

1 Introduction
Heavy metals are known to be hazardous environmental pollutants, with toxic effects on living organisms. In most countries, the contamination of aqueous environments with heavy metals is a daily problem [1–3]. Generally, the presence of this class of pollutants in surrounding water is due to various industrial activities including manufacturing of paints, extraction of metals, fabrication of electronic devices, mining, manufacturing of batteries and oil refining [4–8]. Along with zinc, copper, mercury, nickel and chromium, cadmium and lead are amongst the most important heavy metals from the point of view of water pollution. The simultaneous determination of the two latter pollutants is increasingly investigated in analytical and environmental sciences since they are often present together in the same samples, mostly in those derived from battery manufacturing, fuel oil and smelting activities. In biological systems, lead and cadmium ions lead to several metabolic dysfunctions and, in humans, to pathologies such as anemia, high blood pressure, neurological troubles or even some congenital deformations [3,9–11]. The threshold concentration of lead and cadmium allowed in drinking water set by the World Health Organization is 0.01 mgL−1 (4.82 × 10−8 mol L−1) and 0.003 mg L−1 (2.66 × 10−8 mol L−1), respectively [12]. It is thus evident that the quantification of these metals over these ranges is of particular importance for health care and to ensure a cleaner environment and pollution control.
For the sensitive determination of lead and cadmium, studies based on modified electrodes have raised acute interest during the past decade. Among the reported studies one may mention the detection of both species using a glassy carbon electrode modified by using poly(3,4-ethylenedioxythiophene) and poly (4-styrenesulphonate) [13], a nafion graphene composite film [14] and a hydroxyapatite-modified carbon ionic liquid electrode [15]. Also, the stripping voltammetric detection of both ions has been investigated on a bare bismuth electrode [16], and on the same substrate chemically modified by using a thin film of poly(p aminobenzene sulfonic acid) [17] or a multiwalled carbon nanotube-nafion composite material [18]. For the same analytical purposes, an antimony electrode chemically modified to strongly bind cationic species has also been proven to be a prominent tool. Thus, Gong et al. [19] reported the determination of lead and cadmium with an antimony film-plated glassy carbon electrode, while Toghill and co-workers [20] exploited an antimony nanoparticle modified boron-doped diamond electrode to quantify Pb2+ and Cd2+ ions by linear sweep anodic stripping voltammetry.
In comparison with traditional spectrometric techniques, resorting to electrochemical sensors based on modified electrodes provides many advantages such as easy fabrication, high selectivity, and high sensitivity and accuracy [21–23]. In that field, clay minerals have shown to offer great potential. Due to their microcrystalline structure, their high cation exchange capacity, their low cost and the possibility to easily modify their intrinsic properties, they are continuously and increasingly exploited as electrode modifiers, as recently reviewed by Tonle et al. [24]. For such purposes they may be used either in the native state or after chemical modifications to improve their ability to accumulate target pollutants while allowing the electrochemical detection and quantification of the preconcentrated pollutants. The most used pathways to modify clay minerals involve covalent grafting of functionalized organosilanes onto their –OH surface groups [24] or the intercalation of reactive organic compounds (viz., specific ion ligands) between their layers [5,24].
In previous studies, our group has reported the exploitation of smectite-based composite materials for the individual detection of lead and other heavy metals [5,8,25]. More recently, a smectite modified by co-insertion of cetyltrimethylammonium ions and thiourea was used to electrochemically detect lead [21]. A main result from that study was the demonstration that there was no mutual interference between the signals recorded for lead(II) and cadmium(II). As a continuity of that work, the present study describes the simultaneous detection of lead(II) and cadmium(II) by square wave anodic stripping voltammetry. This was achieved by modifying a glassy carbon electrode with organoclay layers consisting of montmorillonite modified by intercalation of cationic surfactant ions and thiourea. The optimum conditions leading to the sensitive electroanalysis of both analytes were extensively discussed.
2 Experimental
2.1 Clay mineral, chemicals and reagents
The smectite-type clay mineral (sample name: “Sa”) originates from Sabga in the western region of Cameroon (Central Africa). It was previously characterized and its chemical composition and structural formula are provided elsewhere [1,26]. It is characterized by a cation exchange capacity (CEC) of 78.2 meq per 100 g and a specific surface area (N2 adsorption-desorption, BET) of 86 m2 g−1. All chemicals and reagents used in the electrochemical section and the synthesis of materials were of analytical grade, and used as received. Solutions were prepared with deionised water (18 mΩ cm−1) from a Millipore milliQ water purification system. Acidic diluted solutions of Pb2+ and Cd2+were prepared from standard commercial solutions (1000 mg L−1) purchased from Prolabo. Acidic solutions were obtained by diluting concentrated and commercial solutions of HCl (37%, Prolabo), H2SO4 (98%, Prolabo) and HNO3 (70%, Normapur). Cetyltrimethylammonium bromide (CTA+, Br−) from Prolabo and thiourea (Merck) were used for clay modification. Sodium chloride (NaCl, Prolabo) was used for the homo-ionic saturation of the pristine clay.
2.2 Preparation of the organoclay
Upon collection, the raw clay mineral material was crushed, ground and sieved through a 100 μm sieve to remove the larger non-clay fractions. Prior to its chemical modification, the fine particles of the clay mineral were collected by sedimentation following a procedure previously described [27,28]. The starting clay mineral (Sa) was used in sodium saturated homo-ionic form, labeled Sa(Na) throughout the text.
