

1 General details and prospects
Coordination clusters of paramagnetic 3d metal ions continue to attract significant interest owing to their aesthetically pleasing structures and fascinating physical properties as well as their relevance to metalloenzymes [1]. When the intermetallic bridges of this class of compounds create a superexchange pathway, this often leads to isotropic couplings and, in some cases, slow relaxation of the magnetization to make a so-called single-molecule magnet (SMM) [2]. Such compounds (SMMs) are promising candidates for applications in molecular spintronics, high-density data storage and quantum information processing [3]. In the molecular magnetism field, compared to those of other transition metals, complexes of manganese are often characterized by large spin ground states, and this, in conjunction with the presence of Jahn–Teller distorted MnIII ions, makes manganese polynuclear complexes ideal candidates for single-molecule magnet behaviour [4], the archetype of SMMs being the [Mn12O12(AcO)16(H2O)4] reported in 1993 by Christou, Sessoli, Gatteschi, Hendrickson et al. [5].
The design and synthesis of molecular materials with predictable magnetic properties continues to remain a challenging task because structural factors governing exchange coupling between paramagnetic centres are complex and elusive. The use of assisted self-assembly and site-targeted reactions has provided interesting examples of high-nuclearity clusters, mostly characterized by large magnetic moments in the ground state (S). But, to better gain evidence regarding the role played by weak exchange interactions and geometrical factors on the slow dynamics of the magnetization, systems based on small nuclearity are necessary, since they are less complex systems.
In recent decades, a class of complexes characterized by a small number of paramagnetic transition metal ions (<30) connected with each other by simple bridging anions, typically O2–, HO– or CH3O–, i.e. oxo, hydroxo, and methoxo bridges, respectively, has provided valuable insights as simple models of more complex systems [1]. These promising advances in materials science have stimulated us in the research effort on dinuclear complexes, the simplicity of which favours studies of structure–coupling constant relationships and time-consuming theoretical calculations [6].
In the field of coordination chemistry in general and of molecular magnetism in particular, it is noticeable that chemists largely prefer to use either commercially available ligands or those that can be easily synthesized, in order to produce systems based on coordination chemistry. It is worth noting, in passing, that this is unfortunate since it leads to the impression that most compounds resulting from modern coordination chemistry approaches are discovered by luck rather than by an informed knowledge of how a system with a given set of component might behave. In its defence, however, this approach avoids the difficult and often unfruitful step of synthesizing a tailored ligand, which could easily possess coordination properties not predictable from the “paper chemistry” ideas used in its design.
In 2006, we reported the [Mn2] system [Mn2(HLA)4(μ-OCH3)2] C1 {H2LA = 2-salicyloylhydrazono-1,3-dithiolane or N’-(1,3-dithiolan-2-ylidene)-2-hydroxybenzohydrazide}, the core of which consists of two trivalent manganese ions that are connected via two methoxo bridging co-ligands (originating from MeOH solvent). The two MnIII centres are ferromagnetically coupled, with a spin ground state of S = 8/2, which was corroborated by EPR spectroscopy and DFT calculations that confirm also the negative sign of the DS=4 parameter [7]. This system is especially appealing since it displays the largest coupling constant so far reported for a MnIII–MnIII intramolecular interaction: J = 19.7 cm−1 (for the spin Hamiltonian used see section 3.2 below and ref.[7] as well). The strength of this interaction is associated with the peculiar unsymmetrical arrangement of the ligands driven by (the) two non-classical CH-π hydrogen bonds imposed by the dithiolane ligand (see its molecular structure in the solid state, Fig. 1).

(Colour online.) Left: Molecular structure of the [MnIII–MnIII] system framework C1. Ligand = HLA– (H2LA = 2-salicyloylhydrazono-1,3-dithiolane). Solvent molecules have been omitted for clarity. The black arrows represent the CH-π hydrogen bonds. Right: ChemDraw representation of the complex C1.
With all this in mind, we nonetheless felt it worthwhile to explore ways using designed ligands and in which we could prepare 3d-based isostructural analogues of pioneer [Mn2] system, to better understand the roles played by the organic periphery, the bridging ligands, and the remote substituents on the exchange coupling constants between the two coordinated metal centres.
