

1 Introduction
According to the new European regulations, the European countries will have to guarantee that at least 20% of the total energy consumption comes from renewable energy by the end of 2019 [1]. More specifically concerning the transportation sector, 10% of the energy consumption will have to come from renewable energy, including biofuels. The incorporation of renewable feedstocks such as lignocellulosic biomass to petroleum crudes will lead to the treatment of petroleum and biomass sources in the same processes. This means that oxygenated compounds coming from highly oxygenated vegetable oils or lignocellulosic biomass (between 20–40 wt%), such as acid, ester, alcohol functions … [2–5], should be treated in the same hydroprocessing unit as the petroleum fractions to produce ultra-low sulfur fuels (gasoline and diesel). However during hydrodesulfurization processes, oxygenate conversion leads to the formation of by-products such as carbon monoxide (CO), carbon dioxide (CO2) and/or water (H2O) [6,7] depending on the operating conditions and the catalysts. Water is the main by-product in the transformation of alcohols and is also involved in the transformation of carboxylic acids. Carbon monoxide and dioxide are formed by decarbonylation or decarboxylation reactions [8–13]. Therefore, the challenge is to maintain a high catalyst performance for the HDS reaction in order to provide a maximum of 10 wt ppm of sulfur as imposed by the European Union [14], despite the incorporation of new renewable feedstocks. The characteristics of FCC gasoline and straight run gas oil as well as the usual operating conditions are given in Table 1. FCC gasoline is lighter than SR gas oil according to their respective usual distillation range (50–250°C and 180–380°C) and contain much less sulfur compounds. This will result to more severe operating conditions for hydrotreating a diesel fraction, i.e. higher pressure and temperature.
Typical characteristics of FCC gasoline and straight run gas oil and usual operating conditions [34,35].
| Characteristics | FCC gasoline | Straight run gas oils | |
| Distillation range | °C | 50–250 | 180–380 |
| Density at 15 °C | g/cm3 | 0.73 [36] | 0.84–0.86 |
| Sulfur content | wt ppm | 50–7000 | 7000–20 000 |
| Nitrogen content | wt ppm | 20–200 | 50–400 |
| Aromatic content | wt% | 20–40 | 25–40 |
| Diolefins | wt % | 1–2 | – |
| Cetane number | – | 40–60 | |
| Research octane number | 85–95 | – | |
| Motor octane number | 75–85 | – | |
| Bromine number | 15–139 [37] | <3 | |
| Usual operating conditions | |||
| Temperature | °C | 260–320 | 320–380 |
| Pressure | bar | 5–30 | 10–100 |
| H2/HC | L/L | 1–20 | 50–400 |
| HSV | h−1 | 1–6 | 0.5–2.0 |
| Phase | Gas | Gas/liquid | |
| Catalyst | CoMo | CoMoP or NiMoP |
Independent of the incorporation of biomass feedstock in the refinery which will become an extra source of oxygenated compounds, it is known that CO is a main issue for the FCC gasoline HDS process [15]. Indeed CO is known as a main contaminant of this process. It originates from the refinery H2 main stream. When H2 comes from steam reforming or partial oxidation of hydrocarbons, it may contain no negligible amount of CO. Even if H2 is treated before entering the FCC gasoline HDS process, it still may contain more than 100 wt ppm with a detrimental loss of the overall performances.
To the best of our knowledge, CO is not known as an issue on the diesel HDS process. However the incorporation of renewable feedstocks such as vegetable oil into a diesel hydrotreating unit might lead to different kind of problems such as [16]: more exothermic reaction, higher H2 consumption, higher content of contaminants such as Phosphorus, Calcium or Sodium which all might lead to accelerated catalyst deactivation and pressure drop build-up. Moreover carbonic acid could lead to increased corrosion rate and CO and CO2 may limit the S or N conversion by competitive adsorption on the catalyst. This last point has been confirmed by introducing 1 vol% CO in H2 while processing light gas oil with CoMo or NiMo catalyst. For the CoMo catalyst, HDS and HDN respectively dropped to 35 and 65% of the initial activity, while NiMo catalyst was barely sensitive.
Only a few works in the literature are devoted to the effect of the by-products coming from renewable feedstocks hydroprocessing in the HDS processes (gasoline and gas oils). For example, Pinheiro et al. [17,18] showed that, under industrial operating conditions, 2-propanol, cyclopentanone, anisole, and guaiacol, which have been totally decomposed to water and hydrocarbons, were not found to inhibit the CoMo/Al2O3 catalytic performances during straight-run gas oil hydroprocessing. On the contrary, propanoic acid and ethyldecanoate had a strong inhibiting effect on hydrodesulfurization (HDS), hydrodenitrogenation (HDN), and hydrogenation of aromatics (HDAr) reactions due to CO and/or CO2 production. Meanwhile these results have been confirmed by Egeberg et al. [16] who have shown that introducing 15% of rapeseed oil while processing a light gas oil induces a strong decrease of HDS an HDN activity, respectively 20 and 55% on the CoMo catalyst, attributed to the transformation of oxygenated compounds into CO. Philippe et al. [19] showed that oxygenates such as phenol, guaiacol, decanoic acid inhibited the HDS transformation of dibenzothiophene (DBT) and 4,6-dimethyldibenzothiophene (4,6-DMDBT) when these compounds were not totally converted. However, when phenol or guaiacol was totally converted, i.e. to water and hydrocarbons, no effect in HDS was noticed showing that water had no impact on HDS performances. Conversely, the decomposition of decanoic acid to CO led to a strong inhibiting effect [20]. In that respect, density functional theory (DFT) studies have also shown that CO must be considered as a much stronger inhibitor than guaiacol [21], alcohol or carboxylic acid [22–24]. In HDS of a model feed representative of FCC gasoline under various operating conditions, Pelardy et al. [25] confirmed the strong impact of CO on the HDS of 2-methylthiophene and the hydrogenation of 2,3-dimethylbut-2-ene in mixture. The authors explained these results by the mutual competitive adsorption on the catalyst surface and the higher adsorption energy of CO compared to other compounds (sulfur compounds and alkenes) determined by density functional theory (DFT) calculations. CO adsorption is indeed thermodynamically favored on mixed CoMoS sites on the S-edge and M-edge, which explains the loss in HYD and HDS activities.
The present paper summarizes and compares the main results on the impact of CO presence on the transformation of two model feeds of petroleum fractions (FCC gasoline and Straight-Run gas oil). We focus more specifically on the different impact of CO depending on the operating conditions (T, P), the feed (gasoline or gas oil) and the catalysts (CoMoS/Al2O3 or CoMoPS/Al2O3). The model compounds chosen for describing the petroleum fractions were 2-methylthiophene (2 MT) and 2,3-dimethylbut-2-ene (2,3-DMB2N) for the model FCC gasoline and dibenzothiophene (DBT) and 4,6-dimethyldibenzothiophene (4,6-DMDBT) for the model SR gas oil.
2 Experimental part
2.1 Catalysts and chemicals
As reported previously [19,29,25], catalysts used are a CoMo catalyst containing 3 wt% CoO and 10 wt% MoO3 supported on alumina for the HDS of FCC gasoline and a CoMoP/Al2O3 catalyst containing 4 wt% CoO, 19 wt% MoO3 and 2.6 wt% of phosphorus for the HDS of SR gas oil. These catalysts were provided by IFPEN. Both catalysts have been crushed and sieved to a 250–315 μm size range.