The steps involved in the preparation of the organoclay material are summarized in Scheme 1, following an earlier procedure developed by our group [21]: 10 mL of solution containing the modifying agents was prepared by mixing CTABr and thiourea in given proportions. The amount of thiourea used to prepare the solution was kept constant (7.5 times the CEC of the clay) while that of the cationic surfactant was used in concentrations equivalent to 0.25 times the CEC of the pristine clay mineral. Then, 0.1 g of Sa(Na) was suspended in 5 mL of water at pH 7, to which 10 mL of the modifying solution were added. The obtained mixture was then mechanically stirred for 48 h at room temperature. Upon stirring, the dispersion was left to stand for 2 days and the collected product was washed with 10 mL of deionized water, and then with 20 mL of a mixture of water and ethanol (95/5%, v/v). After the second wash, the dispersion was centrifuged and the hybrid material was recovered and stored as a suspension in deionized water (ponderal concentration: 8 g L−1). Twenty four hours later, the suspension was sonicated for 5 min and used to prepare the working electrode. Hereafter and throughout the text, the prepared organoclay is referred to as Sa(CTA0.25,T) where 0.25 represents the amount of CTA+ ions in relationship with the CEC of the clay, and T stands for thiourea.
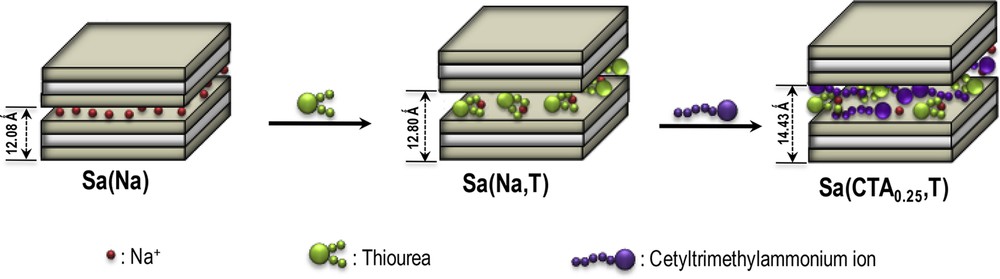
Co-insertion process of neutral thiourea (T) and cethyltrimethylammonium ions (CTA) within the interlamellar space of the Sa smectite sample.
2.3 Physicochemical characterization techniques
Powdered XRD patterns of clay samples were recorded at room temperature using a D5005 diffractometer (Bruker AXS S.A.S) equipped with a Cu Kα1 radiation source (λ = 1.54056 Å) using a generator voltage of 45 kV and a current of 40 mA.
Specific surface areas and pore volumes of the starting clay mineral and the organoclay material were evaluated by the B.E.T. method involving N2 adsorption-desorption measurements. The measurements were performed with a Coulter Omnisorp 100 apparatus (model ASAP 2010 V 3.02 G) at 77 K in the relative pressure range from 10−5 to 0.99. Prior to each measurement, the sample was outgassed at 105 °C under vacuum for 16 h.
The amount of organic compounds retained on the organoclay samples was estimated using spectrophotometry and elemental analysis. For UV analysis, a precise and given amount of the organoclay material was shaken for 24 h in an acidified mixture of water/ethanol (70/30 v/v, pH 1.5) medium to remove the organic compounds and then, the filtrate was analyzed using a UV spectrometer (Agilent Technologies Cary 60 UV–Vis).
C, H, N and S elemental analysis was performed on the pristine clay and on organoclays using a Thermofinnigan Flash EA 1112 instrument (Service of Elemental Analysis, University of Lorraine, France).
2.4 Modification of the glassy carbon working electrode and electrochemical equipment
The glassy carbon electrodes (GCEs) were first polished with alumina pastes of different particle sizes (5, 1, and then 0.5 μm) on a piece of billiard cloth. It was afterwards placed in a 1:1 ethanol-water solution and properly cleaned by sonication for 10 min to eliminate any remaining alumina particles. The thin clay film working electrode was prepared by drop-coating 20 μL of an aqueous dispersion of the Sa(CTA0.25,T) organoclay material onto the active surface (3 mm in diameter) of the GCE. The clay modified electrode, denoted as GCE/Sa(CTA0.25,T), was stored at room temperature for about 4 h to ensure its complete drying before electrochemical experiments.
Square wave voltammetry was used as an electrochemical technique for the quantification of Pb2+ and Cd2+species. Thus, the unmodified smectite and its modified derivative were in turn drop-coated onto the surface of GCEs, and the resulting film-modified electrodes were applied to the simultaneous accumulation and detection of Pb2+and Cd2+ ions in synthetic solutions. The accumulation ability and sensitivity of the organoclay modified electrode was appreciated by comparing the currents measured for both analytes with this electrode to those determined upon using a bare GCE or with a GCE modified by the pristine smectite (sodium saturated form).
The electrochemical procedure for Pb2+ and Cd2+ ions analysis by stripping voltammetry is described elsewhere [5]. Briefly, two successive steps were involved: (i) the preconcentration of either Pb2+or Cd2+, or of both ions simultaneously under open-circuit conditions and (ii) the voltammetric detection in a separate medium. For step (ii), an electrolysis potential was applied for a given time to ensure the reduction of accumulated ions. The potentials were then scanned in anodic direction and the recorded voltammetric peak heights were measured. After each detection test and prior to the next experiment, the working electrode was immersed into an acidic solution and kept under stirring to allow the desorption of the previously accumulated ions.
3 Results and discussion
3.1 Structural and textural characterization of the pristine and modified clay
XRD was used to investigate the structural changes experienced by the smectite clay upon its chemical modification, while N2 adsorption-desorption isotherms were exploited to derive the textural properties of the modified clays. Fig. 1a shows the XRD patterns of the purified smectite clay mineral (Sa(Na)) before modification (ia) and after its modification with thiourea alone (Sa(Na,T), curve iia) and then with CTA+ ions and thiourea (Sa(CTA0.25,T), curve iiia). The pristine clay curve presents a peak around 2θ = 7.36 to which corresponds a basal spacing d001 = 1.20 nm. The broadness of such a peak is characteristic of a poor crystallized smectite-type clay, in agreement with previously reported data on the same material [1,21]. For the Sa(Na,T) material, the basal spacing shifted to 1.28 nm, the d-spacing being not slightly different from that recorded for Sa(Na). These values correspond to slight expansions of the interlayer space probably due to the small quantities of intercalated thiourea which is present in the initial modifying solution.
