

1 Introduction
Because nitrogen oxides (NOx), which are thermally formed by radical chain mechanisms proposed by Zeldovich at high temperatures above 1200 °C [1], emitted from combustion facilities are harmful to human health and the global environment, the removal of NOx from exhaust gas is necessary. Catalysis plays an important role in reducing NOx. For example, three-way catalysts (TWCs) have already been practically applied to the purification of exhaust gases emitted from gasoline-powered vehicles. TWCs can simultaneously reduce NO and oxidize CO and hydrocarbons at a theoretical air/fuel (A/F) ratio of around 14.6. Selective catalytic reduction of NOx using urea or ammonia is a leading technology to reduce NOx emission from diesel exhaust. In this reaction, NOx are efficiently reduced by ammonia in the presence of excess O2.
On the other hand, direct NO decomposition (2NO → N2 + O2), which needs no reductants, is well known as the most desirable reaction for NOx removal. Since the enthalpy of formation for NO is large and positive, = 90.2 kJ mol−1 [2,3], NO molecules are thermodynamically unstable and can be decomposed into N2 and O2. However, despite the thermodynamic instability, direct NO decomposition is a kinetically slow reaction because of its high activation energy (∼335 kJ mol−1) [2,3]. Therefore, the use of a catalyst is an effective strategy for NO decomposition to proceed, and the development of highly active catalysts still remains a challenging subject of study for the solution of NOx problems.
Numerous studies have been performed so far on catalytic NO decomposition, and a wide variety of catalytic materials have been found, such as noble metals, metal oxides, and zeolites [4,5]. In this review article, we overview recent progress in catalytic NO decomposition especially focusing on metal oxide-based catalysts.
2 Historical perspective on catalytic NO decomposition
Direct NO decomposition with catalysts has been extensively studied since the first report in the beginning of the 20th century by Jellinek [6], who revealed that precious metals such as platinum and iridium can catalyze NO decomposition at temperatures as low as 670 °C. Research activities reported before the 1980’s are summarized in a number of excellent reviews [3–5,7,8].
Most of the early studies were performed focusing on the kinetics of NO decomposition. For instance, Amirnazmi et al. [9] studied NO decomposition over Pt/Al2O3, and found that the kinetics follow the equation: r = N k(NO)/[1 + α K(O2)], where k is a reaction rate constant, K is an adsorption equilibrium constant, and α is a conversion factor. This equation means that the reaction is a first order with respect to NO and oxygen inhibits the reaction. As for metal oxide catalysts, Winter [10] measured the NO decomposition rates on 40 metal oxides, and proposed that the NO decomposition rate is closely dependent on the rate of the O2 desorption step. Boreskov [11] also measured the catalytic activities of the 4th-period transition metal oxides for various catalytic reactions including NO decomposition and CH4 oxidation, and revealed a close correlation between the activity for NO decomposition and that for homonuclear exchange of oxygen isotopes. These findings suggest that an effective strategy for the development of highly active NO decomposition catalysts is to create catalytically active sites effectively promoting the O2 desorption step.
Under these circumstances, Iwamoto et al. developed highly active copper ion exchanged Y zeolites (Cu–Y) [12], based on the finding that oxygen species adsorbed on CuII cations exchanged into zeolites can desorb at temperatures as low as 300 °C [13]. They also reported that excessively copper ion exchanged ZSM-5 zeolites (Cu-ZSM5) showed quite high NO decomposition activity in the temperature range of 350–600 °C [14]. According to their further studies [15–17], Cu+ ions in the zeolite produced at elevated temperatures act as catalytically active sites and NO decomposition proceeds via the formation of NOδ− or species on Cu+ and the reversible redox cycle between Cu2+ and Cu+.
Since the discovery of the Cu-ZSM-5 catalyst, extensive studies have been performed to improve its NO decomposition activity [18–21]. For instance, Pârvulescu at el. [22,23] reported that the addition of Sm into Cu-ZSM-5 promotes the O2 desorption step along with the stabilization of the CuI–O–CuII redox sites, resulting in the enhancement of NO decomposition activity. Yahiro et al. [24] also investigated the effect of coexisting rare earth elements such as La, Ce, Pr, Nd, Sm, and Gd on the activity of Cu-ZSM-5. Among the rare earth elements, Sm and Gd were found to act as promoters for NO decomposition over Cu-ZSM-5. They attributed the promotional effect to the stabilization of Cu+ active sites to enhance NO adsorption.
In addition to Cu-ZSM-5 catalysts, metal oxides with weak metal–oxygen bonds, such as cobalt oxide [25–27], oxygen-deficient Sr–Fe oxides [28] and perovskite-type compounds [29–31], have been investigated. Fig. 1 shows a rough comparison of the relative catalytic activities reported so far [32]. It is of interest that the key components for direct NO decomposition are Cu and Co, the combination of which with other components would enhance NO decomposition activity. Although Cu-ZSM-5 is still the most active catalyst as can be seen in Fig. 1, some composite oxides such as perovskite show comparable activity in the high temperature region. Therefore, designing and controlling the catalytically active sites may lead to highly active NO decomposition catalysts because of infinite combinations of metal components for composite mixed oxides. In the next section, recent advances in metal oxide-based NO decomposition catalysts will be overviewed.
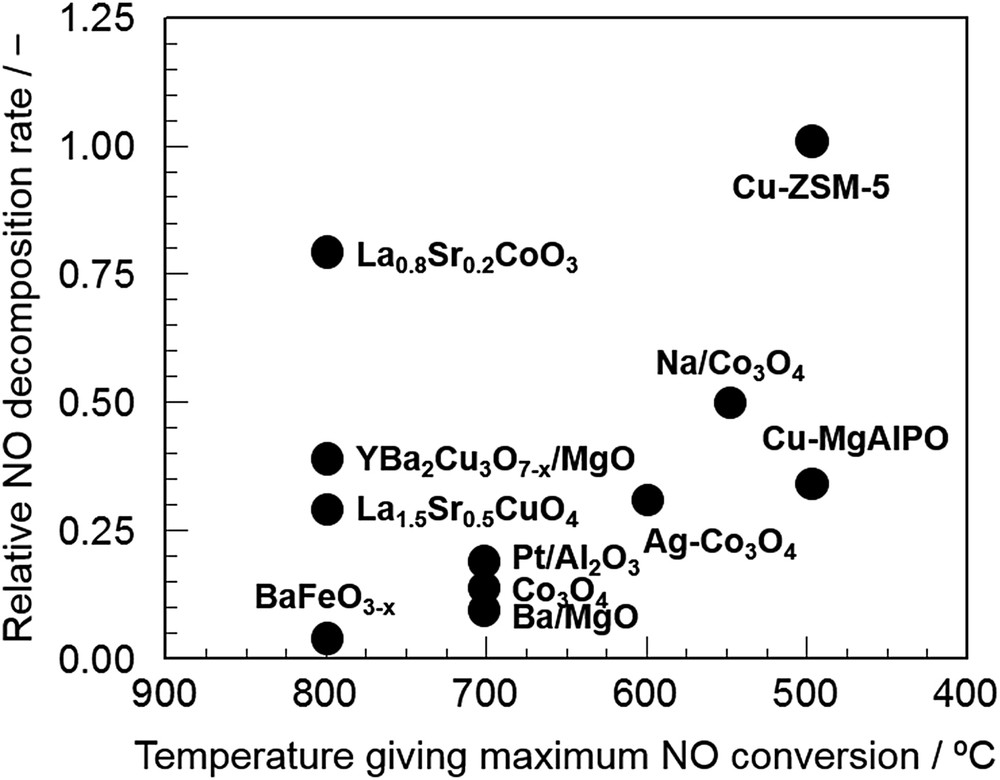
Comparison of the activity of various catalysts reported so far for direct NO decomposition [32].
3 Advances in metal oxide-based NO decomposition catalysts
Many kinds of metal oxides have been reported to effectively catalyze direct NO decomposition. It is of interest that N2 and O2 are always formed at a steady state with an O2/N2 ratio of approximately unity. The formation of N2O is almost negligible in NO decomposition over these metal oxide-based catalysts. Recent advances in four different types of metal oxide-based catalysts will be reviewed.
3.1 Perovskite-type oxides
Perovskite-type materials are the oxides having ABO3 and/or A2BO4 structures shown in Fig. 2, where A is a large cation that is coordinated by twelve O2− ions and B is a small cation located at the center of the octahedron. Perovskite-type oxides show good performance for direct NO decomposition, as reviewed by Zhu and Thomas [33], by Imanaka and Masui [34] and by Royer et al. [35]. Major reports on perovskite-type oxides are summarized in Table 1.
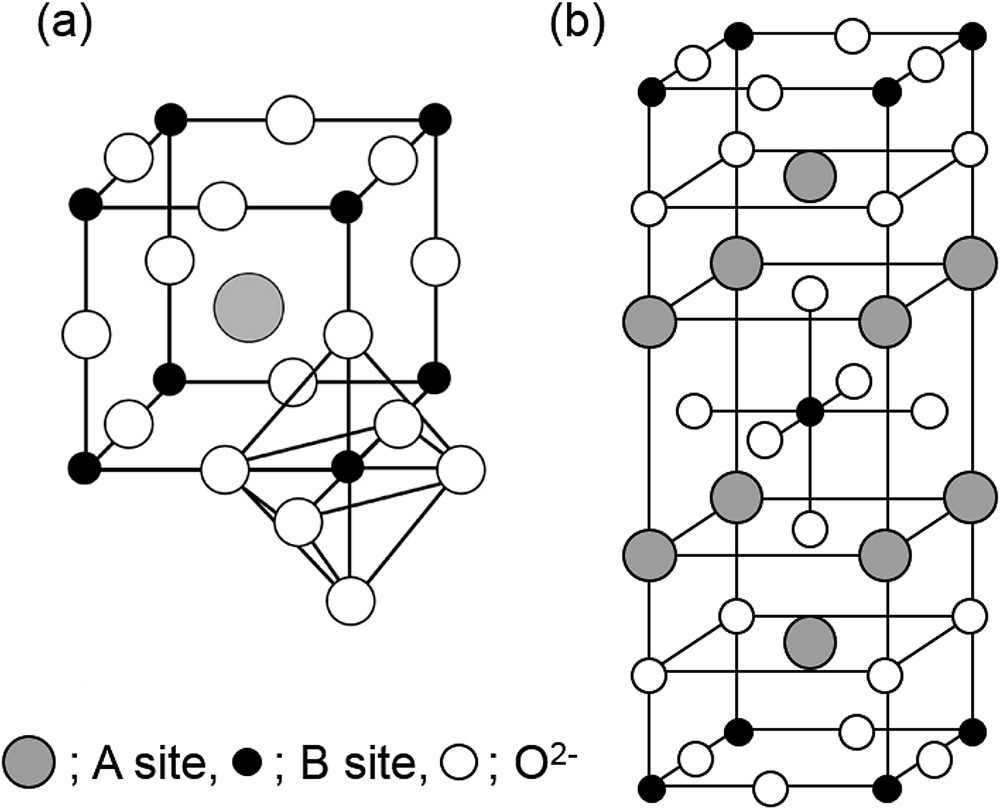
Crystal structure of (a) ABO3-type and (b) A2BO4-type perovskite oxides [34,48].