The catalysts were sulfided in situ under H2S/H2 flow (10 mol% H2S) for 10 h at 673 K at atmospheric pressure before gasoline HDS measurements [24] or using a sulfiding feed made of 4.75% by volume of dimethyl disulfide (DMDS) in toluene as a solvent under a 4.0 MPa of total pressure before gas oil HDS measurements [19,20].
2-methylthiophene (98% purity) and decanoic acid (>98% purity) have been purchased from Alfa Aesar, 2,3-dimethylbut-2-ene (98% purity) from Acros Organics, o-xylene (>99% purity), dimethyl disulfide (>98% purity), toluene (>99% purity) from Fluka, n-heptane (>99% purity) from Carlo Erba, 4,6-dimethyldibenzothiophene (>95% purity) from Eburon Organics and guaiacol (>98% purity) and phenol (>99% purity) from Aldrich Chemicals. They were used without further purification. Carbon monoxide and carbon dioxide (1 or 10 vol% in mixture with H2) have been purchased from Air Liquide.
2.2 Reaction conditions
Reaction conditions are those described in previous papers respectively for HDS of gasoline [25] and for HDS of gas oils [19,20], only the main conditions are reported herein. We report coherent operating conditions with usual industrial ones given in Table 1
For HDS of FCC gasoline, catalytic activity measurements were carried out in a fixed bed reactor at 250 °C under a total pressure of 2 MPa with a ratio H2/feed of 360 NL/L. Using these conditions, all the components are in the gas phase. The model feed FCC gasoline containing 0.3 wt% of 2 MT, 20 wt% of 2,3-DMB2N, and 30 wt% of o-xylene (representing aromatics) diluted in n-heptane was injected in the reactor by a HPLC Gilson pump (307 series, pump's head: 5 cm3) (Table 2).
Partial pressures (MPa) of the different compounds for the sulfidation step and the transformation of the model FCC gasoline feed.
| Pressure (MPa) | Sulfidation | Model FCC gasoline |
| Polefin | 0 | 0.150 |
| 0.01 | 0 | |
| P2MT | 0 | 0.003 |
| Po-xyl | 0 | 0.190 |
| 0.09 | 1.31 | |
| PnC7 | 0 | 0.347 |
| PTOT | 0.1 | 2 |
The impact of CO on the transformation of the model feed has been evaluated according to the experimental procedure reported in a previous paper [25]. The amount of CO varied from 0 to 1.31 kPa.
For HDS of gas oil, the HDS of 4,6-DMDBT (or DBT) was carried out also in a fixed bed microflow reactor at 340 °C and 4.0 MPa of total pressure after an in situ sulfidation of the catalyst according to the procedure described above [19,20]. In these conditions, all reactants and products are totally vaporized. 4,6-DMDBT or DBT (500 wt ppmS) were dissolved in a mixture of toluene to which dimethyl disulfide (DMDS) (9500 wt ppmS) was added to generate H2S. To examine the effect of oxygenates on the 4,6-DMDBT or DBT transformation, the partial pressure of the latter was maintained constant at the standard 1.9 kPa (Table 3) while the pressure of CO varied from 0 to 6 kPa.
Partial pressures of the different compounds for the sulfidation step and the transformation of 4,6-DMDBT or DBT.
| Pressure (MPa) | Sulfidation | DBT or 4,6-DMDBT feed |
| PDBT or 4,6-DMDBT | 0 | 0.002 |
| and | 0.12 | 0.036 |
| Ptoluene | 1.16 | 1.299 |
| 2.6 | 2.627 | |
| PTOT | 4.0 | 4.0 |
For a better precision in the activity measurements, the contact time was chosen such as to keep also the overall conversion of sulfur compounds nearly constant around 25 mol% (24–27 mol%). Hydrogenation (HYD) and direct desulfurization (DDS) activities of the catalyst are defined as the number of moles of sulfur compounds transformed totally by the HYD way or by the DDS way per gram of catalyst and per hour.
The inhibition effect of the CO compound in the feed was measured by the loss of activity measured by the A/A0 ratio where A was the catalyst activity in the transformation of the sulfur compound in the presence of the oxygenated compounds and A0, the catalyst activity in the transformation of the sulfur compound or alkenes without CO.
2.3 Product analysis
The reaction products of the model FCC gasoline have been analyzed online on the fixed bed unit by means of a Varian gas chromatograph equipped with an automatic sampling valve as described in our previous works [19,20,25]. Desulfurized products (mainly pentanes and pentenes), resulting from the transformation of 2-methylthiophene are designated as HDS products. HDS products are the main observed products according to the reaction scheme described in the literature [26,27] (Scheme 1 : Reaction pathway of the 2-methylthiophene hydrodesulfurization Scheme 1). The transformation of the 2,3-dimethylbut-2-ene (2,3-DMB2N) leads to the formation of isomerization products (mainly 2,3-dimethylbut-1-ene, 2,3-DMB1N) and hydrogenation products (mainly 2,3-dimethylbutane, 2,3-DMB) as described previously (Scheme 2). The double bond isomerization of 2,3-DMB2N to 2,3-DMB1N is known to be very fast on transition metal sulfide catalysts so that the mixture composed of 2,3-DMB2N and 2,3-DMB1N is considered as the main reactant [25,26]. The hydrogenation activity has been measured from the formation of 2,3-DMB which was the main hydrogenation product. Skeletal isomers and their hydrogenated products have been obtained with a yield of less than 1%.
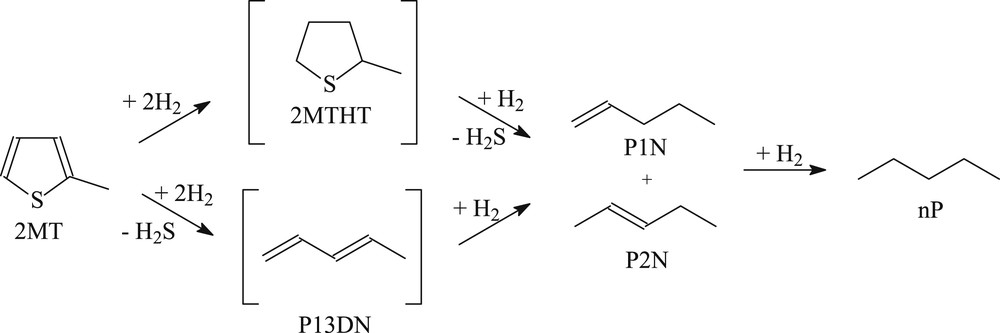
Reaction pathway of the 2-methylthiophene hydrodesulfurization 2 MT: 2-methylthiophene, 2MTHT: 2-methyltetrahydrothiophene, P13DN: pent-1,3-diene, P1N: pent-1-ene, P2N: pent-2-ene, nP : n-pentane.