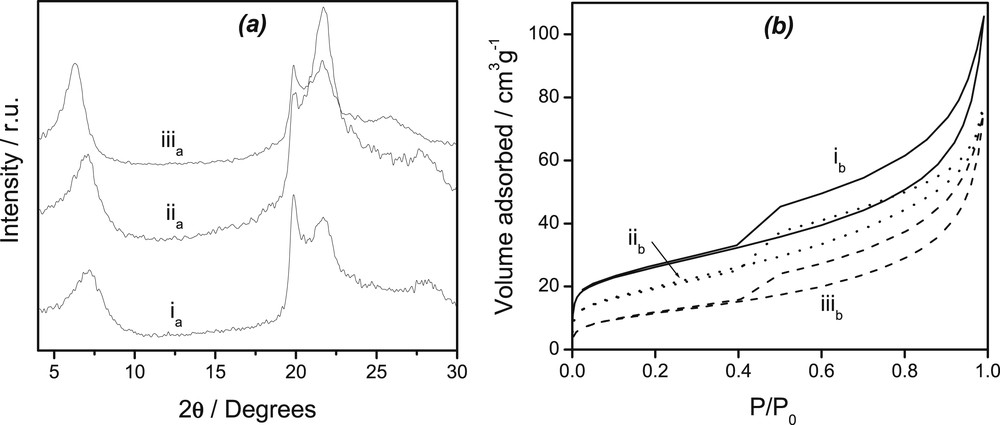
(a): Powder X-ray diffraction patterns of (ia) Sa(Na), (iia) Sa(Na,T) and (iiia) Sa(CTA0.25,T) materials; (b): nitrogen adsorption-desorption isotherms obtained at 77 K for (ib) Sa(Na), (iib) Sa(Na,T) and (iiib) Sa(CTA0.25,T) materials.
On the PXRD curve corresponding to the Sa(CTA0.25,T) organoclay (Fig. 1a, curve iiia), the basal spacing was observed to be 1.44 nm. This significant change suggests a synergistic co-intercalation of the two organic modifiers (CTA+ ions and thiourea) between the layers of the smectite. This can arise by two possible types of interactions: (i) hydrophobic interactions with the surfactant phase, and/or (ii) electrostatic interactions in the surfactant clay layer region (owing to the zwitterionic properties of thiourea [29], which is thus likely to be sandwiched between CTA+ species and the negatively charged clay surface). The arrangement of surfactant molecules within layered clay minerals depends on both the alkyl chain length and the amount of the modifier used for the modification [30-33]. Organoclays with monolayer, bilayer, pseudotrimolecular and paraffin layer configurations of the alkylammonium ions within the interlayer region of smectite are indeed known. In the present study, at a loading level of CTA+ ions equal to 0.25 CEC in the Sa(CTA0.25,T) material, the 1.44 nm d001 value suggests the presence of a mixed arrangement with a monolayer configuration of the surfactant and thiourea between the clay sheets [21,34].
Isotherms determined using N2 adsorption-desorption of the starting clay were recorded and compared to those of the corresponding organoclays (Fig. 1b). This evidenced that for Sa(Na), Sa(Na,T)and Sa(CTA0.25,T) (see curves ib, iib and iiib, respectively), the isotherms displayed a relatively large hysteresis loop indicative of a mesoporous structure with a great contribution from mesopores [28,35,36]. The BET surface areas corresponding to these materials decreased from Sa(Na) to Sa(Na,T), and then to Sa(CTA0.25,T). The equivalent pore volumes followed the same trend (0.1634 for Sa(Na), 0.1186 for Sa(Na,T)), and 0.1154 cm3 g−1 for Sa(CTA0.25,T), respectively.
There was almost a loss of microporosity for the organoclay obtained by the intercalation of only thiourea molecules [Sa(Na,T)] and the organoclay obtained by co-intercalation of thiourea and CTA+ ions [Sa(CTA0.25,T)]. This feature probably results from the adsorption of thiourea molecules in the pores and micropores of the natural clay in the case of Sa(Na,T), and to a compact packing of CTA+ ions and thiourea in the interlamellar spacing that blocks the passage of nitrogen [36] in the case of the Sa(CTA0.25,T) material. All of these observations indicate a change in the textural properties of the clay as nitrogen molecules can no more easily get access to the interlayer space occupied by CTA+ ions and thiourea.
3.2 Electroanalytical application of the organoclay to the simultaneous detection of Lead(II) and Cadmium(II)
3.2.1 General considerations
A set of preliminary investigations were performed to define basic conditions for the electroanalysis of Pb(II) and Cd(II). Since the Sa(CTA0.25,T) possesses an insulating character and involves bulky species due to large metallic centers complexed by thiourea within its structure, the desorption of Pb2+ and Cd2+ ions accumulated during the first step of the electroanalytical procedure (step (i) in Section 2.4) greatly depend on the nature of the supporting electrolyte. Generally, acidic media are convenient for the detection step in electroanalysis involving the formation of complexes between chelating groups and cationic species. The ability of four different acidic solutions (HNO3, HCl, H3PO4 and H2SO4) to desorb Pb(II) and Cd(II) accumulated for 5 min on the GCE/Sa(CTA0.25,T) was therefore evaluated. The results obtained and presented in Fig. 2 show that voltammetric peaks of Pb(II) were observed in all these electrolytes, around −0.5 V vs the SCE. However, it is only in HCl solution that a second signal ascribed to Cd(II) may be observed in addition around −0.8 V. Furthermore it is noticed that HCl led to higher stripping peak currents for both analytes, and to the best electrochemical signals.