Reports on the direct NO decomposition activity of perovskite-type oxides.
| Catalyst | NO conversion (%) | Reaction conditions | Ref. | ||
| NO (%) | Temp. (°C) | W/F (g s cm−3) | |||
| LaNiO3 | 24 | 1.0 | 700 | 3.0 | [36] |
| LaSrNiO4 | 93 | 1.5 | 900 | 1.34 | [37] |
| La0.5Ce0.5SrNiO4 | 70 | 1.0 | 800 | 4.0 | [39] |
| La1.2Ba0.8NiO4 | 89 | 4.0 | 850 | 1.2 | [31] |
| LaSrMn0.2Ni0.8O4 | 85 | 1.0 | 850 | 1.2 | [40] |
| La0.87Sr0.13Mn0.2Ni0.8O3 | 39 | 2.0 | 650 | 4.0 | [41] |
| SrFe0.7Mg0.3O3 | 86 | 1.0 | 950 | 3.0 | [42] |
| SrFe0.7Sn0.3O3 | 79 | 1.0 | 950 | 3.0 | [42] |
| SrFe0.7Zr0.3O3 | 78 | 1.0 | 950 | 3.0 | [42] |
| SrFe0.7Ni0.3O3 | 76 | 1.0 | 950 | 3.0 | [42] |
| BaMn0.8Mg0.2O3 | 89 | 1.0 | 950 | 3.0 | [43] |
| Ba0.9La0.1Mn0.8Mg0.2O3 | 92 | 1.0 | 950 | 3.0 | [43] |
| SrMn0.8Mg0.2O3 | 78 | 1.0 | 950 | 3.0 | [43] |
| La1.867Th0.1CuO4.005 | 65 | 0.3 | 500 | 7,500 h−1 (GHSV) | [44] |
| La1.6Th0.4CuO4 | 42 | 1.0 | 900 | 1.33 | [45] |
| La2CuO4 nanofiber | 100 | 1.0 | 300–450 | 60,000 h−1 (GHSV) | [46] |
| La2CuO4 powder | 78 | 1.0 | 800 | 60,000 h−1 (GHSV) | [46] |
| La1.5Sr0.5CuO4 | 40 | 3.17 | 800 | 2.0 | [48] |
| La0.8Sr0.2CoO3 | 41 | 3.17 | 800 | 2.0 | [48] |
| Ba3Y3.4Sc0.6O9 | 91 | 1.0 | 950 | 3.0 | [49] |
| Sr2.7Ba0.3Fe1.8Zr0.2O7 | 79 | 1.0 | 950 | 3.0 | [50] |
| La0.8Sr0.2CoO3 | 48 | 4.0 | 800 | 4.0 | [84] |
| La0.4Sr0.6Mn0.8Ni0.2O3 | 70 | 4.0 | 800 | 4.0 | [84] |
Since A- and/or B-site cations in the structure are easily replaced by other ions with different ionic sizes and valencies, there are a wide variety of perovskite-type oxides with different elemental combinations. Yokoi and Uchida [36] investigated the influence of transition metals located at the B-site in LaMO3 (M: Cr, Mn, Fe, Co and Ni) on the NO decomposition activity, and found that the activity of LaMO3 was decreased in the order of LaNiO3 > LaCoO3 > LaMnO3 > LaFeO3 > LaCrO3. XPS and molecular orbital calculations suggested that the instability of the adsorbed oxygen species, which has weak interactions with the transition metals, is responsible for high NO decomposition activity. Relatively high NO decomposition performances of Ni-substituted perovskite-type oxides such as LaSrNiO4 [37], La1−xCexSrNiO4 (0 ≤ x ≤ 0.3) [38,39], La2−xBaxNiO4 (x ≤ 1.2) [31], LaSrMn1−xNixO4 (0 ≤ x ≤ 1) [40] and La1−xSrxMn1−yNiyO3 (0.05 ≤ x ≤ 0.6, 0.6 ≤ y ≤ 0.87) [41] were also reported. Ishihara and co-workers [42,43] studied the effect of B-site substitution on the activity of SrFe0.7M0.3O3 and BaMn0.8M0.2O3 (M: Mg, Si, Ti, Cr, Mn, Fe, Co, Ni, Cu, Ga, Ge, Zr, Ru, Sn, Ce, Ta, and Ir), and found that the substitution with Mg, Zr, Sn, Ni, Co and so on in both catalysts is effective in promoting NO decomposition, while that with Cr and Cu was not effective. They explained the positive effect of B-site substitution with Mg by the acceleration of O2 desorption.
It was also reported that Cu-substituted perovskite-type oxides such as La2−xThxCuO4 (0 ≤ x ≤ 0.4) [44,45] and La2CuO4 [46] show good performance for NO decomposition. For example, Yasuda et al. [30,47,48] extensively studied the effect of the valence state of the Cu site in La2−xA′xCu1−yB′yO4, which was controlled by the substitution of A′- and B′-sites with Sr, Ba and Ca, and Zr and Al, respectively, on the NO decomposition activity. They confirmed by XPS that the average oxidation number of Cu varies widely from 1.60 to 2.30 by changing A′- and B′-site elements and their contents. Fig. 3 shows the correlation between the average oxidation number of Cu and the relative catalytic activities at 800 °C, represented by % conversion divided by the surface area. It is of interest that the activities increased sharply with the increase in the average oxidation number of Cu, indicating that controlling the valence state of Cu sites in perovskite-type oxides is one of the effective strategies to develop highly active NO decomposition catalysts.
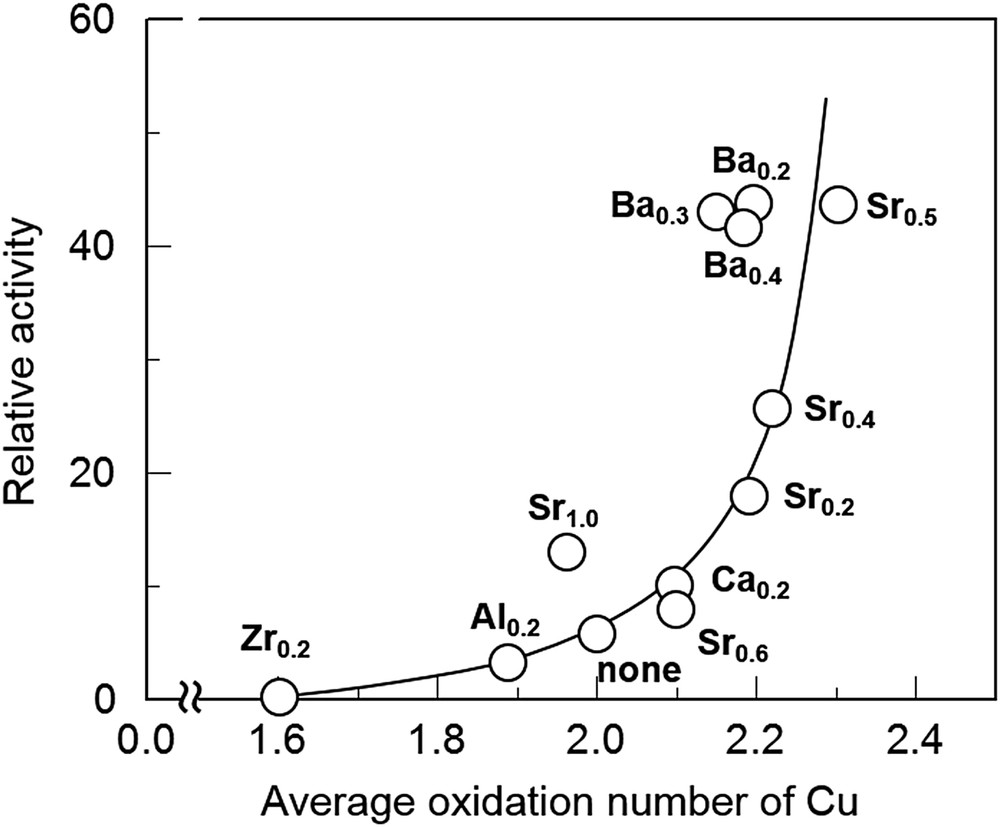
Change in the NO decomposition activity of La2−xA′xCuO4 (A′x = Zr0.2, Al0.2, Sr0.2, Sr0.4, Sr0.5, Sr0.6, Sr1.0, Ba0.2, Ba0.3, Ba0.4, Ca0.2 and none) at 800 °C and average oxidation number of Cu without pretreatment of the catalyst [30,48]. Reaction conditions: NO = 3.17%, He balance, and W/F = 2.0 g s cm−3.