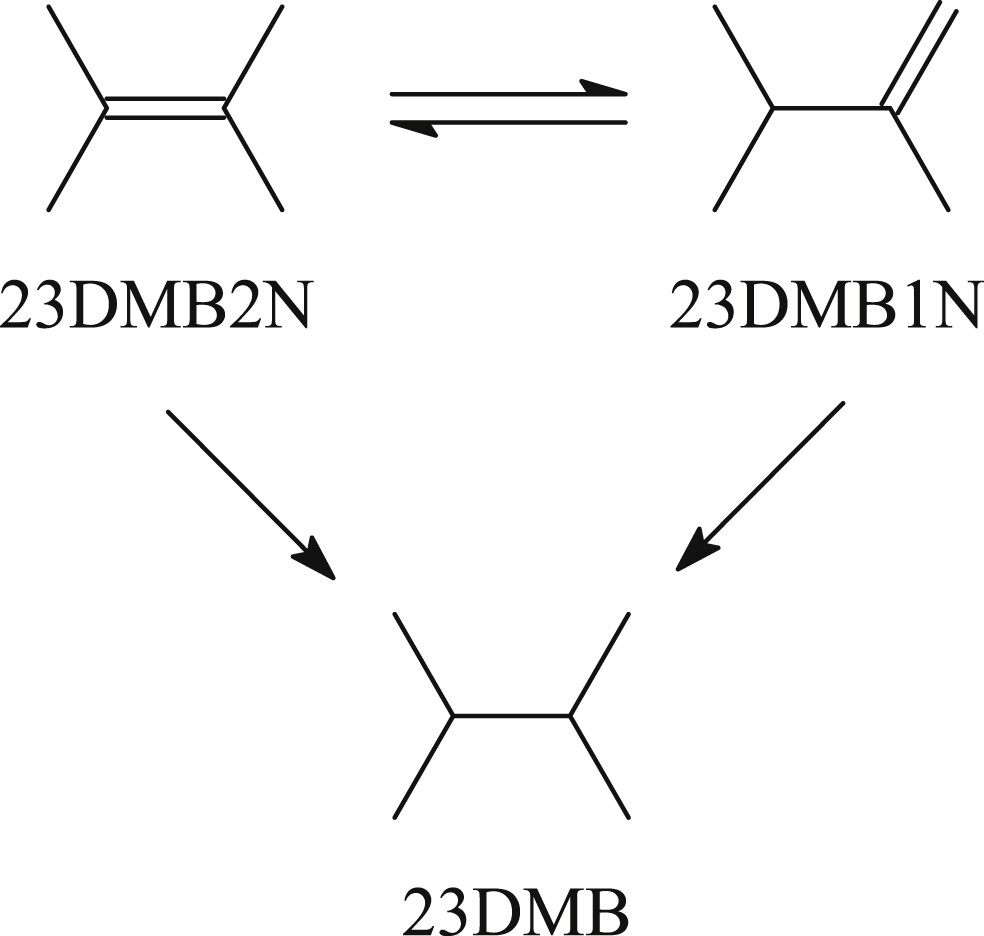
Reaction pathway of the 2,3-dimethylbut-2-ene hydrogenation 2,3-DMB2N: 2,3-dimethylbut-2-ene, 2,3-DMB1N: 2,3-dimethylbut-1-ene, 2,3-DMB: 2,3-dimethylbutane.
The catalyst activity (±2%) in hydrodesulfurization is defined as the number of moles of HDS products formed by total hydrodesulfurization of 2 MT, per hour and per gram of catalyst, and the catalyst activity (±2%) in hydrogenation is defined as the number of moles of 2,3-DMB formed by hydrogenation of 2,3-DMB2N per hour and per gram of catalyst. HDS and HYD activities were measured after stabilization of HDS and HYD product formation. The selectivity of the reaction was calculated by the ratio between hydrodesulfurization (HDS) and olefin hydrogenation (HYD) rate constants (kHDS/kHYD), assuming ideal plug flow reactor in the gas phase and first order reactions, as defined by Dos Santos et al. [28].
Considering the SR gas oil hydrotreating, the reaction products were condensed and liquid samples were periodically collected to be analyzed by gas chromatography. The analyses were carried out with a Varian 3400 chromatograph equipped with a 25 m BP1 (SGE) capillary column (inside diameter: 0.32 mm; film thickness: 5 μm) with a temperature program from 50 to 70 °C (4 °C/min) then from 70 to 250 °C (15 °C/min).
In both cases, carbon monoxide, carbon dioxide and methane have been analyzed online by a Varian 3800 chromatograph equipped with an automatic sampling valve, two Porapack columns 1m × 1/8″ × 2 mm, a methanizer and a flame ionization detector. A backflush procedure has allowed the elimination of H2S and any other organic compounds (which could poison the methanizer Ni catalyst).
3 Results and discussion
3.1 Transformation of the various model feeds without CO incorporation
The transformation of the FCC gasoline model feed (2 MT and alkenes in mixture) is carried out over CoMo/Al2O3 usually used for gasoline hydrodesulfurization. Fig. 1a depicts the evolution of the 2 MT conversion as a function of the contact time. The 2 MT transformation increases linearly until a conversion close to 70%. The complete conversion of 2 MT is obtained for higher contact time. The two main products of complete desulfurization are C5 non-cyclic hydrocarbons: n-pentane and pentenes. As shown in Fig. 1b, pentenes (mainly present in the trans-pent-2-ene, cis-pent-1-ene and pent-1-ene forms) are primary products of the reaction which will be then completely hydrogenated to n-pentane at higher conversion (according to Scheme 1). The transformation of 2,3-DMB2N or more exactly the transformation of the alkenes (2,3-DMB2N+2,3-DMB1N) increases linearly until conversions around 40%, (Fig. 2). The main observed product is the direct hydrogenation product: 2,3-dimethylbutane (2,3-DMB) (Scheme 2). The ratio of the rate constants in HDS over HYD which represents the selectivity and is the key parameter in gasoline hydrotreating reactions is reported in Fig. 3 as a function of HDS yield. As awaited, the selectivity depends on the HDS rate. Indeed, the selectivity decreases linearly while the HDS conversion increases. The conversion of 2 MT is accompanied by a decrease of its inhibiting effect on hydrogenation and therefore by a simultaneous increase of the HYD rate. These results correspond to a competitive adsorption between the various reactants (2 MT, 2,3-DMB2N, H2S) on the catalyst surface as presented in more details by Pelardy et al. [25]. The comparison of the transformation of the model molecule alone and in the mixture demonstrates a mutual inhibition effect of the components in the model FCC gasoline. However, an amount of 0.3 wt% of 2 MT inhibits the olefin saturation, while a much higher amount of olefin (20 wt%) is required to inhibit the conversion of the sulfur organic compound as reported previously [24]. These results are consistent with the DFT work carried out by Krebs et al. [29], which has highlighted the distinct behaviors of the two edges on the CoMoS mixed phase: S-edge which exhibits the preferential adsorption sites for 2 MT and the M-edge, where 2 MT and olefin may compete for the adsorption sites. Moreover 2 MT is more strongly adsorbed than olefin as shown by the adsorption index. It can be thus understood why a small partial pressure of 2 MT strongly inhibits the alkene hydrogenation [25].
