10% during the first three hours under the stream. A stabilization of the value of toluene conversion followed this decrease. Conversely, Pd0.25/HapE showed a fluctuation of the toluene conversion between 10 and 20% during the first three hours of the reaction, and later a stabilization of the conversion at approximately 10%. After about 13 h under the stream, toluene conversion increases gradually to reach 100% over this catalyst at the 25th hour and remained stable till the end of the stability test. The deactivation process observed during the first three hours for both catalysts could be explained by coke formation [82], by the blocking of the active palladium sites by water [83], and by poisoning or fouling of the catalyst [84]. Nevertheless, some researchers stated that the partial deactivation by water vapor of Pd supported on mixed cerium-zirconium-yttrium oxide support is reversible [85]. The characterization of our systems after 3 h under stream should be carried out in order to clarify the reason of the observed deactivation.
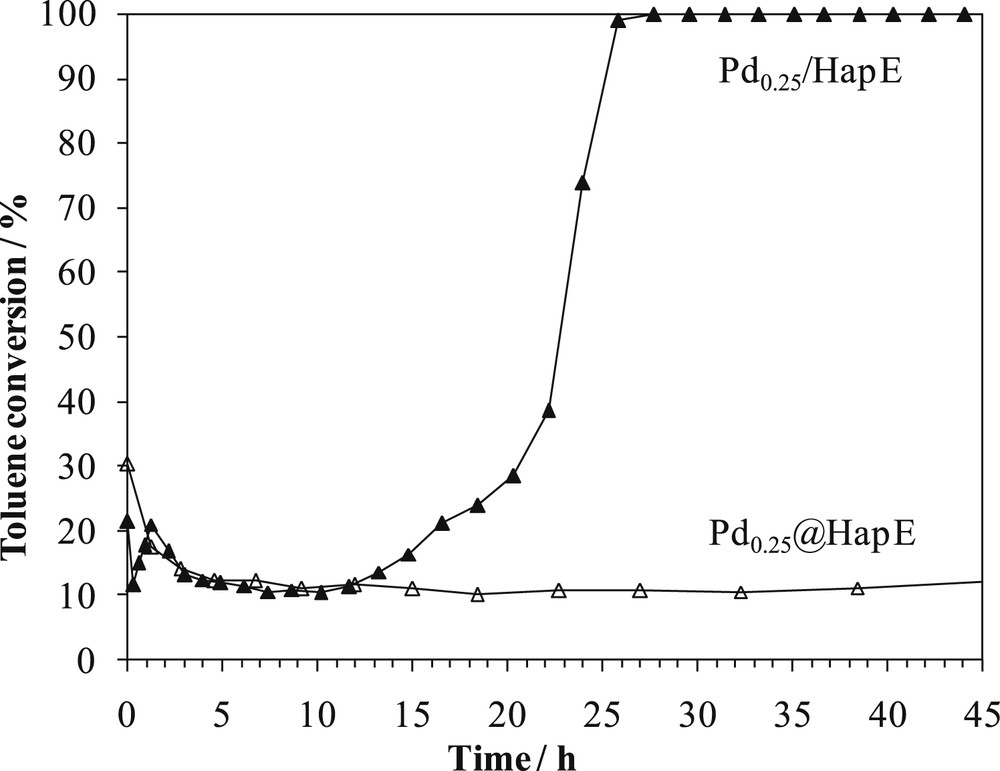
Time-on-stream plot of toluene conversion over Pd0.25/HapE and Pd0.25@HapE catalysts. Feed composition: 800 ppm toluene in air, temperature: 165 °C.
However, after 13 h under the stream, Pd0.25/HapE showed a non-conventional behavior. Such behavior was not very common in literature where either no deactivation or some deactivation was noted for palladium-based catalysts under a stream of VOC diluted in air. As we know, only Li et al. [81] obtained a behavior, under stream of 800 ppm of toluene diluted in air, similar to that of Pd0.25/HapE in our case during their work on Pd/Co3AlO (mixed cobalt-aluminum oxide prepared by hydrotalcite route) where palladium was introduced in the system according to three different methods: impregnation, wet ion exchange, and thermal combustion. These researchers performed an XPS study to determine the states of Pd on the surface before and after the stability tests. Before the test, only PdO species were evidenced and no Pd(0) was detected. After stability tests, both PdO and Pd(0) were evidenced on the catalysts' surfaces. Hence, the higher catalytic reactivity after some time under stream was explained by the existence of Pd(0) besides PdO. Indeed, many researchers demonstrated that the presence of both PdO and Pd(0) is responsible for the good performances in oxidation reactions [83, 86–88]. PdO was partially reduced into Pd(0) under toluene/air flow and therefore the catalytic activity was improved. However, the catalyst was neither fully reduced because of oxygen presence nor fully oxidized because of the continuous feed of toluene [81]. It took Li et al. [81] 5 h at 240 °C and it took us 13 h at 165 °C to reach the chemical equilibrium of PdO–Pd(0) in toluene/air atmosphere.