According to the reports by Yasuda et al. [47,48], cations located at the A-sites contribute indirectly to NO decomposition by affecting the electric state of the B-site cations. In other words, transition metals such as Ni, Cu, and Co located at the B-sites are the catalytically active sites for NO decomposition. On the other hand, Goto and Ishihara [49,50] reported that Ba3Y3.4Sc0.6O9 with a perovskite-related structure, consisting of alkaline earth and rare earth ions, shows high NO decomposition activity in a wide temperature range of 600–850 °C. They also revealed that the addition of BaO into BaY2O4 caused a significant increase in the NO decomposition activity [51]. It should be noted that both catalysts do not contain transition metals as catalytically active sites, and that there are no detectable oxide ion vacancies in the BaO/BaY2O4 catalyst, although Ba3Y3.4Sc0.6O9 includes intrinsic oxygen defects in the structure. Therefore, the mechanism of NO decomposition on Ba3Y3.4Sc0.6O9 and Ba/BaY2O4 seems to be different from that on conventional perovskite-type oxide catalysts.
3.2 Rare earth-based oxides
As described above, one of the strategies to develop highly active perovskite-type oxide catalysts for NO decomposition is to control the valence state of the active site for promotion of O2 desorption. In addition, the creation of oxide anion vacancies would also be effective to promote the dissociation of NO molecules. Non-stoichiometric rare earth oxides such as CeO2, Pr6O11, and Tb4O7 produce oxygen defects easily on their surface by suitable reduction treatment because the metal valency can change between +3 and +4 depending on the environment [52]. This leads us to the consideration that NO is effectively decomposed on the surface of the non-stoichiometric rare earth oxides. In fact, Daturi et al. [53] observed continuous formation of N2 when several pulses of NO were introduced to pre-reduced CeO2–ZrO2 at 500 °C, indicating that oxide anion vacancies are highly reactive toward NO molecules. Haneda et al. [54] also investigated the surface reactivity of pre-reduced several rare earth oxides with NO molecules, and found that NO was decomposed to N2 at 400 °C on CeO2 and Pr6O11 pre-reduced at 500 °C, in which a lot of oxide anion vacancies exist (Table 2). As expected, NO decomposition was not observed over pre-reduced La2O3 and Sm2O3 because oxide anion vacancies are not created in these oxides even after high temperature reduction. Surprisingly, the pre-reduced Tb4O7 did not show activity for NO decomposition, although Tb4O7 gave the largest amount of H2 consumption below 500 °C in H2-TPR, suggesting that the oxide anion vacancy in Tb4O7 is not active for the dissociation of NO molecules. NO decomposition on non-stoichiometric rare earth oxides (REO2, RE: rare earths) may proceed via the following reactions:
| REO2 + xH2 → REO2−x + x“□” + xH2O | (1) |
| NO + “□” → 1/2N2 + O(s) | (2) |
Physical properties and N2 yield in NO decomposition over rare earth oxides at 400 °C [54].
| Catalyst | BET surface area (m2 g−1) | Amount of H2 consumption below 500 °C in H2-TPR (μmol –H2 g−1) | N2 yield in NO decompositiona (μmol –N2 g−1) |
| La2O3 | 7.5 | 2.5 | 0.0 |
| CeO2 | 55 | 75 | 14.7 |
| Pr6O11 | 9.5 | 773 | 19.6 |
| Sm2O3 | 8.0 | 1.5 | 0.0 |
| Tb4O7 | 22 | 1008 | 0.0 |
a Reaction conditions: NO = 970 ppm, weight = 0.1 g, and gas flow rate = 60 cm3 min−1.
Imanaka et al. [34,55] investigated the catalytic performance of rare earth sesquioxides (RE2O3) by focusing on the crystal structure. They speculated that the size of the cavity space of the oxides is an important factor for the NO decomposition activity, because of the improvement of NO accessibility and O2 desorption. There are three types of crystal structures in the polymorphism of the rare earth sesquioxides, namely, A-type (hexagonal), B-type (monoclinic), and C-type (cubic), according to the ionic size of the rare earth elements [34,55,56]. Among them, the C-type structure possesses the largest cavity space, because it is equivalent to a double edge-sharing fluorite structure (REO2) with one-quarter of the oxygen sites vacant and with regular ordering, as can be seen in Fig. 4. In other words, the C-type cubic rare earth oxides include many oxide anion vacancies with large cavity space, which may increase the accessibility of NO molecules and their adsorption.
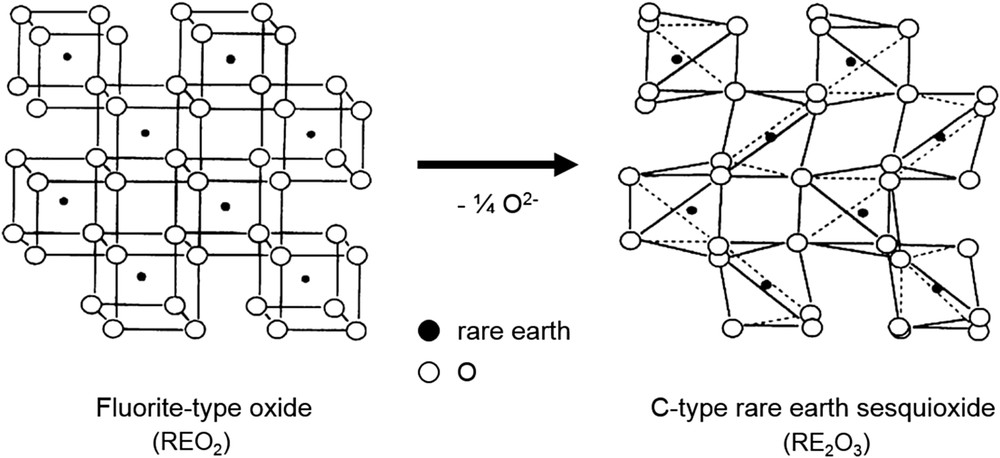
The relationship between the cubic fluorite-type oxide (REO2; RE: rare earths) and the C-type rare earth sesquioxide (RE2O3) [34,55].
Imanaka and co-workers then studied the catalytic performance of C-type cubic rare earth oxides and reported various kinds of effective composite metal oxides with C-type structures such as Gd2O3–Y2O3–BaO [57], Yb2O3–Tb4O7 [58], Y2O3–ZrO2–BaO [59,60], Y2O3–Tb4O7–BaO [61,62], Y2O3–Pr6O11–Eu2O3 [63,64] and Ho2O3–ZrO2–Pr6O11 [65]. As summarized in Table 3, C-type cubic rare earth oxides are highly active catalysts at high temperatures above 800 °C. Among the catalysts reported so far, (Y0.69Tb0.30Ba0.01)2O2.99+δ was found to be the best catalyst with 100% NO conversion at 900 °C (Fig. 5) [55]. The high activity was ascribed to the accelerated oxygen desorption via the redox cycle of Tb3+/Tb4+. In addition, on the basis of mechanistic studies using TPD and in situ FT-IR techniques, the partial substitution of Tb3+ sites with Ba2+ increases the number of basic sites, thus facilitating NO adsorption, and the generation of oxide anion vacancies in the lattice [62]. They proposed a reaction mechanism in which NO adsorbed as nitrosyl on Ba2+ sites reacts with gas-phase NO to form N2 and O2, which is then desorbed to the gas-phase via the redox cycle of Tb3+/Tb4+. Thus, the specific structure promoting NO adsorption and oxygen desorption would be one of the important factors for the development of highly active NO decomposition catalysts.
Reports on the direct NO decomposition activity of C-type cubic rare earth oxides.
| Catalyst | NO conversion to N2 (%) | Reaction conditions | Ref. | ||
| NO (%) | Temp. (°C) | W/F (g s cm−3) | |||
| (Gd0.70Y0.26Ba0.04)2O2.92 | 75 | 1.0 | 900 | 3.0 | [57] |
| (Yb0.5Tb0.5)2O3±δ | 55 | 1.0 | 900 | 3.0 | [58] |
| (Y0.94Zr0.06)2O3.06 | 45 | 1.0 | 1000 | 0.375 | [59] |
| (Y0.89Zr0.07Ba0.04)2O3.03 | 90 | 1.0 | 900 | 3.0 | [60] |
| (Y0.70Tb0.30)2O3±δ | 62 | 1.0 | 900 | 3.0 | [62] |
| (Y0.99Ba0.31)2O3±δ | 76 | 1.0 | 900 | 3.0 | [62] |
| (Y0.69Tb0.3Ba0.01)2O2.99±δ | 100 | 1.0 | 900 | 3.0 | [61] |
| (Y0.90Pr0.10)2O3±δ | 79 | 1.0 | 900 | 3.0 | [63] |
| (Y0.78Pr0.15Eu0.07)2O3±δ | 84 | 1.0 | 900 | 3.0 | [64] |
| (Ho0.87Zr0.05Pr0.08)2O3.05±δ | 71 | 1.0 | 900 | 3.0 | [65] |
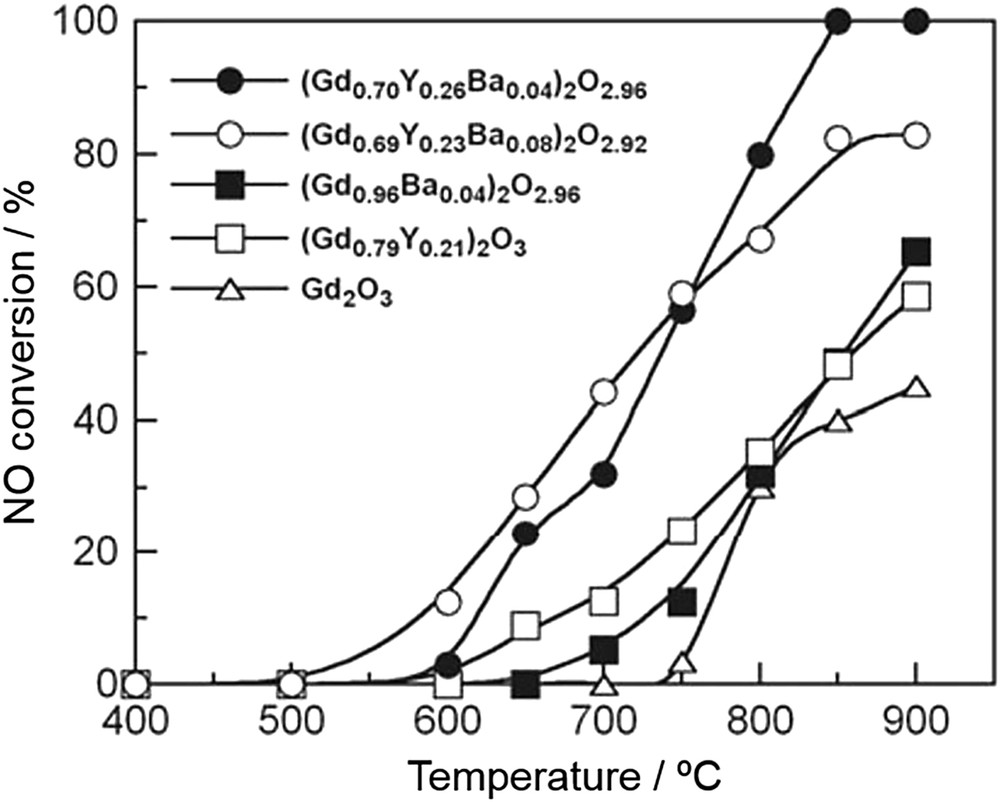
Temperature dependence of NO conversion on (Gd1−x−yYxBay)2O3−y (0 < x < 0.26, 0 < y < 0.08) measured under the conditions of NO = 1% and He balance at W/F = 0.375 g s cm−3 [55].