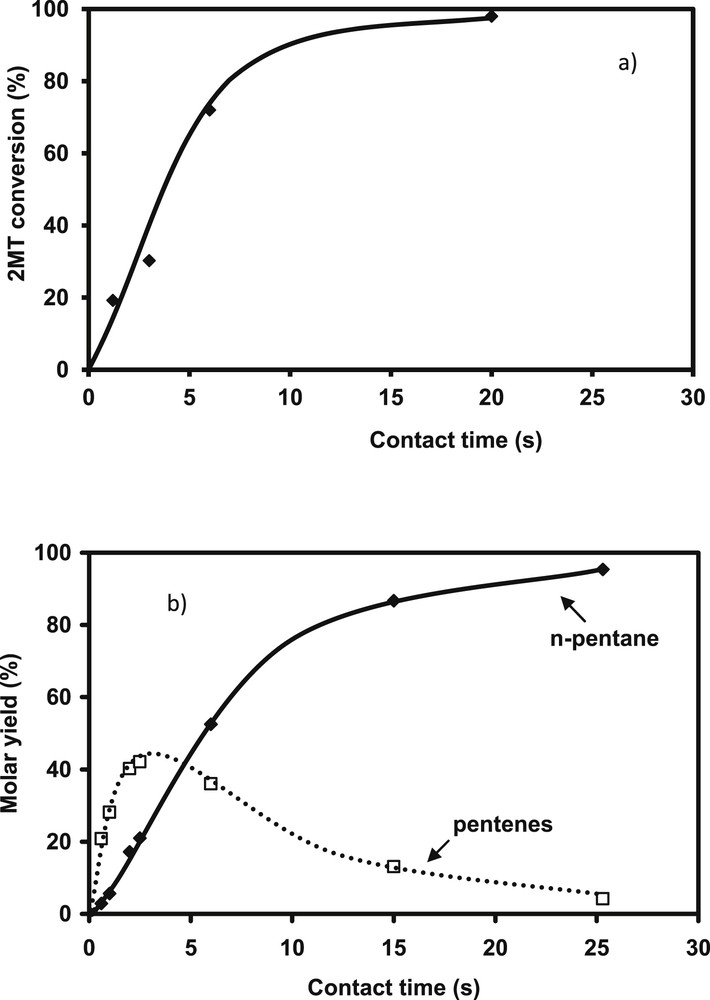
Transformation of the model FCC gasoline. 2 MT transformation a) Conversion versus contact time b) products distribution: n-pentane (♦, full line) and pentenes (□, dotted line) (T = 250 °C, P = 2 MPa, H2/HC = 360 NL/L, CoMo/Al2O3).
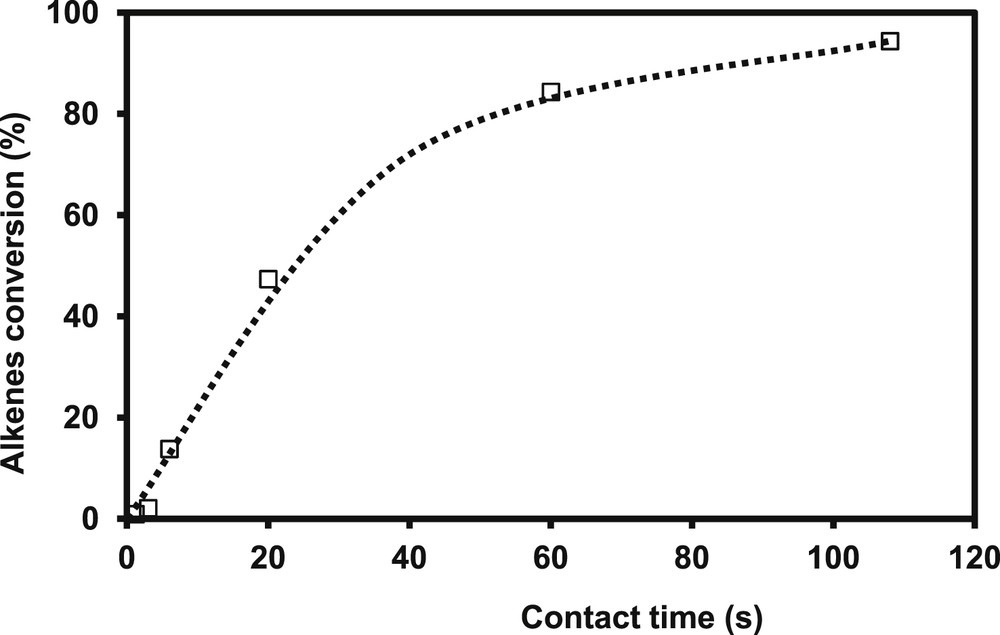
Transformation of the model FCC gasoline. Alkenes (2,3-DMNB2N+2,3-DMB1N) conversion as a function of contact time (T = 250 °C, P = 2 MPa, H2/HC = 360 NL/L, CoMo/Al2O3).
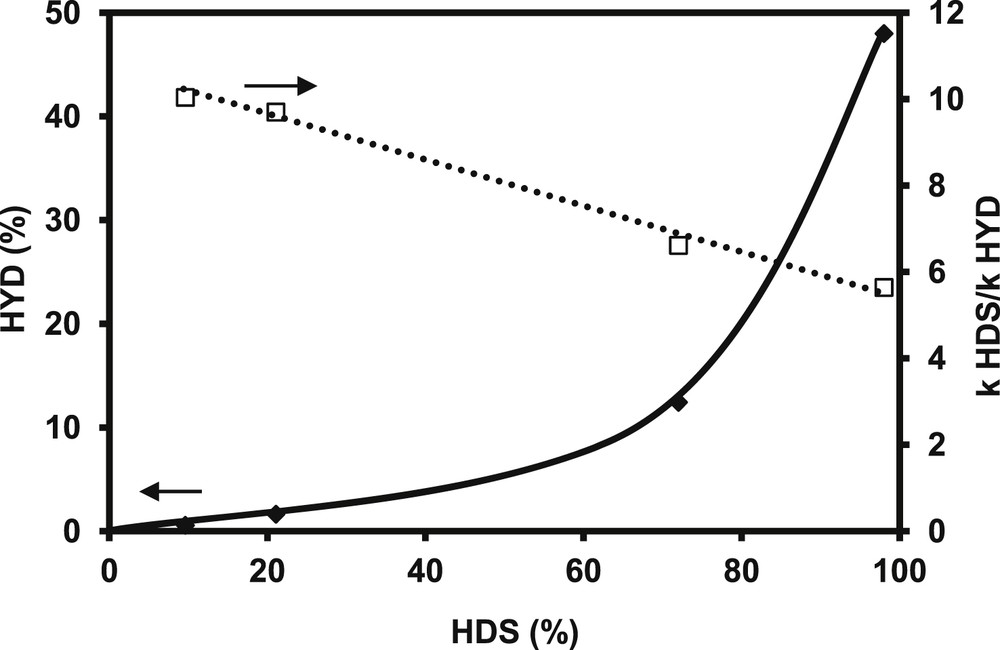
Transformation of the model FCC gasoline. HYD yield (♦, full line) and kHDS/kHYD ratio (□, dotted line) versus HDS yield (T = 250 °C, P = 2 MPa, H2/HC = 360 NL/L, CoMo/Al2O3).