Up till now, we did not have a clear explanation of the difference between the behaviors under time on stream of our two catalysts. We could suggest that Pd0.25@HapE needs more time to reach the equilibrium PdO–Pd(0), probably because palladium and support interaction is different than in Pd0.25/HapE. This hypothesis is suggested by the previously mentioned XPS results where some palladium species is thought to diffuse towards the bulk. Yet it should be verified later by different characterization studies before and after stability tests.
3.7 Comparison of our catalytic systems with literature ones
The reaction temperature, at which 50% of inlet toluene was oxidized, T50, was used to compare the performances of our palladium catalysts with other palladium ones studied in literature in total oxidation of toluene, in conditions similar to ours. The results are summarized in Table 6 from which we concluded that our systems are among the most active found in literature. The palladium catalysts which exhibited a catalytic behavior similar to or higher than ours are mainly composed of mesoporous supports or of zeolite supports with high surface areas (Pd/HFAU, Pd/NaFAU and Pd/CsFAU, with 414–714 m2 g−1) [89,90]. Hence, to enhance our catalytic performances, we suppose to synthesize mesoporous hydroxyapatite. By such a synthesis, an increase in surface area of the apatite supports will occur leading to an increase of palladium dispersion.
Pd weight percentage (wt%), Pd dispersion (%), toluene content in air (ppm), Gas Hourly Space Velocity GHSV (h−1) or Volume Hourly Space Velocity VHSV (m3 h−1), and temperature at 50% toluene conversion, T50 (°C), on our catalysts and different palladium systems from the literature.
| Catalyst | Pd (wt%) (% dispersion) | Toluene (ppm) | GHSV (h−1) (VHSV) (m3 h−1) | T50 (°C) | Reference |
| Pd/HapS | 0.25 | 800 | 15,000 | 168 | The present work |
| Pd/HapD | 0.25 | 800 | 15,000 | 168 | The present work |
| Pd/HapE | 0.25 | 800 | 15,000 | 174 | The present work |
| Pd@HapE | 0.25 | 800 | 15,000 | 168 | The present work |
| Pd/HBEA | 0.5 (18) | 1000 | (60) | 159 | 65,90 |
| Pd/NaBEA | 0.5 (19) | 1000 | (60) | 177 | 65,90 |
| Pd/CsBEA | 0.5 (28) | 1000 | (60) | 230 | 65,90 |
| Pd/HFAU | 0.5 (21) | 1000 | (60) | 162 | 65,89–90 |
| Pd/NaFAU | 0.5 (25) | 1000 | (60) | 155 | 65,89–90 |
| Pd/CsFAU | 0.5 (12) | 1000 | (60) | 152 | 65,89–90 |
| Pd/ZrO2 | 0.5 (15) | 1000 | (60) | 196 | 65,90 |
| Pd/TiO2 macro mesoporous | 0.5 | 1000 | (60) | 230 | 5,65,90 |
| Pd/TiO2–ZrO2 (80/20) | 0.5 | 1000 | (60) | 251 | 63,65 |
| Pd/TiO2–ZrO2 (50/50) | 0.5 | 1000 | (60) | 256 | 63,65 |
| Pd/TiO2–ZrO2 (20/80) | 0.5 | 1000 | (60) | 280 | 63,65 |
| Pd/TiO2 (non porous) | 0.5 | 1000 | (60) | 240 | 73 |
| Pd/Nb2O5(5%)–TiO2 | 0.5 | 1000 | (60) | 210 | 73 |
| Pd/V2O5(5%)–TiO2 | 0.5 | 1000 | (60) | 201 | 73 |
| Pd/Ta2O5 prepared from Ta(OEt)5 | 0.5 | 1000 | (60) | 207 | 91 |
| Pd/mesoporous ZrO2 | 0.5 (54) | 1000 | (60) | 250 | 29 |
| Pd/mesoporous ZrO2-400 | 0.5 (64) | 1000 | (60) | 240 | 29 |
| Pd/mesoporous ZrO2-600 | 0.5 (40) | 1000 | (60) | 155 | 29 |