3.3 Alkali or alkaline earth metal-doped cobalt oxide
According to the early work by Boreskov[11], Winter [10], Hightower et al. [7] and Shelef et al. [4,25], cobalt oxide (Co3O4) is regarded as one of the most active single metal oxides for NO decomposition. The high activity of Co3O4 would be due to the relatively low ΔH of vaporization of O2 [66]. This means that the Co–O bond strength of Co3O4 is relatively weak, making the lattice oxygen to be more easily desorbed. Hamada et al. [26] investigated the additive effect of Ag on the NO decomposition activity of Co3O4, based on its weak affinity for oxygen. The addition of a small amount of Ag (Ag/Co atomic ratio = 0.05) was found to significantly increase the catalytic activity of Co3O4. On the other hand, Haneda et al. [67,68] reported that pure Co3O4 is entirely inactive for direct NO decomposition and that alkali metals remaining as impurities in Co3O4 are essential for NO decomposition to occur. Namely, Co3O4 prepared by a precipitation method using ammonia, urea, (NH4)2CO3 and (COOH)2 as precipitation agents was virtually inactive, whereas Co3O4 prepared using Na2CO3 effectively catalyzed the NO decomposition reaction. This clearly indicates that the presence of residual Na is important to achieve high NO decomposition activity. This interesting effect of residual Na was first reported by Park et al. [27].
Haneda et al. [67,68] studied the additive effect of alkali metals on the activity of Co3O4 for NO decomposition. They prepared Co3O4 by a precipitation method using urea and then alkali metals (Li, Na, K, Rb, and Cs) were doped into Co3O4 by impregnation. As summarized in Table 4, the specific activity of Co3O4 at 600 °C, which was calculated by dividing the formation rate of N2 by the BET surface area, was significantly increased by addition of a small amount of alkali metals (M/Co atomic ratio = 0.035). Fig. 6 shows the dependence of the activities of alkali metal-doped Co3O4 on the M/Co atomic ratios at 600 °C. For each catalyst, the activity was increased with the M/Co atomic ratio, and the optimal atomic ratio was at around 0.02–0.05. In addition to alkali metals, promotional effects of alkaline earth metals such as Sr and Ba on the activity of Co3O4 was also reported by Haneda et al. [69] and Iwamoto et al. [70]. Since the addition of alkali or alkaline earth metals into Al2O3, SiO2 and ZrO2 did not generate NO decomposition activity [27,68,70,71], the interaction between Co3O4 and alkali or alkaline earth metals must be an essential factor.
BET surface area and activity of alkali-doped Co3O4 with an M/Co atomic ratio of 0.035 for direct NO decomposition to N2 at 600 °C [67].
| Catalyst | BET surface area (m2 g−1) | Activitya at 600 °C | |
| molN2 min−1 g−1 | molN2 min−1 m−2 | ||
| Co3O4 | 9.0 | 1.35 × 10−8 | 1.50 × 10−9 |
| Li/Co3O4 | 8.3 | 4.04 × 10−7 | 4.87 × 10−8 |
| Na/Co3O4 | 23 | 1.72 × 10−6 | 6.90 × 10−8 |
| K/Co3O4 | 34 | 2.17 × 10−6 | 6.39 × 10−8 |
| Rb/Co3O4 | 38 | 1.91 × 10−6 | 6.60 × 10−8 |
| Cs/Co3O4 | 33 | 1.44 × 10−6 | 4.38 × 10−8 |
a The reaction rate was determined under the differential conditions obtained at an NO conversion level of below 30%.
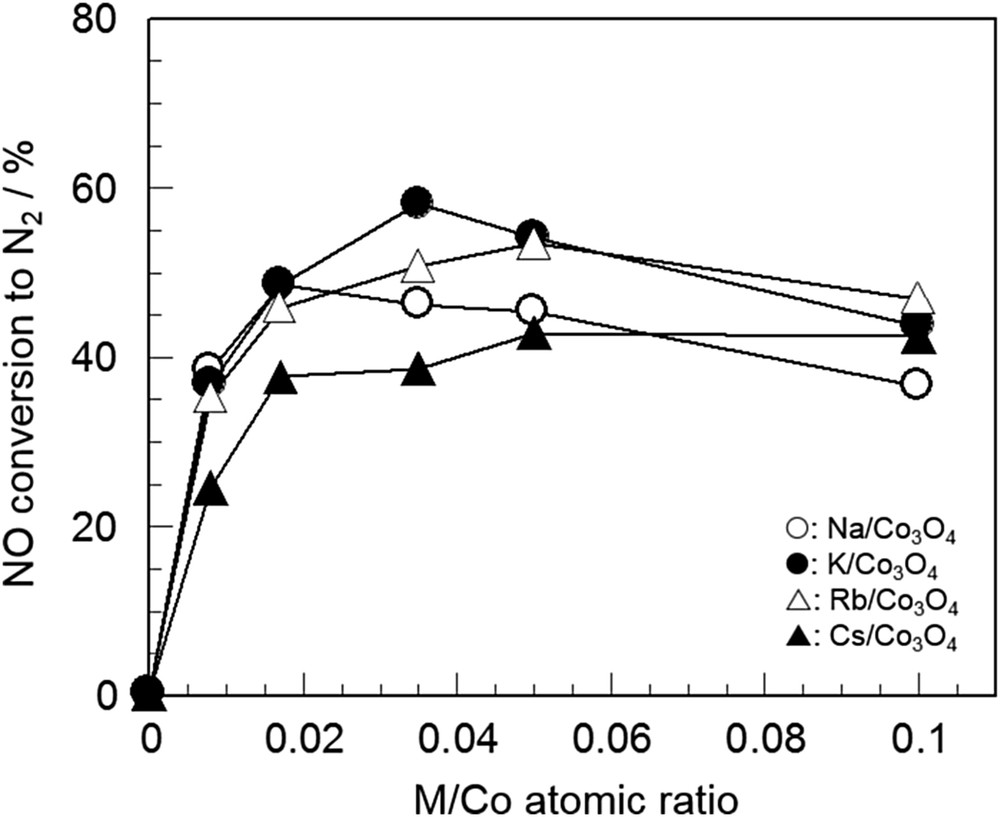
Change in the NO decomposition activity of alkali-doped Co3O4 as a function of the M/Co atomic ratio [67]. Reaction conditions: NO = 0.1%, He balance, and W/F = 0.5 g s cm−3.
Nakamura et al. [68,72] investigated the role of potassium in NO decomposition over cobalt oxide by a surface science approach. Low energy electron diffraction (LEED) analysis of K-doped CoO/Co(0001) surface suggested that K atoms strongly interact with neighboring Co atoms, leading to a slight reduction of Co atoms to lower oxidation states. A quite similar phenomenon was also reported for K/Co3O4 powder catalysts by Haneda et al. [67]. They observed a slight shift of the Co 2p3/2 binding energy to a lower value in the XPS spectrum when potassium was added to Co3O4, indicating a lower valence electronic state of cobalt. The role of alkali metals is probably to weaken the Co–O bond strength, promoting oxygen desorption from Co3O4, as shown by the O2-TPD profiles of alkali metal-doped Co3O4 in Fig. 7. Park et al. [27] also reported that the presence of Na facilitated the desorption of oxygen from Co3+ to form Co2+ via the formation and decomposition of NaNO3 in NO decomposition. On the basis of these reports, it can be concluded that alkali metal additives possess an essential role to create the catalytically active Co2+ sites for NO decomposition.
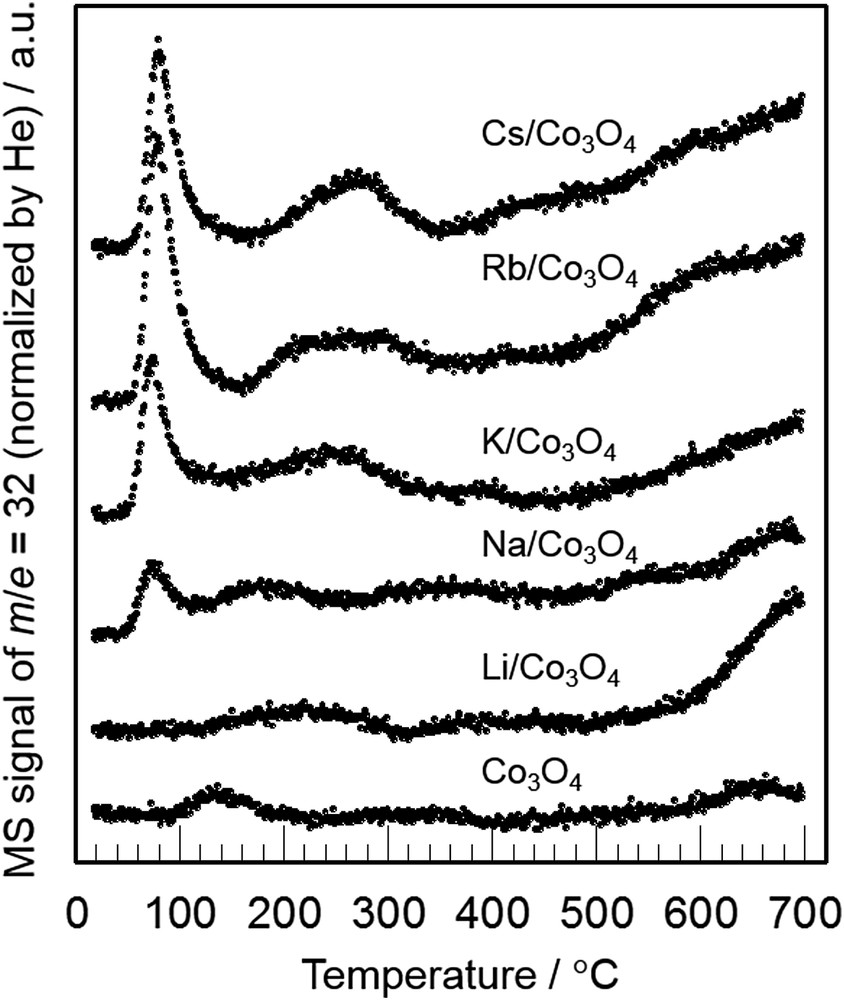
O2-TPD profiles of alkali-doped Co3O4 with an M/Co atomic ratio of 0.035 [67].