Concerning the SR gas oil hydrotreating, the transformation of DBT and 4,6-DMDBT model molecules were studied at 340 °C, 4.0 MPa over a commercial CoMoP/Al2O3. In the same operating conditions, a linear increase of the conversion of both model compounds (Fig. 4 for 4,6-DMDBT and Fig. 5 for DBT) is observed at low contact times and a slope-change is noticed at higher. Indeed, the linear part is observed until a conversion of 50% of 4,6-DMDBT and around 60% for the DBT. The main products formed are respectively reported in Fig. 6 for the transformation of 4,6-DMDBT and in Fig. 7 for the transformation of DBT. As reported in previous works [30,31,19,20], the main product of the transformation of 4,6-DMDBT is the methylcyclohexyltoluene (MCHT) resulting due to the hydrogenation of an aromatic ring followed by C–S bond rupture (HYD way). Lower amount of dimethylbiphenyl (DMBP) (representative of the direct desulfurization way: DDS way) and dimethyltetrahydrodibenzothiophene (DMTHDB) were also observed. Conversely, the main product of the transformation of DBT is the biphenyl resulting from the direct C–S bond rupture (DDS way). The formation of lower amount of cyclohexylbenzene (CyHBz) and tetrahydrobenzothiophene (THDBT) is also noticed.
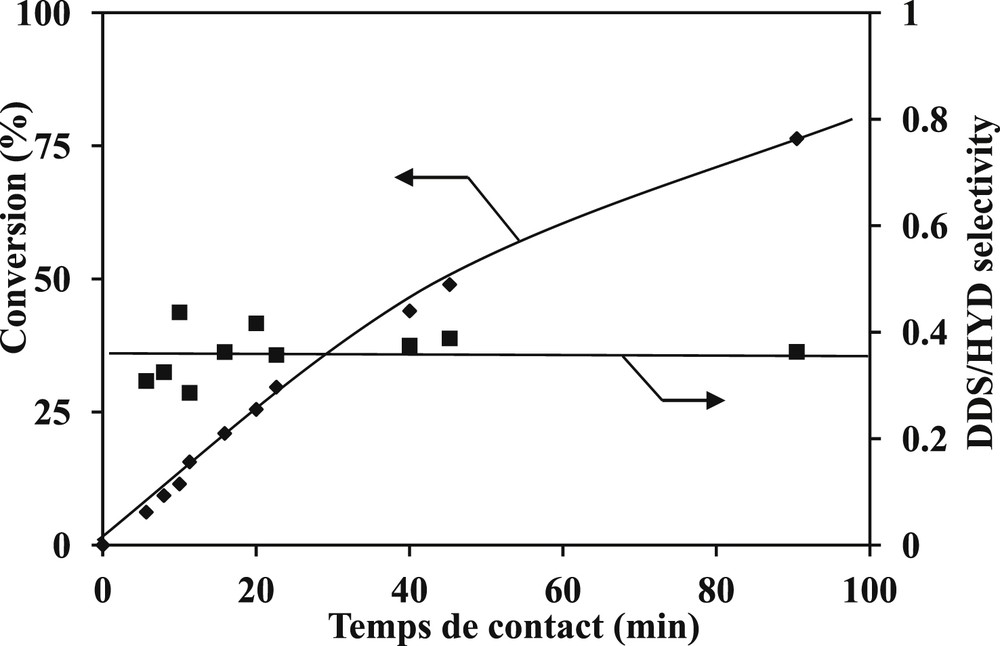
Transformation of 4,6-DMDBT. Conversion and DDS/HYD selectivity (T = 340 °C, P = 4 MPa, H2/HC = 468 NL/L, CoMoP/Al2O3).

Transformation of DBT. Conversion and DDS/HYD selectivity (T = 340 °C, P = 4 MPa, H2/HC = 468 NL/L, CoMoP/Al2O3).
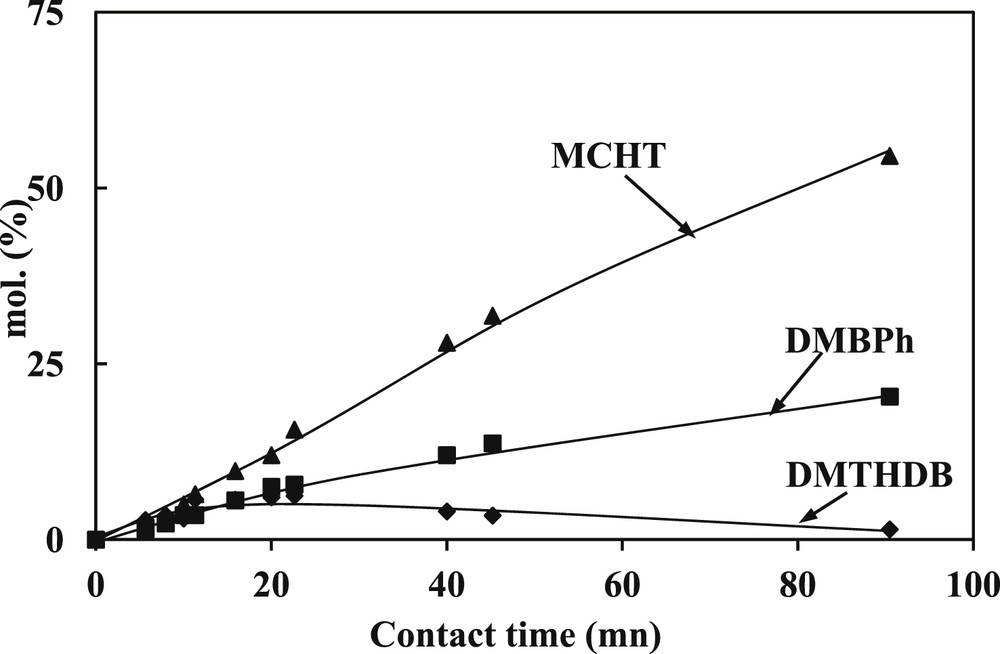
Transformation of 4,6-DMDBT. Distribution of the main products (T = 340 °C, P = 4 MPa, H2/HC = 468 NL/L, CoMoP/Al2O3).