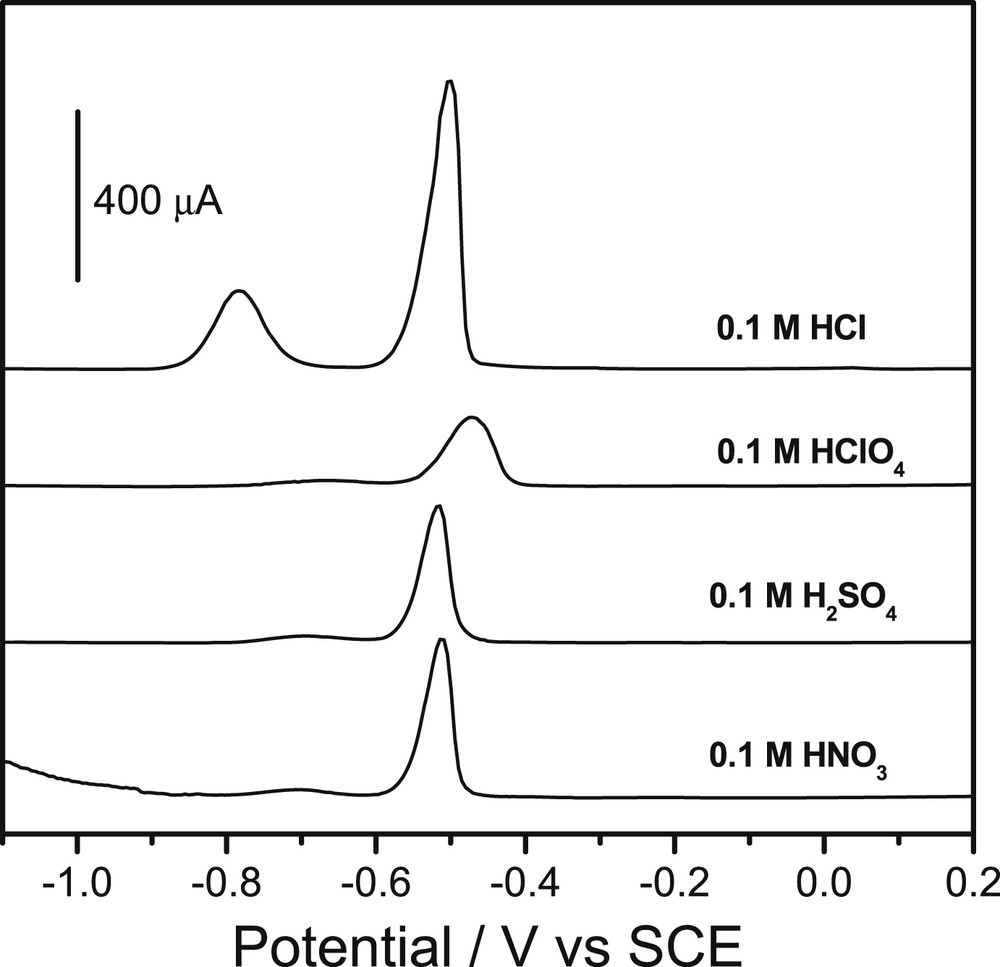
Square wave voltammograms recorded for a mixture of 10−6 M Cd2+ and 10−6 M Pb2+on a GCE modified by using a thin film of Sa(CTA0.25,T), in each acidic medium reported on the graphs. Accumulation was performed for 5 min in an aqueous solution at pH 4 containing both analytes.
Since the concentrations of all tested acids were the same (0.1 M), this feature is due to an anion effect and it is actually explained by the ability of Cl- to form stable complexes with Pb2+ and Cd2+ as previously observed in other studies [21,37,38], leading to better desorption efficiencies. Therefore, a 0.1 M HCl solution was used in further experiments as the detecting medium.
The organoclay modified electrode was also tested for the individual and simultaneous detection of both Pb2+ and Cd2+ ion mixtures at the same concentration. Fig. 3 presents the square wave voltammetric curves recorded for a solution containing either Pb2+ (Fig. 3a) or Cd2+ (Fig. 3b) alone or both metal ions (Fig. 3c). Upon preconcentration, the signals were obtained in 0.1 M HCl under the same conditions (i.e. after 5 min of accumulation in an aqueous solution containing analytes at 10−6 M at pH 4). The curve in Fig. 3c shows that the GCE/Sa(CTA0.25,T) sensor can be successfully applied to the detection of Cd2+ and Pb2+cations in a given solution, although there is a slight decrease (of ca. 5%) in the signal of Cd2+ when compared to the peak relative to this ion alone (Fig. 3b).
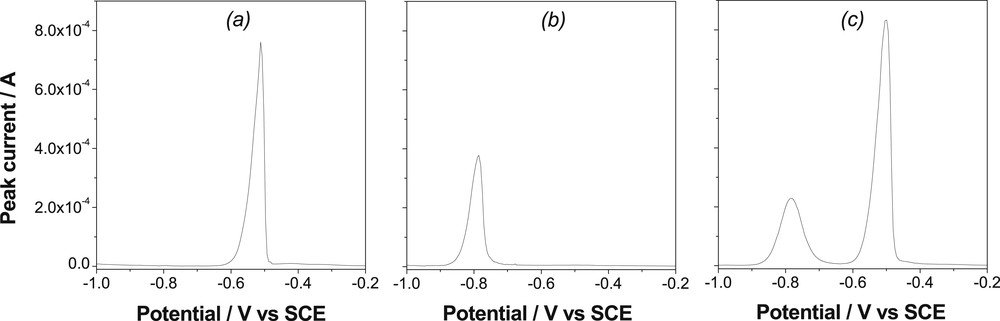
Square wave voltammograms recorded on the GCE/Sa(CTA0.25,T) in 10−1 M HCl for (a): 10−6 M Cd2+, (b): 10−6 M Pb2+and (c): 10−6 M Cd2++10−6 M Pb2+. Electrolysis time: 60 s; electrolysis potential: −1 V; preconcentration duration: 5 min in an aqueous solution at pH 3.5.