3.4 Barium oxide supported on rare earth oxides
It has been generally accepted that oxide anion vacancies play an important role in direct NO decomposition. Accordingly, extensive studies have been performed to construct oxygen deficient sites in perovskite-type oxides, C-type cubic rare earth oxides and cobalt oxides, as described before. On the other hand, it should be noted that alkaline earth oxides containing no oxygen deficient sites also catalyze NO decomposition [73]. Their activity decreases in the order of BaO > SrO > CaO. In addition, Vannice et al. [74,75] and Xie et al. [76,77] also reported the activity of supported-alkaline earth oxide catalysts such as Sr/La2O3, Sr/Sm2O3 and Ba/MgO for NO decomposition. They proposed that barium oxide serves as an adsorption site for nitro intermediate species [76]. Because alkaline earth oxides are known to be solid base materials, the basicity of the catalysts seems to be an important factor to determine the NO decomposition activity.
Haneda and co-workers [71] investigated the catalytic performance of supported alkaline earth metal oxides and found that the activity is strongly dependent on the metal oxide support. In Table 5, the NO decomposition activities of supported alkaline earth metal oxides (10.9 wt%) are summarized. As for the supported Ba catalysts, Y2O3 is the most effective support. Ba/Y2O3 showed the highest NO decomposition activity, which decreased in the order of Ba/Y2O3 > Sr/Y2O3 > Ca/Y2O3 > Mg/Y2O3 > Y2O3. There was no relationship between the BET surface area and NO decomposition activity. They also revealed that the activity of Ba/Y2O3 increases with increasing Ba loading up to 5 wt% and that the surface basic sites participate in NO decomposition. These findings suggest that a clarification of the role of surface basic sites is essential for the development of highly active catalysts.
BET surface area of and NO conversion on supported alkaline earth metal oxide (10.9 wt%) [71].
| Catalyst | BET surface area (m2 g−1) | NO conversion to N2 (%) | ||
| 700 °C | 800 °C | 900 °C | ||
| Y2O3 | 18.9 | 2.7 | 4.7 | 8.7 |
| MgO | 32.0 | ≤1 | ≤1 | ≤1 |
| La2O3 | 7.5 | ≤1 | ≤1 | 3.5 |
| Sm2O3 | 8.0 | 4.7 | 10.8 | 13.9 |
| Al2O3 | 190 | ≤1 | ≤1 | ≤1 |
| SiO2 | 286 | ≤1 | ≤1 | ≤1 |
| Ba/Y2O3 | 13.8 | 4.4 | 15.7 | 41.0 |
| Ba/MgO | 9.4 | 8.4 | 12.2 | 35.5 |
| Ba/La2O3 | 6.4 | ≤1 | ≤1 | 21.5 |
| Ba/Sm2O3 | 12.8 | 5.6 | 14.5 | 36.1 |
| Ba/Al2O3 | 170 | ≤1 | ≤1 | ≤1 |
| Ba/SiO2 | 144 | ≤1 | ≤1 | ≤1 |
| Mg/Y2O3 | 2.2 | 4.6 | 7.6 | |
| Ca/Y2O3 | 2.8 | 6.6 | 15.1 | |
| Sr/Y2O3 | 15.9 | 2.0 | 3.4 | 26.4 |
After the report by Haneda and co-workers, many researchers investigated various kinds of Ba catalysts supported on rare earth oxides. For example, Ishihara et al. [51,78] found that BaO catalysts supported on Y2O3 and Sc2O3 show relatively high NO decomposition activity with an N2 yield as high as 80% at 950 °C under the conditions of NO = 1% and W/F = 3.0 g s cm−3. It was suggested that BaO nano-particles dispersed on the surface of Y2O3 and Sc2O3 play an important role in NO adsorption. The additive effect of Ba into CeO2-based mixed oxides such as CeOx–MnOx and CeOx–FeOx was also reported by Iwamoto et al. [79–83]. They prepared Ba catalysts supported on CeOx–FeOx and CeOx–MnOx mixed oxides by impregnation combined with glycothermal or solvothermal methods, and measured NO decomposition activity under the conditions of NO = 0.6% and W/F = 1.0 g s cm−3. Among the catalysts tested, Ce0.8Mn0.15Ba0.05Ox prepared by a solvothermal method showed the highest activity with an N2 yield of 77% at 800 °C. On the basis of XRD, Raman and XPS analyses, the additive effect of FeOx and MnOx was attributed to the increase in the population of oxygen deficient sites in the CeOx lattice on which NO dissociation and subsequent O2 desorption take place. The catalysis of Ba-doped CeOx–MnOx and CeOx–FeOx seems to be very similar to that of perovskite-type oxides and C-type cubic rare earth oxides as described before.
Haneda et al. performed detailed investigation on the importance of surface basicity of supported-Ba catalysts [84–86]. They prepared Ba-doped rare earth oxides (REOs), which are known to show different surface basicity depending on the radius of the rare earth cations [87], by a coprecipitation method and examined the relationship between NO decomposition activity and surface basicity [85]. Table 6 summarizes the BET surface area and NO decomposition activities of REOs and Ba–REOs evaluated under the conditions of NO = 0.1% and W/F = 1.0 g s cm−3. Although REOs without Ba catalyzed NO decomposition, the addition of 8 mol% Ba significantly enhanced the NO decomposition activity. The activity of Ba–REOs was decreased in the order of Ba–Dy2O3 ≈ Ba–Sm2O3 ≈ Ba–Y2O3 > Ba–Nd2O3 > Na–Tb4O7 > Ba–La2O3 >> Ba–Pr6O11 > Ba–CeO2. Sesquioxides of trivalent rare earth metals such as Y2O3 and Dy2O3 seem to be effective supports for Ba. No correlation was observed between the BET surface area and catalytic performance for all the catalysts. Fig. 8 shows the relationship between the amount of desorbed CO2 in the temperature range of 300–600 °C in CO2-TPD, which corresponds to the amount of Ba species exposed on REO surfaces, and the rate of N2 formation at 800 °C. It is noteworthy that the N2 formation rate increases with the amount of CO2 desorption, indicating that the amount of Ba species exposed on REO surfaces determines the NO decomposition activity of Ba-doped REO catalysts.
BET surface area and NO decomposition activity of REOs and Ba (8 mol%)–REOs prepared by a coprecipitation method [85].