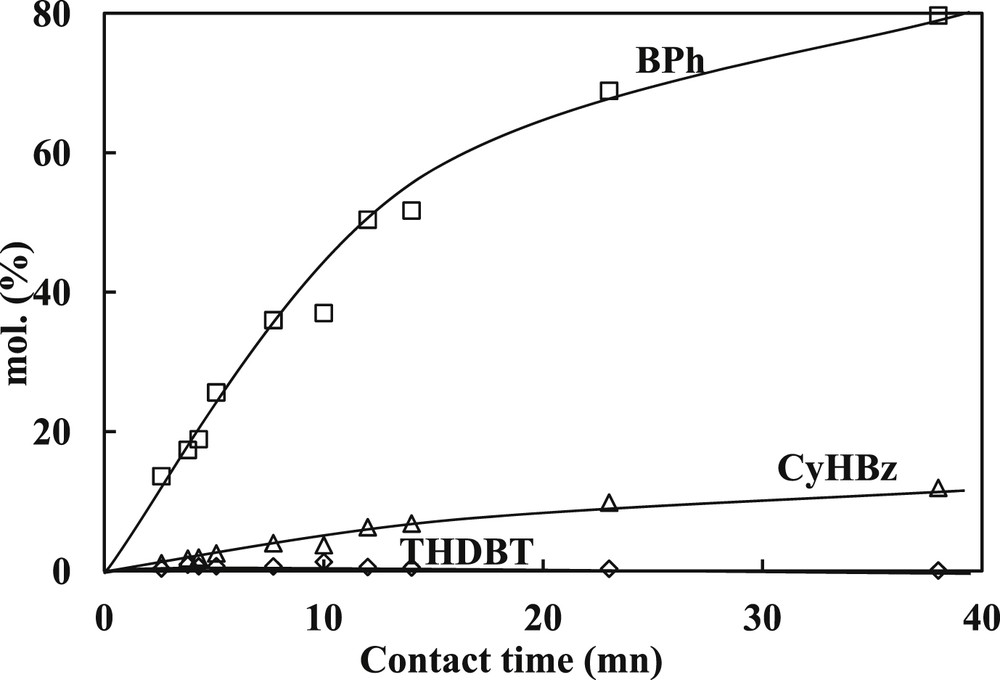
Transformation of DBT. Distribution of the main products (T = 340 °C, P = 4 MPa, H2/HC = 468 NL/L, CoMoP/Al2O3).
Regarding the selectivity towards the different two ways (DDS and HYD), measured by DDS/HYD ratios based on the conversion of both sulfur compounds (DBT and 4,6-DMDBT), we noticed that the ratios are constant whatever the conversions and contact times (Figs. 4 and 5). The DDS/HYD ratio which is around 0.3 for the transformation of 4,6-DMDBT confirms that the HYD is the main way, conversely for the transformation of DBT where the DDS way is the main way (DDS/HYD = 6.5). Moreover, these results confirm also that DBT is more reactive than 4,6-DMDBT as reported in Fig. 8 where the conversion of DBT is higher than the conversion of 4,6-DMDBT whatever the contact time and as reported in Table 4 where the activity of the catalyst and the DDS/HYD selectivity were compared. These results which are in accordance with previous studies [29,30,18,19] are in agreement with the generally accepted reaction scheme of the transformation of DBT (Scheme 3) and 4,6-DMDBT (Scheme 4).
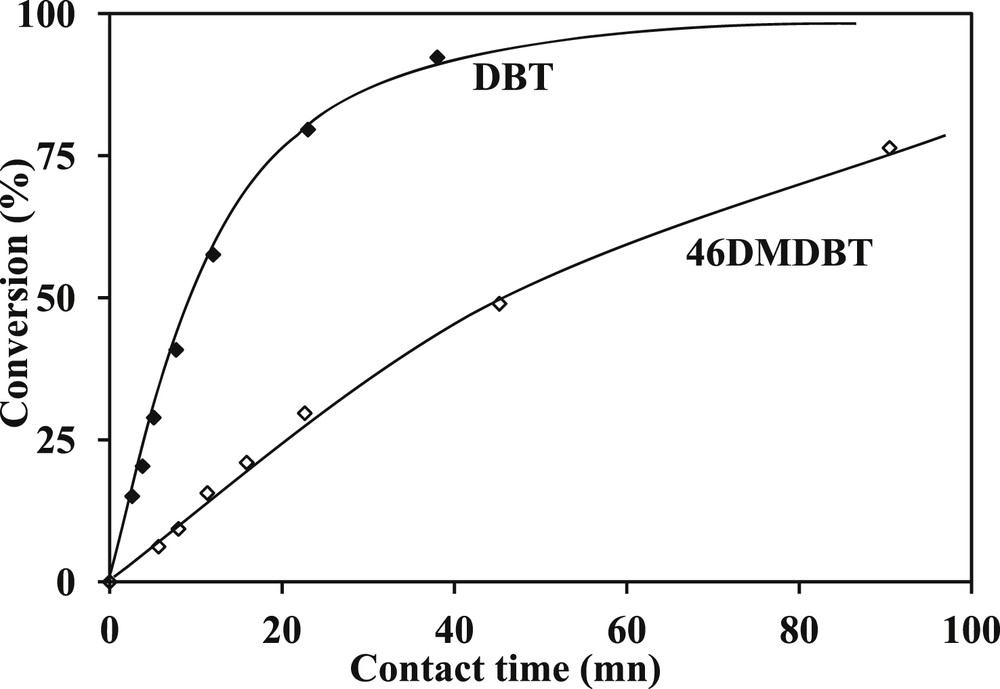
Comparison of the transformation of 4,6-DMDBT and DBT (T = 340 °C, P = 4 MPa, H2/HC = 468 NL/L, CoMoP/Al2O3).
Transformation of DBT and 4,6-DMDBT. Comparison of the activity and the DDS and HYD selectivity (T = 340 °C, P = 4 MPa, H2/HC = 468 NL/L, CoMoP/Al2O3).
| Activity (mmol/g/h) | DDS/HYD | C (%)pds) | S (% pds) | |||
| Global | DDS | HYD | Selectivity | Amount | ||
| DBT | 3.5 | 3.1 | 0.3 | 6.5 | 2.5 | 7.1 |
| 4,6-DMDBT | 0.8 | 0.2 | 0.6 | 0.3 | 2.6 | 6.8 |
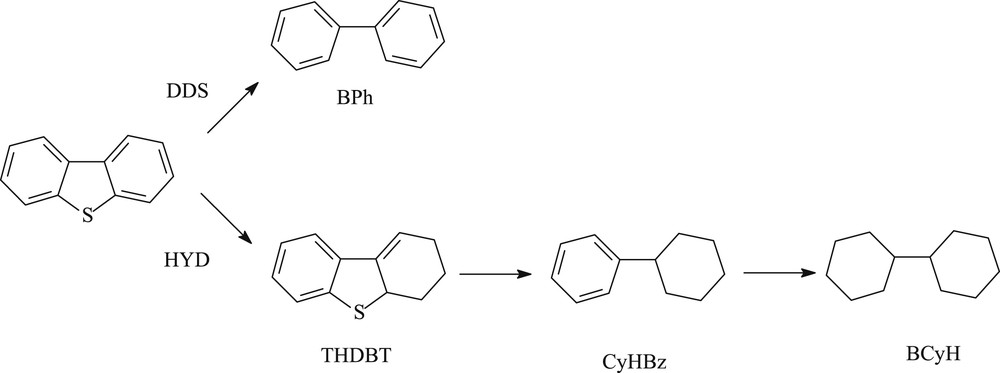
Reaction pathways of the hydrodesulfurization of DBT.

Reaction pathways of the hydrodesulfurization of 4,6-DMDBT.
As reported previously by Bataille et al. [29] and by Cristol et al. [32], the main reason for this reactivity difference is the aromaticity of DBT and DMDBT that prevents the adsorption through the thiophene ring. In conjunction with steric hindrance, this aromaticity imposes the adsorption of DMDBT by the benzene ring (Scheme 4). A direct correlation between the adsorption geometry/energy and the reactivity of the sulfur compounds is not straightforward but, on the basis of the present results, Cristol et al. [32] proposed that the hydrogenation route (HYD) proceeds via a benzene adsorption on the molybdenum edge of the MoS2 crystallites (considering a non-promoted catalyst) whereas the direct desulfurization (DDS) proceeds by adsorption on the sulfur edge.