For comparison purposes, the GCE surface was also modified by using a thin film of unmodified purified smectite, and used for experiments similar to those shown in Fig. 3c. The anodic stripping square wave voltammograms obtained (Fig. S1 in Supporting Information) displayed anodic peak currents for lead(II) and cadmium(II) ca. 6.5 and 11.5 times, respectively, smaller than that those obtained under the same conditions for the modified smectite (compared to Fig. 3b). This result illustrates the important role of modifying the natural clay Sa(Na). This evolution in peak currents of Pb(II) and Cd(II) on the GCE/Sa(CTA0.25,T) can be explained by the enhanced complexation capacity of the clay mineral due to the presence of thiourea ligands [1,5,39] and the presence of the cationic surfactant CTA+ in the bulk of the organoclay. Both factors confer to the starting clay mineral new reactive sites with strong chelating effects towards lead(II) and cadmium(II).
The reproducibility and stability of the sensor based on the GCE/Sa(CTA0.25,T) was also checked. Thus, a freshly prepared electrode was pretreated following a series of 7 runs, all involving a complete “open-circuit”/“accumulation” and “detection” sequence, for lead(II) and cadmium(II). As shown in Fig. S2 (Supporting Information), reproducible signals were obtained from repetitive experiments. For this analysis, the relative standard deviation (RSD) of the anodic peak current was found to be 3% for Cd(II) and 2% for Pb(II). This indicates that thin films of Sa(CTA0.25,T) deposited on the GCE are quite stable and can be used for several successive measurements.
In order to determine the reproducibility of the modification procedure, six different organoclay modified GCEs were prepared by drop-coating and tested for the electrochemical stripping analysis of Pb2+ and Cd2+ ions (5 min accumulation in 10−6 M Pb2+ + 10−6 M Cd2+ followed by detection in 0.1 M HCl). A good reproducibility was observed since the relative standard deviation in peak current was less than 2% for both analytes. This fact could be ascribed to the stability of the organoclay dispersion used for the preparation of film-modified electrodes, as well as to the simple and rigorous procedure of modification of the GCEs (typically 20 μL of organoclay drop-coated on each GCE).
3.2.2 Effect of depositing potential and duration on the detection of Pb2+ and Cd2+
For metal trace determinations based on anodic stripping voltammetry, the value (Eel) of the potential applied to metal ion capture and accumulation as well as the duration of its application are important parameters. Fig. 4a depicts the effects of the Eel value on the stripping peak currents after 5 min of accumulation on the GCE modified by Sa(CTA0.25,T). One notices on curve ii in Fig. 4a that from 0 to −0.5 V (vs. the saturated calomel reference electrode) the stripping response for Pb2+ was quite low. It became higher from −0.5 V to −1 V and reached a plateau at more negative values. Such a behavior was also observed for Cd2+ (Curve i, Fig. 4a) but comparatively lower responses were obtained, revealing that the sensor under study was more sensitive to lead than to cadmium. It is important to note that when the Eel value was made more negative (under −1.2 V), the reduction of water molecules and protons started to interfere, inducing a destabilization of the organoclay film at the electrode and poor reproducibility stripping peaks for Cd(II) and Pb(II). Hence, for both ions and for further experiments, Eel was imposed at −1 V vs. the saturated calomel reference electrode. It is well known that the electrolysis duration also greatly affects the detection limit and thereby the sensitivity of a sensor [21,40]. The effect of electrolysis duration (telec) on the stripping peak currents of lead and cadmium was investigated while keeping Eel at −1 V vs. the saturated calomel reference electrode (Fig. 4b): a rapid increase in peak heights for both Pb2+ and Cd2+ upon increasing telec was observed on the GCE/Sa(CTA0.25,T) when preconcentration electrolysis durations were limited over the range 0–20 s for Cd(II) and between 0 and 40 s for Pb(II). At higher deposition times the plots for Pb2+and Cd2+ slightly levelled off, indicating that the accumulation equilibria were kinetically achieved. A good compromise between both ions then involved an accumulation time of 60 s which is hereafter used.
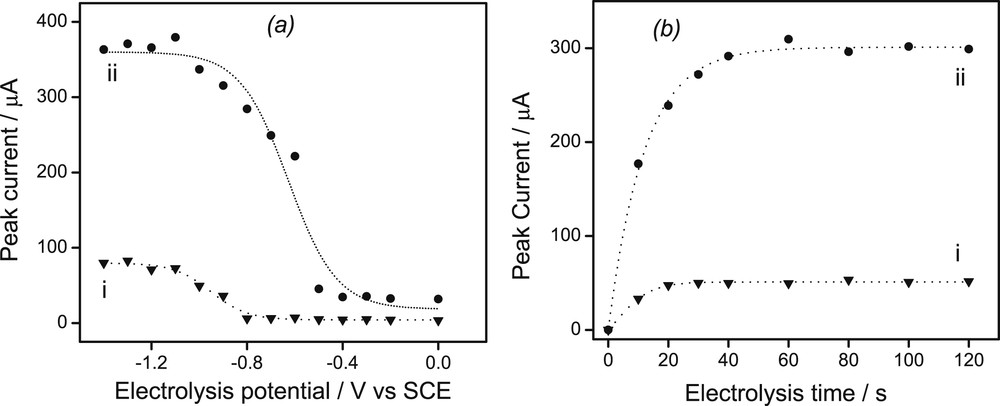
(a) Variation of the peak currents recorded on the GCE/Sa(CTA0.25,T) for(i) Cd2+ and (ii) Pb2+ ions; (b) Variation of the electrode response for (i) Cd2+ and (ii) Pb2+ ions as a function of electrolysis time. For (a) and (b), detection was performed in 0.1 M HCl after 4 min of accumulation in an aqueous medium (at pH 3.5) containing both ions at 10−6 M.