It is shown that the [Mn2] system framework is both magnetically and structurally robust in the solid state, indicating that the [Mn2] system is an excellent platform for peripheral chemical engineering in the development of magnetic materials and devices. We have therefore embarked on a program of exploring the coordination chemistry of various novel aroylhydrazone ligands (representative examples are given in Fig. 2), so far with a range of functional groups, aiming at accessing new dinuclear compounds with a strong ferromagnetic exchange coupling scenario, with, expectedly, the preparation of a compound with two transition metals showing a SMM behaviour and/or an intramolecular coupling constant larger than the record value J = 19.7 cm−1 (so far held by our [Mn2] system C1). Such ferromagnetically coupled systems could therefore be incorporated into crystal lattices along with conducting molecules, and be addressed on a variety of surfaces including biological molecules, which may lead to the formation of innovative magnetic/conducting bifunctional materials [8]. One should note, however, that to better get insights in the coordination chemistry of magnetic materials, synthetic strategies must be developed to access well-formed single molecules (and not only crystalline powders), whose structures could be elucidated using X-ray diffraction crystallography.

(Colour online.) Molecular structures (obtained from X-ray diffraction) of the 2-salicyloyl-hydrazono-1,3-dithiolane ligand H2LA and related ligands described in this article (HLB, HLC, H2LD, H2LE and HLF) and explored successfully in the preparation of the dinuclear single molecules of Mn, Fe, Co, (Ni) and Cr metal ions. The number of hydrogen atoms indicated in the formula corresponds to the potentially acid hydrogen on the phenol or amine groups; C brown, H grey, N blue, O red, S yellow. Below, the corresponding ChemDraw representation of the ligands.
2 Synthetic strategies for the preparation of the ligands and the dinuclear complexes
Due to the dependence of coupling pathways on the structure and symmetry of the organic ligands, dinuclear metallorganic systems offer interesting possibilities to tune metal-to-metal interactions by subtle structural changes within the organic periphery. Macrocycles of a ring large enough to accommodate two metal ions in it have often been selected for this purpose, because they allow one to fix the position and therefore the distance between the two metal ions in an easy way [9]. The idea of large rings has also been developed into macrobicyclic systems with axial, lateral, and cylindrical topologies [10]. A different approach is found in the case of bis-macrocycles in which two rings are connected either by a carbon–carbon [11] or a nitrogen–nitrogen [12] linker. Depending on its length, the metal–metal interaction can be controlled, and thus tuned. But, open-chain ligands being less rigid than macrocyclic ligands, the design of non-macrocyclic ligands to carry out this specific task to combine two metal ions within a coordination compound becomes de facto a real synthetic and crystallization challenge: an overview of the strategy and of the main results achieved in this way by our group is given in the following sections.
2.1 Ligand design
Numerous recent works in manganese carboxylate chemistry describe stimulating directions for the synthesis of new polynuclear Mn complexes with various sorts of capping/bridging ligands whilst displaying promising magnetic properties [13]. Thus, for instance, a ligand can be designed to provide a coordination environment favoured by a 3d metal such as MnIII (relatively N rich) as well as a site (relatively O rich) more favoured by a “hard” metal ion such as FeIII. Our approach was to prepare new open-chain compartmentalized ligands with the aim of capturing different types of paramagnetic centres to produce magnetically interesting dimeric systems in which their interconnection would be potentially left (in case of intramolecular OH…N bonding) to the appreciation of the bridging ligands.
Aroylhydrazone ligands are useful in this approach since they are easy to tailor. Hydrazones are a class of organic molecules with the general structure R1R2C = NNH2 [14]. They are related to ketones and aldehydes by the replacement of the oxygen with the NNH2 functional group (see Scheme 1). They are formed usually by the reaction of hydrazine (H2N–NH2) on ketones or aldehydes [15]. Hydrazones can also be synthesized by the Japp–Klingemann reaction via β-keto-acids or β-keto-esters and aryl diazonium salts. Hydrazones have found (from the 1970s) useful applications in synthetic organic chemistry, e.g., Shapiro and Bamford–Stevens reactions, Wolff–Kishner reduction, synthetic inorganic chemistry, and in the medical field where the hydrazone-based coupling methods are used to couple drugs with targeted antibodies against a certain type of cancer cell, where the hydrazone-based bond is stable at neutral pH (in the blood) and cleaved in the acidic environment of lysosomes of the cell, therefore releasing the drug in the cell, where it exerts its function [16].

2-salicyloyl-hydrazono-1,3-dithiolane ligand H2LA: keto-enol tautomerism (solution state).
We first investigated many reaction conditions between acetate and/or acetylacetonate manganese sources [Mn(OAc)x vs. Mn(acac)x] with salicyl derivative ligands bearing various different coordination sites in terms of Lewis base hardness and softness. The first compound that we explored in this way is the 2-salicyloyl-hydrazono-1,3-dithiolane ligand H2LA (see Scheme 1). This ligand is especially appealing for the following reasons:
- • the ligand system is rich of two N- and two O-donating atoms, i.e. N,N,O,O coordination pocket, for site targeted reactions;
- • it is particularly versatile as a result of the tautomerization, with potential reaction in the keto form or through the enol form;
- • owing to the rigidity of the phenyl backbone and the close(st) disposition of the keto and phenol groups, and the distance-range of remote sulphur substituents, the ligand favours interactions with a single metal centre;
- • it offers numerous possibilities for hydrogen bonds, and/or Van der Waals ‘weak’ interactions, and deposition on Au surfaces;
- • the H2LA ligand system should lend itself to synthetic modification through derivatization at the phenyl ring.