3.2 Transformation of the various model feeds with CO incorporation
The impact of various amount of CO in the feed was more particularly studied for the transformation of two model feeds (representing of FCC gasolines and SR gas oils) in various operating conditions (see Tables 2 and 3) and over two catalytic systems (CoMo/Al2O3 for FCC gasoline and CoMoP/Al2O3 for gas oil).
Under all the operating conditions tested, no transformation of CO into methane, CO2 or water was observed, even if methanation and water–gas-shift reactions are thermodynamically favored under these conditions. In all cases, a strong and reversible inhibiting effect is observed on HDS activities, whatever the operating conditions, the catalysts and the model compounds involved. Indeed, a strong and similar negative effect is noticed from the addition of small amount of CO. The effect is similar for the transformation of 2 MT and alkenes in the FCC model feed (Fig. 9a). For example, the loss of activity, measured by the A/A0 ratio between the activity in presence of various amount of CO (A) and the initial activity (A0), is equal to 53 for a partial pressure of CO introduced of 1.2 kPa (Fig. 9b). Nevertheless, no modification of the selectivity represented by the kHDS/kHYD is noticed for various initial conversion of 2 MT (Fig. 10). This result shows that the active sites involved in C–S rupture and the hydrogenation of 2,3-DMB2N are sensitive to the presence of CO in the same way.
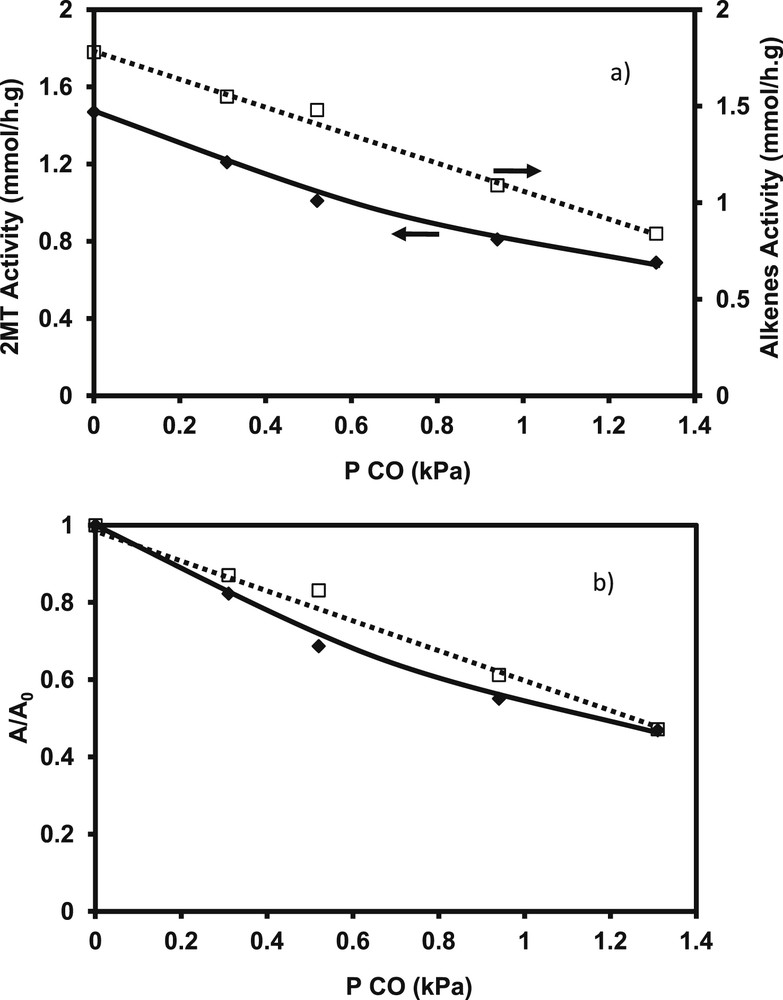
Transformation of the model FCC gasoline. Impact of the CO partial pressure a) 2 MT and the alkenes activity b) A/A0 loss of activity (T = 250 °C, P = 2 MPa, H2/HC = 360 NL/L, CoMo/Al2O3).
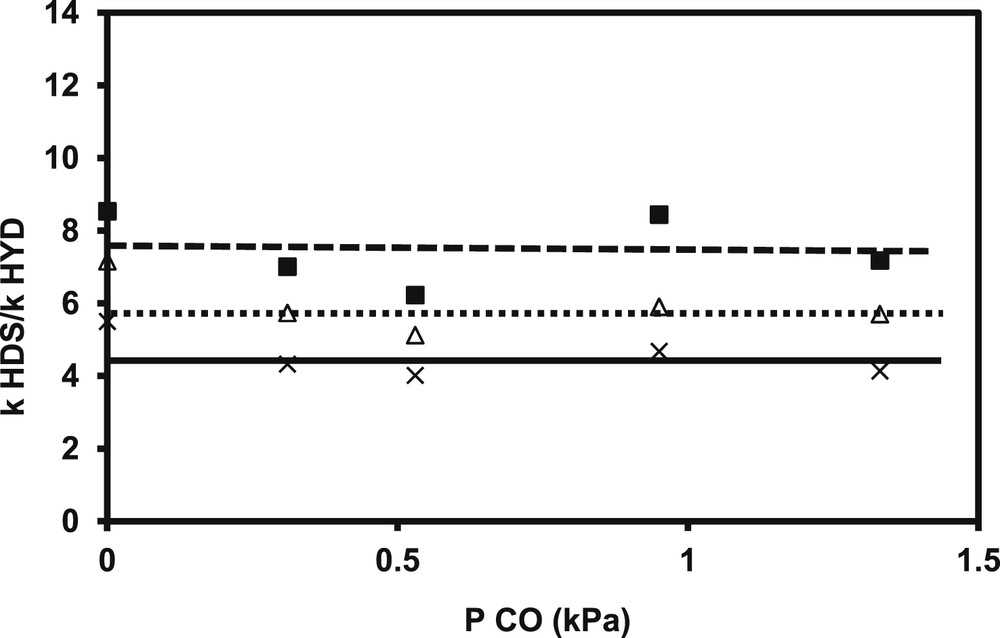
Transformation of the model FCC gasoline. Impact of different CO partial pressures and initial conversion of 2 MT (C0,2MT) on the kHDS/kHYD ratio (C0,2MT = 50%: ■, C0,2MT = 59.9%: Δ, C0,2MT = 79.6%: ×) (T = 250 °C, P = 2 MPa, H2/HC = 360 NL/L, CoMo/Al2O3).
Regarding the model sulfur compounds (DBT and 4,6-DMDBT) of gas oils, some differences can be considered between 4,6-DMDBT and DBT under the same operating conditions (Fig. 11a and Fig. 11b). In fact, the loss of activity for the transformation of 4,6-DMDBT is slightly lower than for the transformation of DBT. For example, for a partial pressure of CO of 6.0 kPa, the loss of activity is around 47% (A/A0 = 0.53) for 4,6-DMDBT and 56% (A/A0 = 0.44) for DBT. This corresponds also to a difference in the hydrogenation and direct C–S bond rupture routes, the main reaction involved in HDS of model gas oils.