3.2.3 Optimization of the accumulation step for the detection of Pb2+ and Cd2+
Accumulation of Pb2+ and Cd2+ ions under open-circuit conditions can be influenced by two main factors: the pH of the accumulation medium and the duration of preconcentration of analytes. In fact, the diffusion of Pb(II) and Cd(II) to the binding sites at the modified electrode is likely to be influenced on one hand by the speciation of the analyte which is related to the pH, and on the other hand by the time necessary for the analyte to reach these sites located in the bulk of the organoclay. Using the preselected optimal parameters (Eel: −1 V, tel: 60 s), the pH of the preconcentration medium (on the stripping peak currents of 1 μM Pb(II) and 1 μM Cd(II) obtained upon 4 min of accumulation) was varied from1 to 7 to avoid the precipitation of metal hydroxides which may occur in basic media. The obtained results are shown in Fig. 5: upon increasing the pH from 1 to 3.5, the peak current increased and then reached a maximum value around pH 3.5 for both ions. Above this value, the electrode response decreased upon increasing the pH. The low sensitivity of the organoclay sensor at pH values < 2 could be related to a protonation of amine groups of thiourea that may reduce its complexing ability [41]. Therefore, an optimal pH value of 3.5 was chosen for further study.
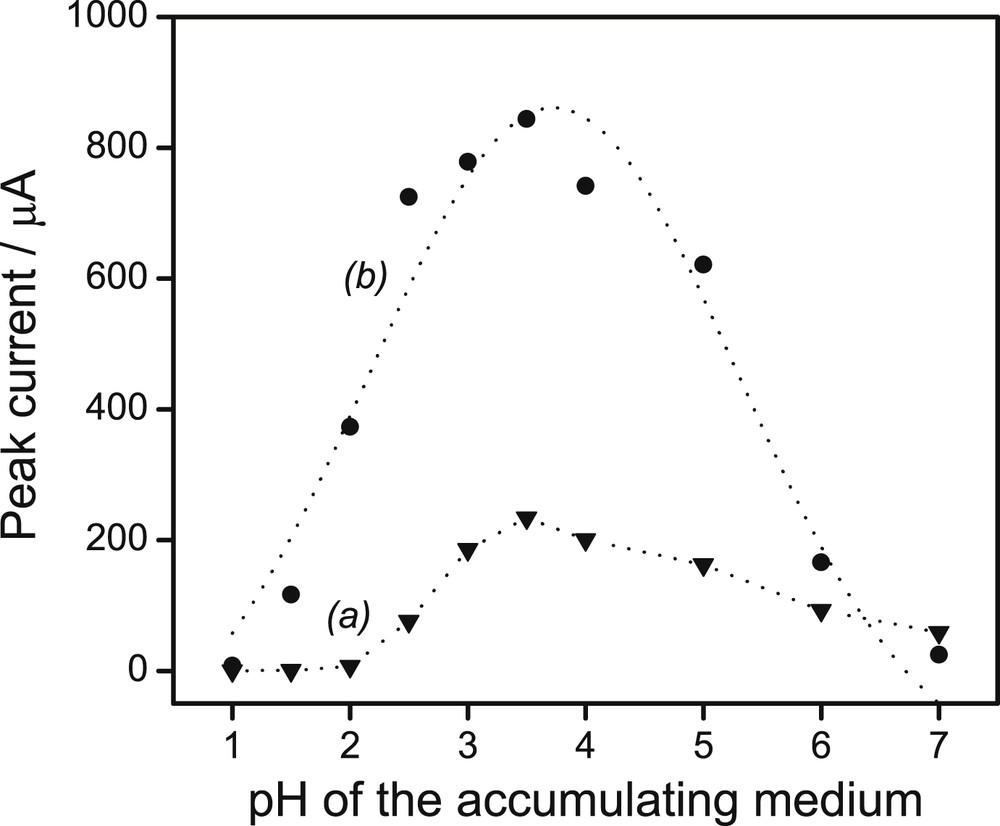
Effect of the pH of the preconcentration medium on the peak current of 1 μM of both Cd2+ and Pb2+ in 0.1 M HCl on the GCE/Sa(CTA0.25,T). (Electrolysis potential: −1 V, electrolysis duration: 60 s, and preconcentration duration: 4 min in aqueous solution at pH 4).
The effect of preconcentration time (taccum) was examined for both heavy metals diluted to reach the same concentration (10−7 M) and in the same accumulation solution. Fig. 6 depicts the SWV curves obtained: the electrode response gradually increased in each case with the time of accumulation. As shown in the inset of Fig. 6, the relationship between the peak currents of each ion and taccum varying from 0 to 20 min was linear. This simply indicates the presence, even after 20 min of accumulation, of a large number of adsorption sites unoccupied on the surface of the electrode [5,8,25,39]. In the meantime, for a more concentrated solution (containing Pb2+ and Cd2+ at 10−6 M), the obtained SW voltammetric peaks also increased with the preconcentration time duration but stabilized after 15 min accumulation for Pb(II) and 8 min for Cd2+ (II) (see Fig. S3, Supporting Information).