| Catalyst | BET surface area (m2 g−1) | NO conversion to N2 (%) | |||
| 600 °C | 700 °C | 800 °C | 900 °C | ||
| La2O3 | 6.9 | 0.0 | 1.1 | 1.9 | 2.6 |
| CeO2 | 15.2 | 1.2 | 1.3 | 1.4 | 1.7 |
| Pr6O11 | 9.2 | 1.1 | 2.0 | 3.2 | 4.4 |
| Nd2O3 | 5.0 | 0.7 | 1.7 | 2.1 | 2.8 |
| Sm2O3 | 9.1 | 0.9 | 3.5 | 9.2 | 14.6 |
| Gd2O3 | 5.7 | 0.5 | 2.6 | 6.6 | 11.2 |
| Tb4O7 | 3.6 | 0.5 | 3.6 | 8.7 | 13.1 |
| Dy2O3 | 9.9 | 0.7 | 3.3 | 6.9 | 10.5 |
| Y2O3 | 16.6 | 3.0 | 8.1 | 11.8 | 14.5 |
| Ba–La2O3 | 3.2 | 0.7 | 10.4 | 29.6 | 43.5 |
| Ba–CeO2 | 8.8 | 1.5 | 6.7 | 12.4 | 14.2 |
| Ba–Pr6O11 | 2.7 | 1.1 | 8.0 | 18.0 | 26.7 |
| Ba–Nd2O3 | 7.1 | 1.4 | 14.4 | 35.9 | 48.8 |
| Ba–Sm2O3 | 6.0 | 2.1 | 17.5 | 42.8 | 54.0 |
| Ba–Gd2O3 | 5.3 | 1.5 | 13.0 | 38.2 | 51.2 |
| Ba–Tb4O7 | 6.1 | 3.9 | 15.6 | 30.9 | 44.6 |
| Ba–Dy2O3 | 6.7 | 3.6 | 19.0 | 45.1 | 56.5 |
| Ba–Y2O3 | 14.6 | 2.8 | 17.8 | 40.5 | 47.6 |
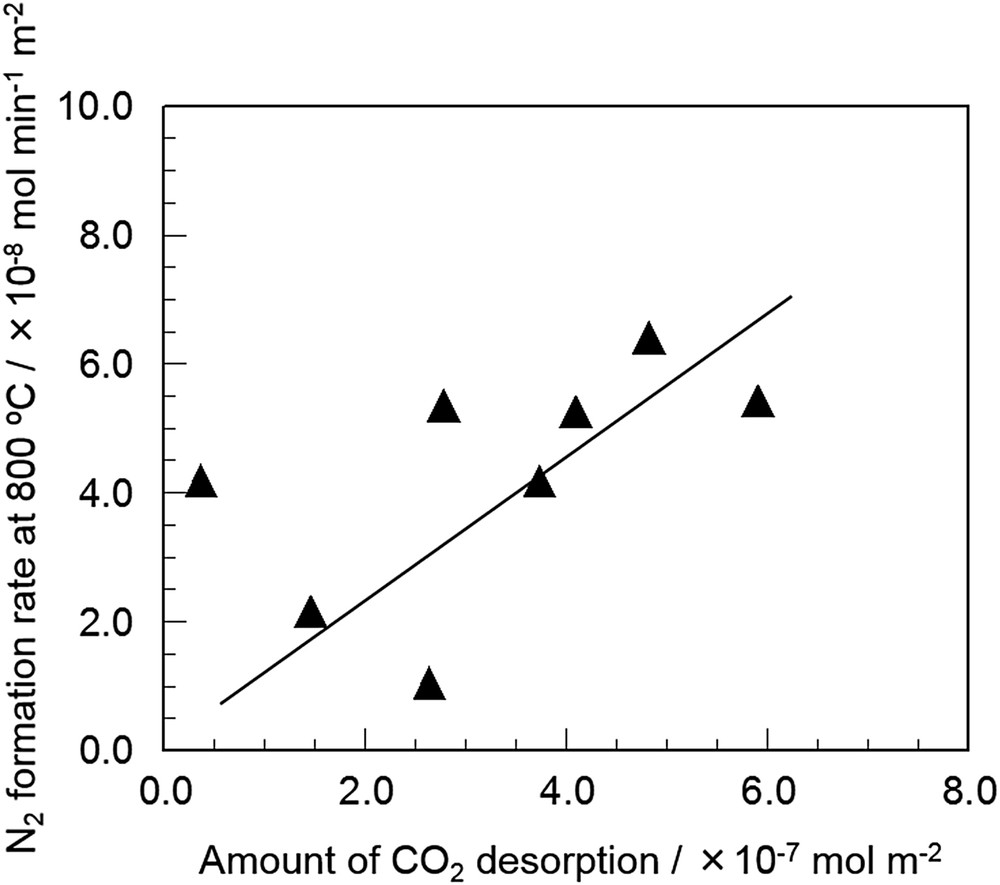
Relationship between the amount of CO2 desorption and the rate of N2 formation at 800 °C over Ba-doped REOs. [85].
Haneda et al. also revealed that catalytically inactive BaCO3 is formed when the Ba species was supported on rare earth oxides with strong basic sites, such as La2O3 [85]. In other words, the selective formation of catalytically active, highly dispersed Ba species can be expected by controlling the surface basicity of the support oxide. They investigated the additive effect of CeO2 having weak basicity on the catalytic activity of Ba–Y2O3 [86], and found that the addition of CeO2 decreased the surface basicity of Y2O3, resulting in the enhancement of highly dispersed Ba species. In accordance with their assumption, the NO decomposition activity of 8 mol% Ba–Y2O3 was improved by the addition of CeO2 and reached a maximum at a CeO2 content of 10 mol% (Fig. 9). The predominant role of the CeO2 additive is not to catalyze NO decomposition but to effectively create the highly dispersed Ba species as catalytically active sites. In contrast to the catalyst materials including oxygen deficient sites such as perovskite-type oxides, Ba-doped REOs are quite unique NO decomposition catalysts, where highly dispersed basic Ba species act as catalytically active sites.
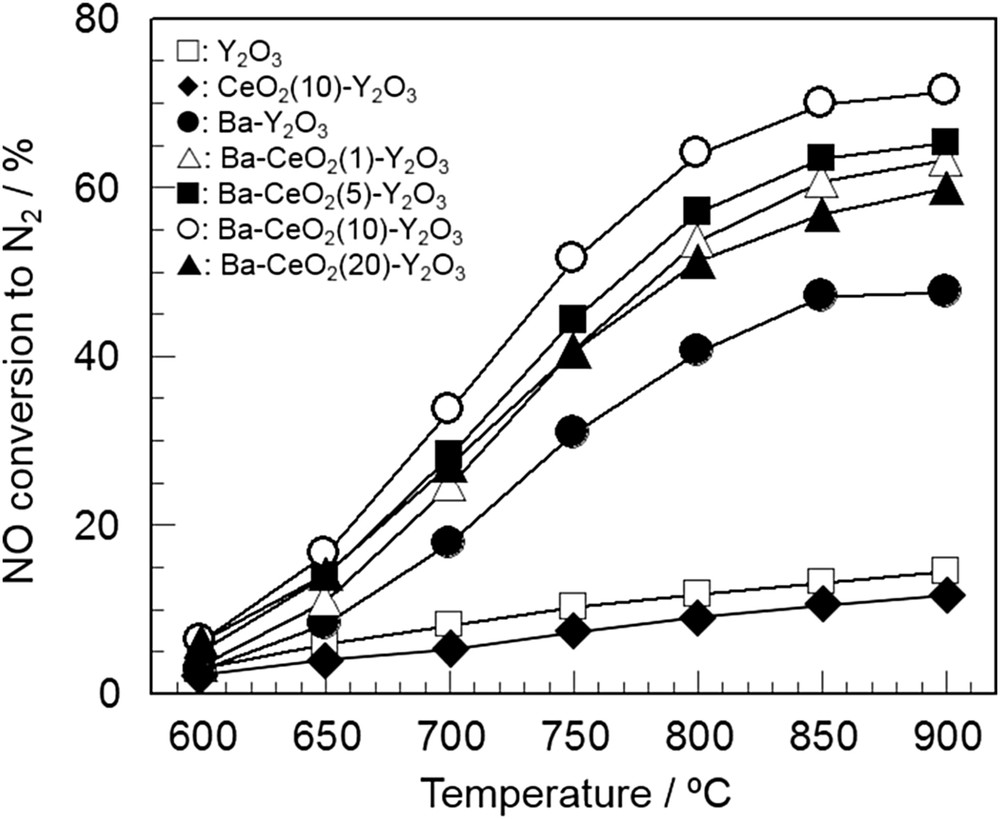
Activity of Ba (8 mol %) doped CeO2–Y2O3 with various CeO2 contents for NO decomposition [86]. Reaction conditions: NO = 0.1%, He balance, and W/F = 1.0 g s cm−3.
4 Mechanism of NO decomposition on metal oxide catalysts
Kinetic studies of NO decomposition over various catalysts have been extensively performed to reveal the reaction mechanisms. Amirnazmi et al. [9] reported that the kinetics of NO decomposition over Pt/Al2O3 and Co3O4 catalysts follow the equation: r = N k(NO)/[1+αK(O2)], suggesting that oxygen inhibition is due to equilibrated chemisorption of oxygen on sites required for the rate-determining process of NO chemisorption. Winter [10] proposed a mechanism in which the reaction is initiated by the attack on NO molecules of surface R2 centers, which are pairs of adjacent oxygen vacancies with one electron (F+ centers). A similar reaction mechanism has also been proposed over various metal oxides having oxygen defect sites. For example, Teraoka et al. [88] determined the kinetics of NO decomposition over perovskite-type oxides, and proposed a mechanism in which two NO molecules are adsorbed onto a pair of adjacent oxide ion vacancies as the rate-determining step, followed by desorption of N2 and O2. Tsujimoto et al. [62] proposed that direct NO decomposition over C-type cubic rare earth oxides, such as (Y0.69Tb0.30Ba0.01)2O2.99+δ, proceeds via the formation of nitrosyl species on oxide anion vacancies and subsequent reaction with gas-phase NO (Fig. 10).
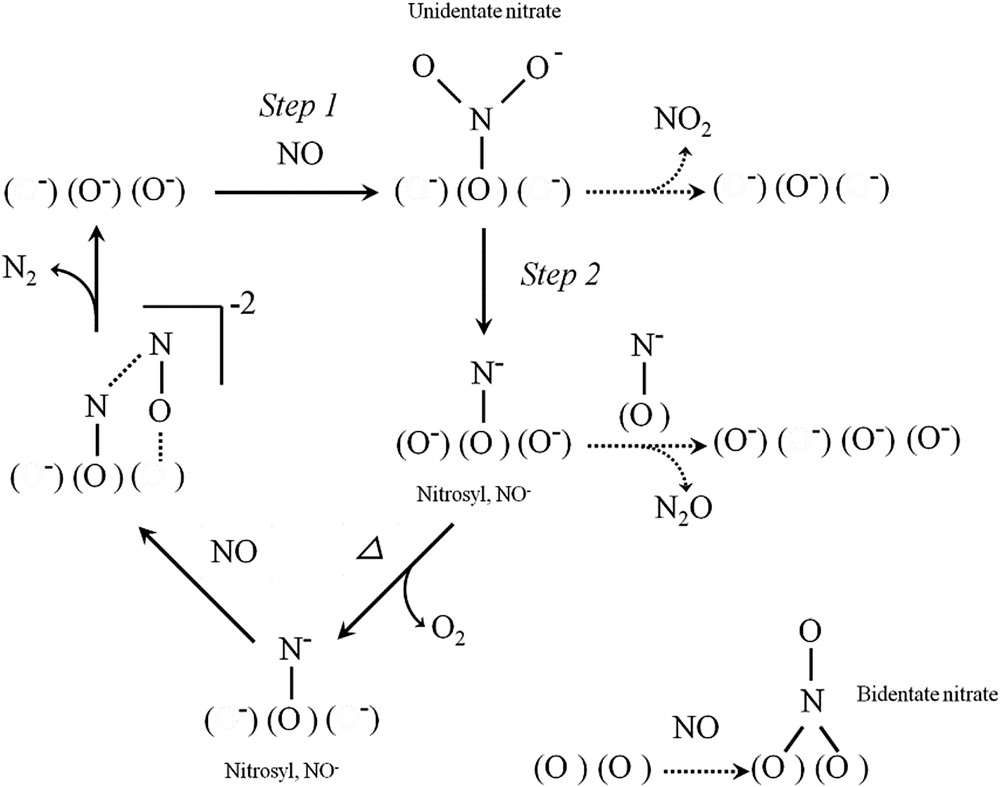
Proposed mechanism of NO decomposition over C-type cubic rare earth oxides [62].