Thereafter, we worked on subtle derivatization on this ligand. We have been able to synthesize a large library of compounds (>50), under classical conditions or by applying microwave irradiation (MW).
2.2 Synthesis of the organic compounds
We will review herein the six ligands that made us being successful to prepare dinuclear homometallic single molecules of manganese (Mn), iron (Fe), chromium (Cr) and cobalt (Co) metal ions (see Fig. 2). One should note that we were also capable to isolate a large series of single crystals of mononuclear complexes, where the metal centre is in a mononuclear environment, as would be found in siderophore and similar chelating agent complexes, together with dinuclear species based on the nickel(III) metal centre as well. The purpose of the present review being the focus on dinuclear coordination molecules with potential application in the materials field, the latter are not herein reviewed and all details for their synthesis, crystal structures, and applications (e.g., catalysis of ethylene) can be found in our published work [17].
Ligand H2LA, which contains heterocyclic sulphurs allowing potential anchoring on gold surfaces (Au), was synthesized in a 71% yield, according to the procedure described in 2000 by H.C. Chen et al. [18]. One should note that from its design, we expected a tetradentate bridging ligand for connecting two manganese centres [19]. But intramolecular hydrogen bonding, i.e. O–H…N (see solid-state structure), took the more on this bridging by forming mononuclear complexes, which have been characterized by elemental analysis, IR spectroscopy, and single-crystal X-ray diffraction [17a]. Noteworthy, the addition of a base, i.e. KOAc, to a solution of isolated monomer has allowed us to “double” the number of Mn metal centres, thus resulting in the targeted dimeric species [7].
In comparison with H2LA, the parent N’-cyclopentylidenebenzohydrazide ligand HLC lacks both addressable heterocyclic sulphur atoms and the phenolic hydroxyl moieties offering a N,N,O tridentate pocket. Its synthesis was first described in 1990 by M. Okimoto and T. Chiba [20]. Compared to H2LA, the N’-(1,3-dithiolan-2-ylidene)benzohydrazide ligand HLB is missing only the phenol moiety, while the N’-cyclopentylidene-2-hydroxybenzohydrazide ligand H2LD lacks the heterocyclic sulphur atoms, allowing a variability in electronic density, steric hindrance, and secondary coordination site availability.
In 2010, we reported the straightforward and quantitative synthesis of HLC and H2LD compounds by applying MW irradiation for a few minutes, and the preparation in a 50% yield of HLB molecule at room temperature from the equimolar mixing of salicylhydrazide, carbon disulphide (CS2) and 1,2-dibromoethane, in EtOH basified with NaOH. Well-formed colourless to light-yellow crystals of the three ligands allowed their structure determinations in the solid state using the X-ray diffraction technique [21].
As for the H2LA ligand, the 2-salicyloyl-hydrazono-1,3-dithiane ligand H2LE displays two heterocyclic sulphurs for potential deposition on Au surfaces. But this analogue bears a larger remote 1,3-dithiane ring with, however, no impact on the targeted construction of dimeric complexes. It was synthesized in a 65% yield, following the same procedure as that employed in the synthesis of H2LA [22].
Thiophene-based compounds have found tremendous applications in materials science in general and in the optoelectronics field in particular [23]. Thus, in view of developing a new class of optically and magnetically interesting bifunctional materials, we have synthesized the novel (E)-2-hydroxy-N’-(1-(thiophen-2-yl)ethylidene)benzohydrazide ligand HLF, tailored with a remote thiophene group. As for its analogues HLC and H2LD, a quantitative preparation is possible by applying a MW irradiation for a few minutes on an equimolar mixture of benzydrazide and 2-acetylthiophene, following conditions optimisations reported by Andrade et al. [17d,24].
2.3 Synthesis and molecular structure of the dinuclear complexes
As mentioned earlier, the credibility of complexes of manganese was gained in the field of molecular magnetism in general and single molecule magnets field in particular, with the discovery of the first example of SMMs [5]. Our synthetic strategy is to design ligands that provide coordination pockets capable of strongly coordinating the metal ion centres, with, however, the assistance of bridging alkoxides. Precisely, we have been working on slight-to-subtle modifications on the aroylhydrazone backbone as well as on the bridging ligands or other metal centres (in addition to Mn), such as chromium [25], iron [21,26] and cobalt [22]. Our efforts were successful and several mononuclear and dinuclear complexes have been synthesized, structurally and magnetically characterized using X-ray diffractometry, magnetic susceptibility, EPR, and DFT techniques.