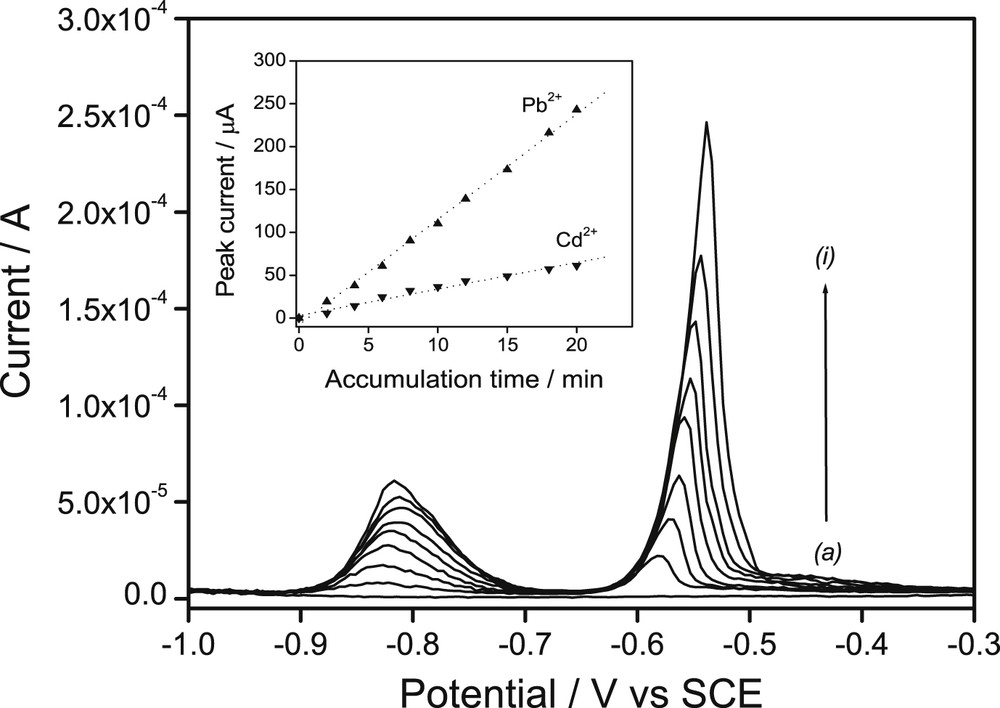
Square wave voltammograms recorded on the GCE/Sa (CTA0.25,T) as a function of accumulation time for a mixture of Cd2+ and Pb2+ (concentration of each ion: 1 × 10−7 M). Inset shows the dependence of peak currents of both ions as a function of accumulation time. Other experimental parameters were kept at their optimized values.
This behavior indicates that a steady–state equilibrium between adsorption/complex formation and desorption/complex dissociation was reached faster with higher analyte concentrations, typical of stripping methods based on adsorptive or complex formation accumulation [24,42].
3.2.4 Calibration graph and direct simultaneous detection of Pb(II) and Cd(II) ions
The results obtained in the previous sections clearly indicate that the GCE/Sa(CTA0.25,T) sensor is adequate for quantifying lead(II) and cadmium(II) simultaneously, viz., when both ions are present in the analyte solution. Thus, both heavy metal ions were introduced into an aqueous solution with concentrations varying between 10 and 7 and 10 and 6 M for Cd2+, and between 10 and 8 and 10 and 7 M for Pb2+ ions. Calibration curves for both analytes were recorded (Fig. 7) upon using the above preconcentration and optimized detection conditions, evidencing a linear dependence between the electrode responses and the concentrations of each ion.
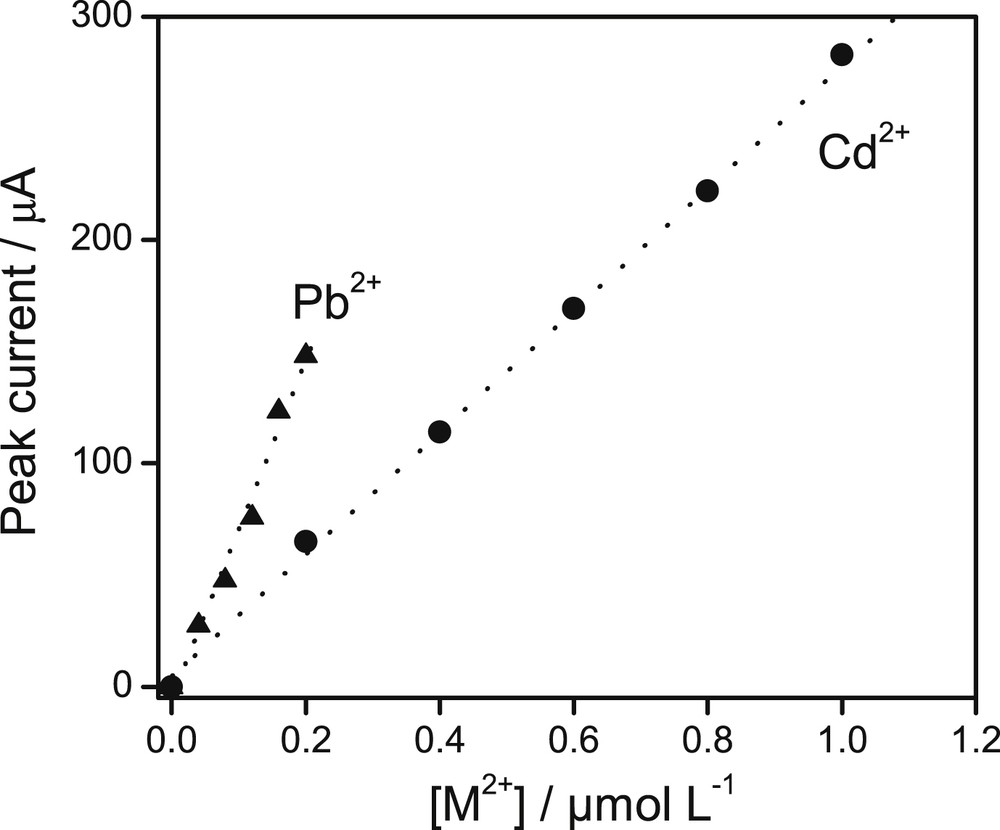
Calibration curves obtained for Pb2+ and Cd2+ when the concentration of Pb(II) was varied between 10−8 and 10−7 M, and that of Cd(II) in the range from 10−7 to 1 × 10−6 M. Other experimental parameters are those optimized in the accumulation and detection steps.