Vannice et al. [75] suggested a reaction mechanism of NO decomposition over several alkaline earth and rare earth oxides (e.g., La2O3 and Sr/La2O3) in which oxygen defect sites do not serve as active sites. They proposed a Langmuir–Hinshelwood model in which a quasi-equilibrated NO adsorption step is assumed and a reaction of adsorbed NO leading to the formation of N2O is the rate-determining step. On the other hand, Xie et al. [76,77] explained NO decomposition over Ba/MgO by a modified Eley–Rideal mechanism, in which the reaction between the gas-phase NO and a surface reactive species, presumably ionic NOx species such as , is assumed to be the rate-determining step. As given in Fig. 11, a Ba2+ – NO2 species, detected by in situ Raman spectroscopy, is proposed to be an intermediate in NO decomposition over Ba/MgO, suggesting that the active sites are not oxygen defects but Ba sites.
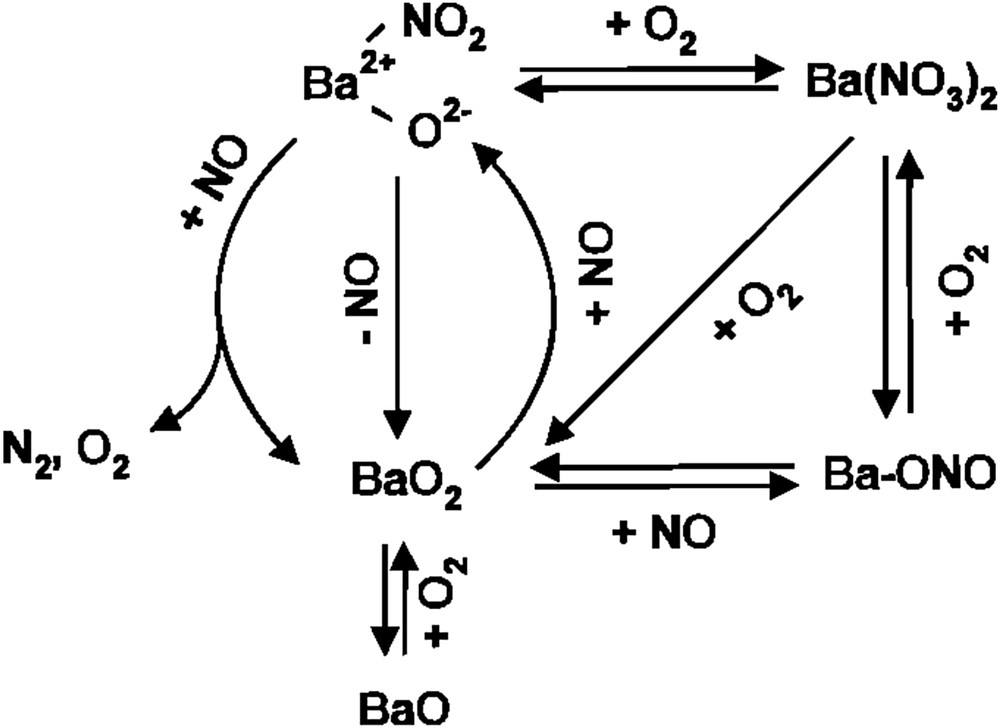
Proposed reaction scheme of NO decomposition over Ba/MgO [76].
Haneda et al. [68,89] investigated the kinetics of NO decomposition over alkali metal-doped Co3O4 catalysts, and revealed that NO decomposition proceeds via a bimolecular reaction because the reaction order with respect to NO was higher than unity. The formation of nitrite species () on the catalyst surface was observed during NO decomposition by in situ FT-IR spectroscopy. Isotopic transient kinetic analysis performed by 14NO → 15NO switch revealed the formation of mixed labeled 14N15N (Fig. 12), suggesting that a surface-adsorbed NO2− species serves as an intermediate in the NO decomposition reaction. Because the gas switch from 14NO/He to 15NO/He caused a slow decrease in the formation of 14N2, a Langmuir–Hinshelwood model is likely to be a better description of the process. They proposed the following reaction mechanism for NO decomposition over alkali metal-doped Co3O4 catalysts:
| NO (g) + * ⇄ NO* | (3) |
| NO * + O2-→NO2− * + [ ]− | (4) |
| (5) |
| N2Ox *+(x−1)*→N2 (g) + xO*(x < 3) | (6) |
| 2O *⇄O2 (g) + 2 * | (7) |
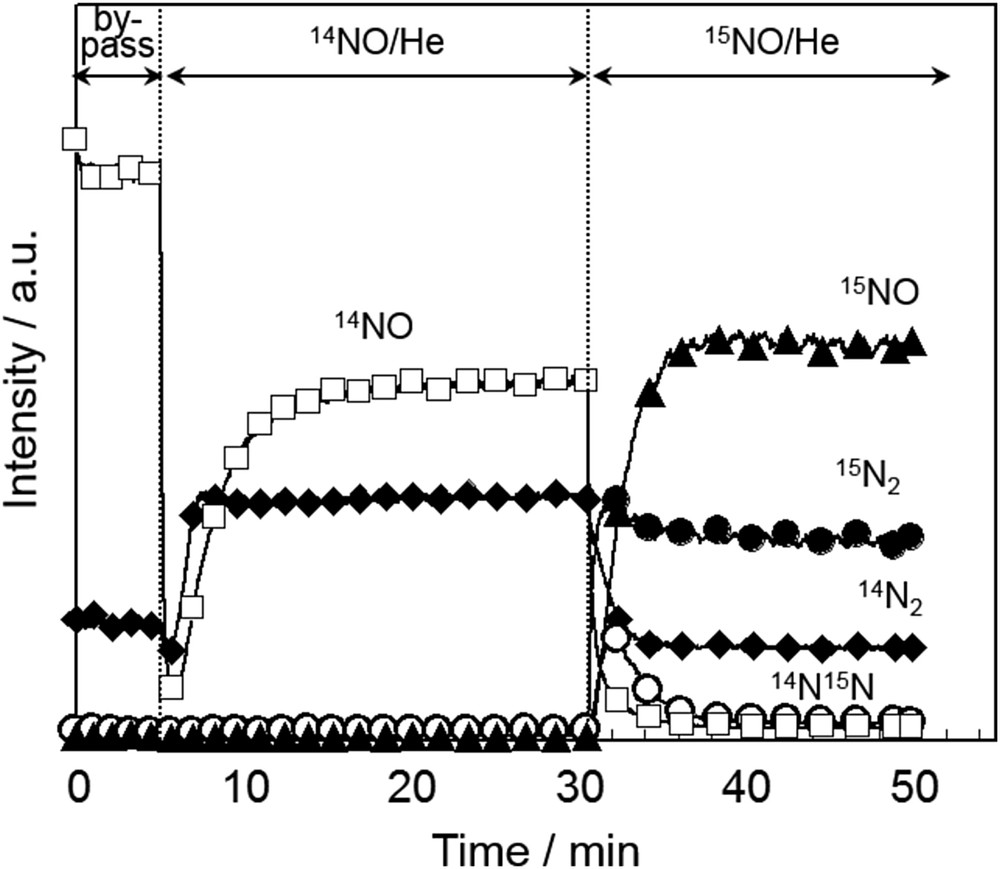
Product responses (14N2, 14N15N, 15N2, 14NO, and 15NO) following the replacement of 14NO/He with 15NO/He in the reaction stream over K/Co3O4 at 600 °C [68,89]. Conditions: NO = 3000 ppm, gas flow rate = 30 cm3 min−1, and catalyst weight = 0.1 g.
Haneda et al. [84–86] also performed a mechanistic study of NO decomposition over Ba–Y2O3 and Ba–CeO2–Y2O3 catalysts by in situ FT-IR spectroscopy and isotopic transient kinetic analysis. In accordance with the reaction over alkali metal-doped Co3O4 catalysts, a reaction mechanism was proposed in which NO is adsorbed onto Ba sites as NO2- species, which migrates to the interface between Ba and Y2O3, and then reacts with the adsorbed NO species to form N2.
5 Influence of coexisting gases on NO decomposition
Although a wide variety of catalysts have been found for direct NO decomposition as mentioned before, coexisting gaseous O2 significantly decreases the activity of almost all the NO decomposition catalysts. This is due to the strong adsorption of O2 on the catalytically active sites, inhibiting NO adsorption. Amirnazmi et al. [9] reported that the reciprocal rate of NO decomposition over Pt/Al2O3 is linearly proportional to the O2 concentration, indicating that the reaction order with respect to O2 is ca. −1.0. Iwamoto et al. [15] examined the influence of coexisting O2 on the NO decomposition activity of Cu-ZSM-5 with a Cu ion exchange level of 122% under the conditions of NO = 0.5% and W/F = 4.0 g s cm−3, and found that the maximum NO conversion was slightly decreased from 55% to 40% when 8% O2 was added to the reaction gas. On the other hand, they also revealed that the maximum NO conversion over Cu-ZSM-5 was drastically decreased from 23% to 2% by the addition of 1% O2 into 0.1% NO/He reaction gas. The influence of coexisting O2 on the NO decomposition activity seems to be dependent on the NO concentration.