| Pd/ZrO2 ref. | 0.5 (30) | 1000 | (60) | 180 | 29 |
| Pd/Hierarchically porous Ta2O5 | 0.5 | 1000 | (60) | 210 | 92 |
| Pd/Hierarchically porous Nb2O5 | 0.5 | 1000 | (60) | 210 | 92 |
| Pd/MgO | 0.5 (14.1) | 9500 | (1200) | 525 | 28 |
| Pd/Al2O3 | 0.5 (4.1) | 9500 | (1200) | 325 | 28 |
| Pd/ZrO2 | 0.5 (16.5) | 9500 | (1200) | 280 | 28 |
| Pd/SiO2 | 0.5 (21.5) | 9500 | (1200) | 325 | 28 |
| Pd/SnO2 | 0.5 (1.2) | 9500 | (1200) | 375 | 28 |
| Pd/Nb2O5 | 0.5 (3.6) | 9500 | (1200) | 325 | 28 |
| Pd/WO3 | 0.5 (3.1) | 9500 | (1200) | 425 | 28 |
| Pd/ZrO2 | 0.5 (16.5) | 9500 | (1200) | 277 | 28 |
| Pd/TiO2 | 0.5 | 1000 | (60) | 200 | 64 |
| Pd/Nb2O5 (3%)–TiO2 | 0.5 | 1000 | (60) | 235 | 64 |
| Pd/CeO2 (0.5%)–TiO2 | 0.5 | 1000 | (60) | 220 | 66 |
| Pd/CeO2 (5%)–TiO2 | 0.5 | 1000 | (60) | 217 | 66 |
| Pd/TiO2 IMP | 1 (14-16) | 975 | 16000 | 220 | 62a |
| Pd/TiO2 LPRD | 1 (28-38) | 975 | 16000 | 220 | 62a |
| Pd–Mg3Al calcined at 290 °C | 0.5 | 1000 | (30) | 210 | 14b |
| Pd–Mg3Al calcined at 450 °C | 0.5 | 1000 | (30) | 220 | 14b |
| Pd/Mg3Al | 0.5 | 1000 | (30) | 240 | 14b |
| Pd/Al2O3 | 0.5 | 1000 | (30) | 250 | 14b |
| Pd/γ–Al2O3 | 1 | 1000 | 15000 | 220 | 80 |
| L–Pd | 0.5 (24.8) | 1000 | 19000 | 360 | 93c |
| LT–Pd | 0.5 (12.6) | 1000 | 19000 | 335 | 93c |
| L–PdT | 0.5 (9.6) | 1000 | 19000 | 290 | 93c |
| Pd/SBA-15 | 0.5 (29) | 1000 | 10000 | 167 | 94d |
| Pd/LaSBA-PS | 0.5 (12) | 1000 | 10000 | 178 | 94d |
| Pd/MCM-41 | 5 (78) | 170 | [6.36]e | 450 | 95 |
| Pd/ZSM-5 | 0.3 | 1500 | 26000 | 230 | 96 |
| Pd extracted solvent/γ-Al2O3 | 0.1 | 270 | 50000 | 225 | 97 |
| PdCl2 aqueous solution/γ-Al2O3 | 0.1 | 270 | 50000 | 250 | 97 |
| Pd extracted solvent/cordierite | 0.1 | 270 | 50000 | 170 | 97 |
| PdCl2 aqueous solution/cordierite | 0.1 | 270 | 50000 | 180 | 97 |
| Pd/LaMnO3 | 0.1 | 1220 | 18000 | 165 | 71 |
| Pd/LaMnO3 | 0.5 | 1220 | 18000 | 175 | 71 |
| Pd/ZSM-5 | 0.48 (21) | 1000 | 32000 | 206 | 86,98 |
| Pd/KIT-6 | 0.49 (30) | 1000 | 32000 | 213 | 86,98f |
| Pd/ZK-3% | 0.49 (41) | 1000 | 32000 | 193 | 98f |
| Pd/ZK-6% | 0.48 (49) | 1000 | 32000 | 191 | 98f |
| Pd/ZK-12% | 0.50 (47) | 1000 | 32000 | 194 | 98f |
| Pd/ZK-24% | 0.47 (35) | 1000 | 32000 | 200 | 98f |
| Pd/ZK-mixed | 0.49 (27) | 1000 | 32000 | 211 | 98f |
| Pd/SBA-15 | 0.49 (31) | 1000 | 32000 | 207 | 99g |
| Pd/SBA-15-T | 0.48 (29) | 1000 | 32000 | 206 | 99g |
| Pd/SC-8.74 | 0.49 (64) | 1000 | 32000 | 183 | 99g |
| Pd/SC-17.3 | 0.51 (53) | 1000 | 32000 | 187 | 99g |
| Pd/SC-29.6 | 0.47 (42) | 1000 | 32000 | 199 | 99g |
| Pd/SC-38.2 | 0.48 (38) | 1000 | 32000 | 198 | 99g |
| Pd/SC-79.1 | 0.5 (34) | 1000 | 32000 | 205 | 99g |
| Pd/FeCrAl | 0.1 | ∼975 | 10000 | 215 | 72h |
| Pd/FeCrAl | 0.2 | ∼975 | 10000 | 206 | 72h |
| Pd/FeCrAl | 0.5 | ∼975 | 10000 | 203 | 72h |
| Pd/Co3AlO (COP) | 0.90 | 800 | 30000 | 220 | 81i |
| Pd/Co3AlO (IMP) | 0.99 | 800 | 30000 | 263 | 81i |
| Pd/Co3AlO (WIE) | 1.01 | 800 | 30000 | 253 | 81i |
| Pd/Co3AlO (TCB) | 0.95 | 800 | 30000 | 280 | 81i |
| Pd/CeO2 | 0.5 | ∼2090 | 10000 | 159 | 87 |
a Pd deposited on commercial TiO2 either by impregnation (IMP) or by Liquid Phase Reduction Deposition (LPRD).