Based on a minimum signal to noise ratio of 3, the detection limits were estimated to be 4.2 × 10−10 M for lead(II) and 12 × 10−10 M for cadmium(II). These analytical performances are comparable to [10,16,43,44], or better than some data reported in previous literature for both metals as shown in Table 1 [17,45,46]. The advantages of the sensor proposed herein are that the organoclay exploited as the electrode modifier is easy to prepare while the procedure used to modify the electrode is quite simple.
Comparison of the performance of various modified electrodes, towards the voltammetric detection of Cd2+ and Pb2+ ions.
| Nature of electrode | Analytes | Limit of detection | Reference |
| aCPE modified with zirconium phosphated amorphous silica | Cd2+ | 2.31 × 10−9 M | [10] |
| Bismuth-film electrode | Pb2+ | 9.66 × 10−10 | [16] |
| Cd2+ | 1.77 × 10−9 M | ||
| Bismuth/poly(p-aminobenzene sulfonic acid) film electrode | Pb2+ | 3.86 × 10−9 M | [17] |
| Cd2+ | 5.6 × 10−9 M | ||
| bMWCNTs-Nafion/Bismuth Composite Electrode | Pb2+ | 1.2 × 10−10 | [18] |
| Cd2+ | 3.57 × 10−10 M | ||
| Graphite-Polyurethane composite electrode, including organofunctionalized SBA-15 Nanostructured Silica | Cd2+ | 2.9 × 10−8 M | [43] |
| Pb2+ | 8 × 10−10 M | ||
| Carbon-based mercury film electrode | Cd2+ | 2.2 × 10−9 M | [44] |
| Pb2+ | 3.86 × 10−10 M | ||
| cGCE coated by nafion film containing Ag-Hg Amalgam | Cd2+ | 1.65 × 10−9 M | [45] |
| Pb2+ | 4.90 × 10−9 M | ||
| GCE modified with a Langmuir–Blodgett film of p-tert-butylthiacalix[4]arene | Pb2+ | 2 × 10−8 M | [46] |
| Cd2+ | 8 × 10−9 M | ||
| GCE covered by a film of smectite intercalated by CTA+ ions and thiourea | Cd2+ | 1.2 × 10−9 M | Present work |
| Pb2+ | 4.2 × 10−10 M |
a CPE: Carbon Paste Electrode.
b MWCNTs: Multiwalled carbon nanotubes.
c GCE: Glassy Carbon Electrode.
The application of the GCE/Sa(CTA0.25,T) sensor prepared herein to real environmental samples remains the main future perspective of this work but the present results established that the organoclay materials prepared are sensitive electrode modifiers for such purposes.
4 Conclusion
A new chemically modified composite electrode for the stripping anodic square wave voltammetric detection of Cd2+ and Pb2+ has been developed. It was achieved upon drop-coating a thin film of an organoclay modified by a cationic surfactant and thiourea onto a glassy carbon electrode. The results obtained indicated that there was a real analytical interest in modifying natural clay with a synergistic combination of thiourea and cetyltrimethylammonium ions to drastically improve its complexation properties. Considering the excellent reproducibility, linearity and sensitivity of the results obtained in the present study, the application of the new sensor to industrial waste containing mixtures of Cd2+ and Pb2+ ions can be envisaged for further investigation.
Acknowledgements
The financial support by the Academy of Science for the Developing World (Grant No. 12-117 RG/CHE/AF/AC-G allowed to I. K. Tonle; and Grant No. 07-052-LDC/CHE/AF/AC allowed to E. Ngameni) is acknowledged. I. K. Tonle also acknowledges the financial support from the Alexander von Humboldt Foundation (Germany). The authors equally acknowledge the International Union of Crystallography (IUCr) and Bruker AXS S A S France for the donation of a diffractometer (Siemens D5005) to the University of Dschang (Cameroon).
Appendix A Supplementary data
The following is the supplementary data related to this article:
Fig. S1: Square wave voltammograms recorded in 10−1 M HCl for (a): GCE/Sa (Na) and (b): GCE/Sa (CTA0.25,T). Accumulation was performed for 5 min in an aqueous solution at pH 3.5 containing1 × 10−6 M Cd2+ and 1 × 10−6 M Pb2+. Other experimental conditions: electrolysis time 60 s; electrolysis potential −1 V.
Fig. S2: Square wave voltammograms recorded on the GCE/Sa (CTA0.25,T) in 0.1 M HCl for a series of seven parallel experiments. Operating conditions: 5 min of accumulation in an aqueous solution at pH 4 containing 1 × 10−6 M Cd2+ and 1 × 10−6 M Pb2+.
Fig. S3: Square wave voltammograms recorded on the GCE/Sa (CTA0.25,T) as a function of accumulation time for a mixture of Cd2+ and Pb2+(concentration of each ion: 1 × 10−6 M). The inset shows the dependence of peak currents of both ions as a function of accumulation time. Other experimental parameters were kept at their optimized values.