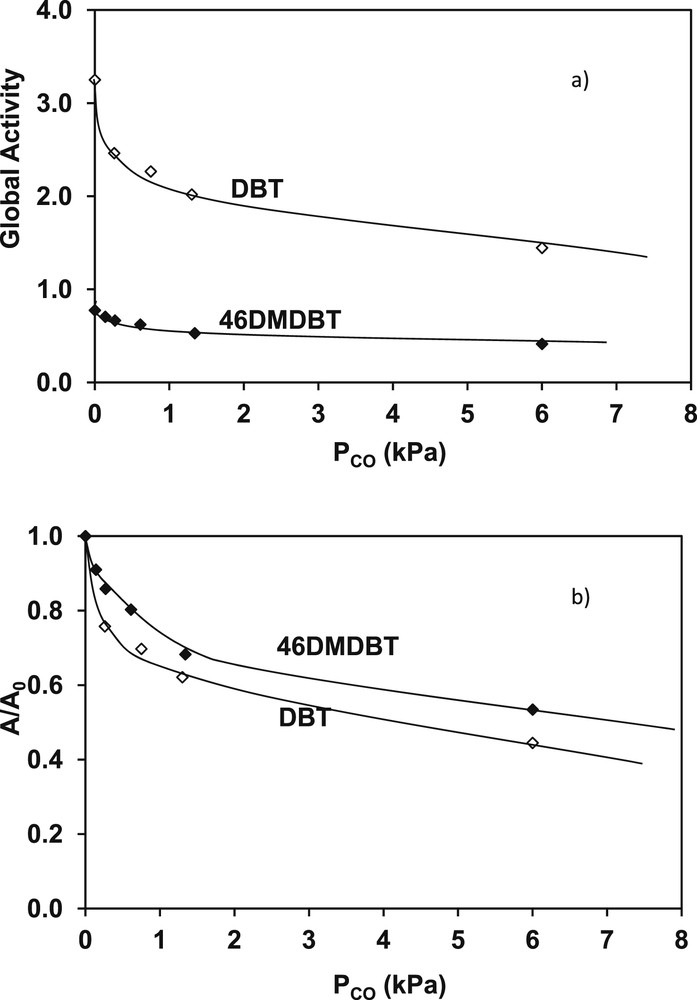
Transformation of DBT and 4,6-DMDBT. Impact of different CO partial pressures a) global activity b) A/A0 loss of activity (T = 340 °C, P = 4 MPa, H2/HC = 468 NL/L, CoMoP/Al2O3).
From our data, we are able to compare the effect of CO on the transformation of the model FCC gasolines and the model SR gas oils. An inhibiting effect is noticed whatever the model compounds involved (2 MT, 2,3-DMB2N, DBT and 4,6-DMDBT). Nevertheless, this effect is more or less significant depending on the model compounds and the operating conditions. Regarding the results obtained for a same partial pressure of CO of 1.3 kPa, the inhibiting effect, which is the same for 2 MT and 2,3-DMB2M, is more significant (50%) than for 4,6-DMDBT and for DBT (respectively 32 and 38%). In order to explain these experimental results, it is necessary to consider the structure of the active sites involved in the transformation of these various compounds as described by Krebs and al [29] by DFT calculations. The S-edge sites, more favorable for 2 MT transformation, and the M-edge sites, more favorable for olefin HYD, [33] are both poisoned by CO. Hence, the simultaneous inhibition of the same amount of HDS S-edge sites and HYD M-edge sites (of the hexagons) will result in the same activity loss for both reactions. As a consequence, the selectivity (HDS/HYD) remains unchanged either in absence or in presence of CO. Considering the HDS of DBT and 4,6-DMDBT, the inhibiting effect of CO could be also the result of a higher adsorption energy of CO than for DBT and 4,6-DMDBT. Nevertheless, this inhibiting effect of CO for a same partial pressure (1.3 kPa) was lower for the transformation of DBT (38%) and 4,6-DMDBT (32%) than for the transformation of 2 MT (53%) and 2,3-DMB2N (50%). Considering mutual competitive adsorptions of CO, alkenes, sulfur compounds (2 MT, DBT and 4,6-DMDBT) on the catalyst surface, these differences may correspond to a difference of CO adsorption energy in the operating conditions of HDS of gasolines and gas oils (higher temperature and pressure in the latter case, for example a lower temperature could increase CO adsorption energy compared to the one of 2 MT) and/or a higher adsorption energy of DBT and 4,6-DMDBT.
From the experimental point of view, it is difficult to explain the higher inhibiting effect of CO of CoMoS catalyst performances during model FCC gasoline hydroprocessing than during model gas oil HDS since various parameters are varied simultaneously: lower temperature, lower partial pressure of reactant; however it merits to be representative of industrial ones.
This comparison is helpful to consider the expecting CO effect variation on industrial conditions between both hydrotreating processes: ultra-low sulfur gasoline and diesel hydroprocessing.
Finally, a way to prevent this negative impact would be to use a non-sensitive catalyst to CO as reported by Bouvier et al. [24] under various applications. Indeed, the NiMo/Al2O3 catalyst was less sensitive than the CoMo/Al2O3 to the presence of CO during the HDO of 2-ethylphenol, a model compound of lignocellulosic materials. These results were coherent with previous work of Egeberg et al. [16], for straight run gas oil hydroprocessing, showing that NiMo is barely sensitive to oxygenated compounds while the CoMo activity is highly impacted.
4 Conclusion
The presence of CO strongly inhibits the transformation of the sulfur compounds and the alkenes used as model compounds representative of FCC gasolines and gas oils in this study. Nevertheless, this negative effect depends on the feed nature and the operating conditions. Thus this effect is more significant in presence of 2 MT and 2,3-DMB2N representative of an FCC gasoline than in presence of DBT and 4,6-DMDBT representative of a gas oil, even if the operating conditions are not the same. In the case of FCC gasoline hydrotreating, the effect is due to phenomena of competitive adsorptions between sulfur compounds or alkenes and CO on the catalyst surface. Considering that CO is adsorbed on both S-edge and M-edge considered as active sites for HDS of 2 MT and the hydrogenation of 2,3-DMB2N, this could explain why the inhibiting effect is the same for the transformation of both molecules in mixture. In the case of gas oil hydrotreating, the negative effect of CO is less pronounced for the transformation of DBT and 4,6-DMDBT, probably due to a lower adsorption energy of CO and/or to higher adsorption energy of DBT and 4,6-DMDBT.
Finally, carbon monoxide appears as a strong reversible inhibitor for HDS of various thiophenic compounds even at very low partial pressures. Considering co-processing of renewable feedstocks and petroleum fraction, we thus expect stronger difficulties in the synthesis of ultra-low sulfur gasoline. However, additional experiments are mandatory to compare thiophenic reactivity in presence of CO in closer operating conditions (in matter of temperature and total pressure) in order to distinguish the effect of the operating conditions and the effect of the nature of sulfur compounds. To transform feedstocks containing traces of CO, it therefore seems important to adapt the catalyst properties regarding CO adsorption and/or modify operating conditions.