Effects of the O2 concentration on the NO decomposition activity of metal oxide-based catalysts including perovskite-type oxides have also been examined by many researchers. For instance, Ishihara et al. [43,90] investigated the effect of coexisting O2 on NO decomposition over BaMnO3-based perovskite-type oxides. They revealed that the activity of La0.7Ba0.3Mn0.8In0.2O3 was significantly inhibited by the presence of O2 with a reaction order of −0.53, while the substitution of Mn sites with Mg (BaMn0.8Mg0.2O3) relieved the inhibiting effect of coexisting O2 with a reaction order of −0.18. Haneda et al. [86,89] performed a kinetic study on NO decomposition over alkali metal-doped Co3O4 and CeO2-doped Ba–Y2O3 catalysts and found that the presence of O2 decreased the NO decomposition activity with reaction orders between −0.26 and −0.40 depending on the types of catalysts. They also compared the catalytic activities of K/Co3O4 (K/Co = 0.035), Cu-ZSM-5 (Cu, 3.2 wt%), Pd/Al2O3 (Pd, 1 wt%) and Ba–CeO2(10)-Y2O3 (Ba, 8 mol%) for direct NO decomposition in the absence and presence of 1% O2 under the conditions of NO = 0.1% and W/F = 0.5 g s cm−3 [67,86]. As is presented in Fig. 13, the effective temperatures giving high NO decomposition activity changed depending on the catalysts: 550–700 °C for K/Co3O4, 350–550 °C for Cu-ZSM-5, above 650 °C for Pd/Al2O3 and above 650 °C for Ba–CeO2(10)–Y2O3. The extent of activity depression by O2 also changed depending on the catalysts; namely, the activity depression of Cu-ZSM-5 and Pd/Al2O3 was noticeable, while that of Ba–CeO2–Y2O3 and K/Co3O4 was not so large. It is noteworthy that the maximum NO conversion on Ba–CeO2–Y2O3 was higher than that over Cu-ZSM-5 and K/Co3O4.
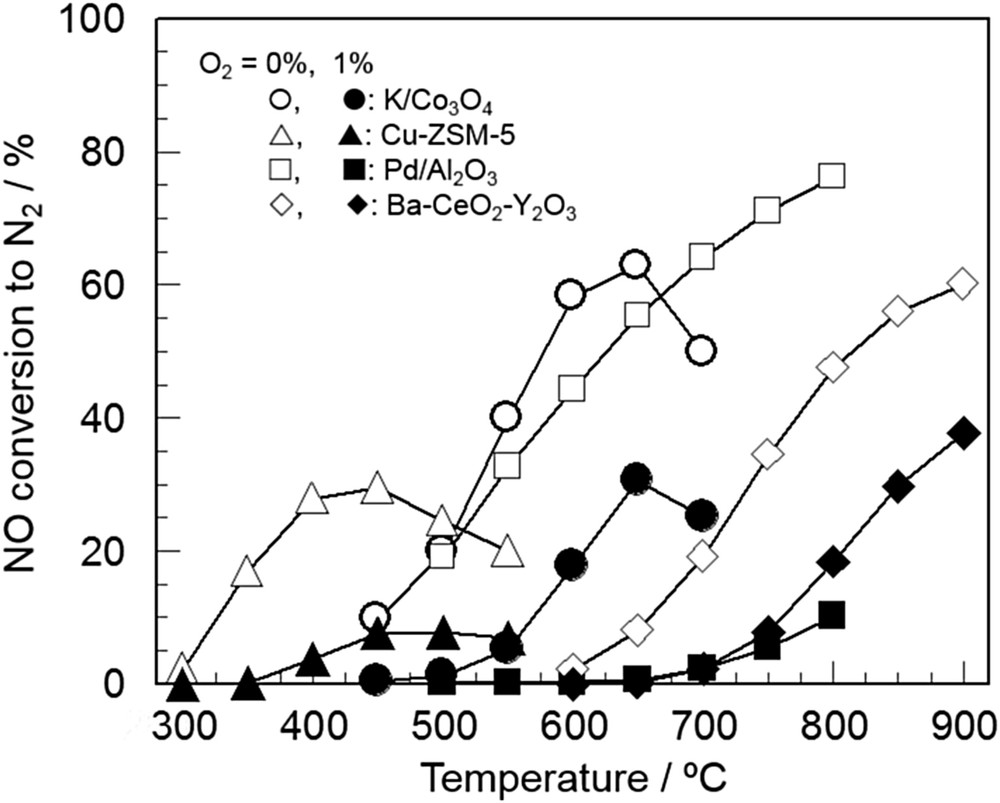
Comparison of the catalytic activity of K/Co3O4 (K/Co = 0.035), Cu-ZSM-5 (Cu, 3.2 wt%), Pd/Al2O3 (Pd, 1 wt%) and Ba–CeO2(10)-Y2O3 (Ba, 8 mol%) for NO decomposition [67,86]. Conditions: NO = 1000 ppm, O2 = 0 or 1%, He balance, and W/F = 0.5 g s cm−3.
In addition to O2, CO2 is another important constituent which may affect direct NO decomposition because the NO decomposition process would be used for exhaust gas treatment of combustion facilities. Teraoka et al. [91] investigated the effect of CO2 on NO decomposition over La-based perovskites, using La0.8Sr0.2CoO3 as a representative catalyst sample. The N2 yield on La0.8Sr0.2CoO3 was drastically decreased from ca. 58% to ca. 10% by the coexistence of 5% CO2 under the conditions of NO = 0.9% and W/F = 4.0 g s cm−3 at 800 °C. It is noteworthy, however, that the NO decomposition activity was almost completely recovered after CO2 removal from the reaction gas. They considered that the CO2 impact is related to the formation of carbonate species, which may hinder the access of NO molecules to the catalytically active oxide anion vacancies. Iwakuni et al. [92] also reported that the N2 yield on the BaMnO3-based perovskite, Ba0.8La0.2Mn0.2O3, decreased from 70% to 30% by the addition of 1% CO2 into the reaction gas composed of 1% NO/He (W/F = 3.0 g s cm−3) at 850 °C, indicating a much larger negative effect than that seen with O2. The negative effect of CO2 was even stronger in NO decomposition over the catalysts including alkaline earth elements such as Ba. This is probably because CO2 strongly adsorbs on alkaline earth metals, forming thermally stable carbonate species.
On the other hand, Imanaka et al. [58,63–65] reported relatively high tolerance toward CO2 for C-type cubic rare earth oxides not including alkaline earth elements. As given in Fig. 14, the N2 yield on (Y0.90Pr0.10)2O3 + δ was decreased from 80% to 58% by the coexistence of 1% CO2 under the conditions of NO = 1.0% and W/F = 3.0 g s cm−3 at 900 °C. However, further increase in the CO2 concentration up to 10% did not cause significant decreases in the NO decomposition activity [63]. CO2 poisoning seems to be much smaller for C-type cubic rare earth oxides than that observed for other conventional catalysts such as perovskite-type oxides. Long-term durability tests are necessary to understand the potential of C-type cubic rare earth oxides for practical applications.
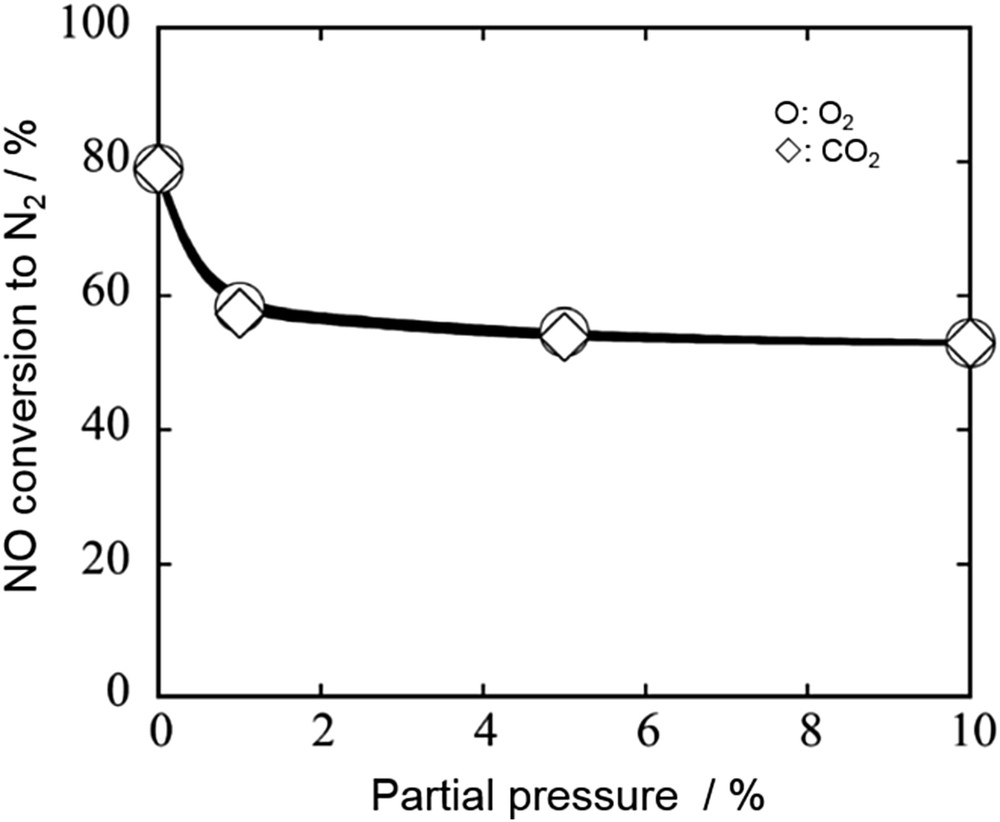
Effect of partial pressures of O2 and CO2 in the feed gas on the conversion of NO over the (Y0.90Pr0.10)2O3+δ catalyst at 900 °C [63]. Conditions: NO = 1.0%, O2 = 0–10%, CO2 = 0–10%, He balance, and W/F = 3.0 g s cm−3.
6 Conclusions
The direct NO decomposition, which needs no reductants, is the most desirable NOx abatement technology. Since the discovery of Cu-ZSM-5 as an epoch-making NO decomposition catalyst [5], various kinds of catalysts have been reported to effectively catalyze this reaction. However, no catalysts exhibit high activity under realistic conditions in the presence of coexisting gases (O2, CO2, H2O) and at high space velocities. Recently, various kinds of metal oxide-based catalysts such as perovskites, C-type cubic rare earth oxides and alkaline earth based oxides have been developed as new types of catalysts for possible practical applications, although the NOx removal performance needs more improvement. Further studies should be continued on the basis of the current knowledge on the structure of catalytically active sites and reaction mechanisms described in this article.
Acknowledgments
This study was partially supported by a Grant–in–Aid for Scientific Research (No. 22350068) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.