b Pd-Mg3Al signifies Pd incorporated into a Mg3Al support prepared by hydrotalcite route. Pd/Mg3Al signifies Pd impregnated on the same support.
c Pd impregnated on Kraft lignin activated by H3PO4 without thermal treatment (L-Pd) and after thermal treatment (LT-Pd). L-Pd after thermal treatment is designated by L-PdT.
d PS signifies post-synthesis; a solution of lanthanum nitrate was added to a calcined SBA-15.
e This value corresponds to the concentration of toluene (gtoluene m−3) divided by the catalyst weight (g).
f KIT-6 is a porous material prepared from the initial sol without addition of ZSM-5. ZK-x% corresponds to biporous materials ZSM-5 and KIT-6 with different weight percentages, x, of ZSM-5. ZK-mixed designs a mechanical mixture of ZSM-5 (12 wt%) and KIT-6.
g T signifies that the sample was not reduced by H2. All the other samples of this reference were reduced by H2 after calcination under air. SC-x signifies SBA-15 with different x = Si/Al ratios.
h FeCrAl are wire mesh monolithic catalysts.
i Co3AlO is a cobalt-aluminum mixed oxide prepared by hydrotalcite route. COP: Coprecipitation, IMP: Impregnation, WIE: Wet Ion Exchange, TCB: Thermal Combustion Method.
4 Conclusion
Three hydroxyapatites were synthesized with three different Ca/P ratios: Ca/P = 1.64 (stoichiometric HapS), Ca/P = 1.56 (deficient HapD) and Ca/P = 1.71 (rich HapE). The potential of these materials as catalyst support for palladium and gold was tested in the reaction of toluene total oxidation. Four parameters were investigated: the support’s nature, the active phase nature, its content, and the way of incorporating it on the support.
Hydroxyapatite supports lead to more efficient catalysts than classical alumina one does, because at 150 °C, the specific rates on apatites supports were four to six times higher (for Pd content of 0.24–0.30 wt%) than the specific rate on alumina support (0.4 wt% of Pd). Even if palladium improved toluene conversion obtained on the support, increasing palladium loading above 0.25 wt% did not result in further improvement, Pd1/HapS giving a specific rate, at 150 °C, ∼7 times lower than Pd0.25/HapS. Furthermore, it was shown that palladium colloidal suspension with HEA16Cl as surfactant can be successfully used to deposit Pd nanoparticles on a hydroxyapatite support; similar catalytic performances have been obtained in the presence of Pd-based catalysts prepared using either this methodology approach or the conventional metal salt impregnation. PdO species were evidenced on the support surfaces of both preparation methods and no Pd(0) was detected. It was also found that palladium is more active than gold towards the studied reaction. Finally, it is concluded that the catalytic performances are mainly related to the high dispersion of active species as well as to support–metal interaction. Palladium supported on hydroxyapatites by conventional method showed an increase of toluene conversion after 13 h under stream whereas palladium impregnated via a colloidal suspension, did not show such behavior. It is thought that Pd(0) formation after a given time under the air-toluene mixture is responsible for this catalytic reactivity improvement. However, after impregnating a colloidal suspension, some interaction between palladium and support might probably exist inhibiting such catalytic reactivity enhancement. It is also shown that our catalytic systems compete with the best palladium systems studied in literature, evidencing that hydroxyapatite could be an alternative support to conventional ones (alumina, zeolite…) for catalytic VOC removal.
Acknowledgments
Mrs. D. Chlala acknowledges the award of a doctoral fellowship by the Agence Universitaire de la Francophonie (AUF) – Région du Moyen-Orient. Mrs. M. LABAKI is also grateful to this institution for research fellowship. This work was also supported by a grant from the project PHC CEDRE 2015 N° 32933QE.
Chevreul institute (FR 2638), Ministère de l'Enseignement Supérieur et de la Recherche, Région Nord – Pas de Calais and FEDER are acknowledged for supporting and funding this work.
The authors also want to warmly thank Dr. Rose-Noëlle VANNIER for Rietveld refinements, Mrs. Martine TRENTESAUX for her technical help in XPS spectra recording and Mrs. Laurence BURYLO for XRD measurements. Special thanks to Mrs. Ferial SROUR NEMR to her excellent proofreading.