From the synthetic point of view, it was not straightforward to establish the appropriate conditions for the synthesis of the dinuclear complexes. Therefore, various conditions such as the reaction composition, stoichiometry, concentration, time, pH and temperature were investigated, and the optimal conditions are herein discussed. We focus reviewing in this section the preparation, using the six above discussed ligands, of 15 new or novel dimers based on Mn, Fe, Cr and Co metal centres.
2.3.1 Manganese complexes: [MnIII2(HLA)4(μ-OCH3)2], [MnIV2(Lc)4(μ-O)2]·THF, [MnIV2(LF)4(μ-O)2]·2MeOH, and [MnIV2(LF)4(μ-O)2]·H2O·THF
The first dimer that we prepared is the [Mn2(HLA)4(μ-OCH3)2] C1 (see Fig. 3). After 1-h standing at room temperature of two equivalent of H2LA with Mn(III) acetate dihydrate in a MeOH/pyridine solution (10/1 v/v), well-formed brown rectangular crystals of the target [MnIII–MnIII] molecule were isolated in an overall 67% yield. Upon optimization of the synthesis, it was observed that the same reaction in pure MeOH yields a brown powder that can be recrystallized into C1 in proper solvent systems, e.g., diffusion of MeOH in CHCl3 or THF solutions, allowing for a better control of the crystallization parameters. Although EPR analysis in the solid state and in solution points to a reversible dimerization mechanism of unspecified mononuclear complexes, the reaction of three equivalents of the ligand in MeOH yields the monomer [MnIII(HLA)3], from which no subsequent dimerization has been observed [7,27].
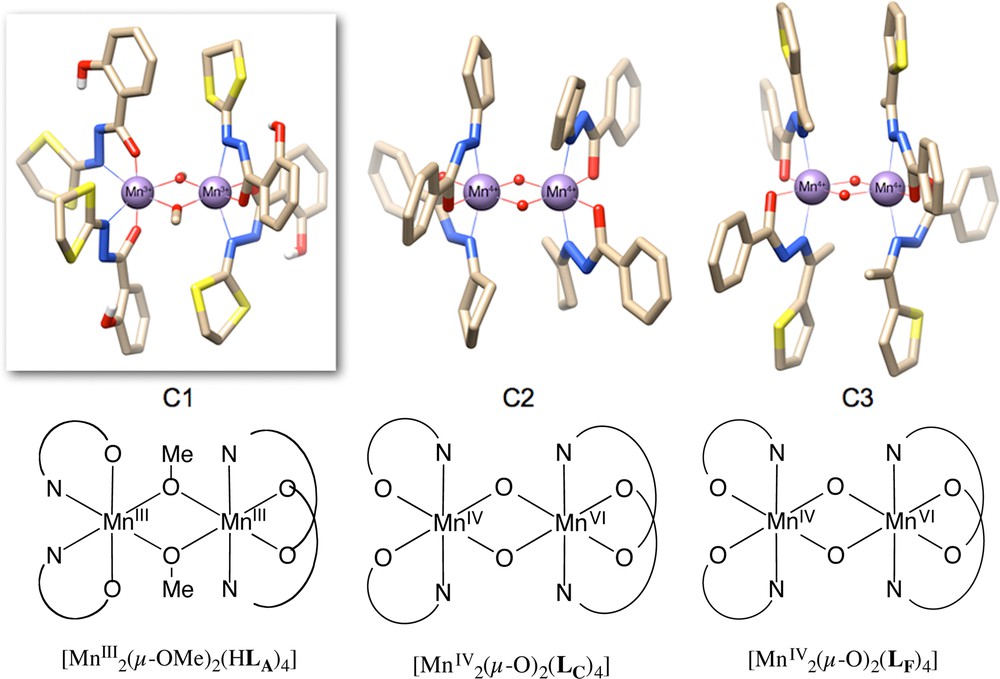
(Colour online.) Molecular structures of the dinuclear manganese +III and +IV compounds C1, C2 and C3, using ligands HLA–, LC– and LF–, respectively. Solvent molecules and hydrogen atoms (except those of the phenol groups) have been omitted for clarity; O, red, C, brown. Below, ChemDraw representations of the complexes.
In an effort to gain more insight into the structural factors controlling and influencing the formation and the stabilization of this dimer C1 in the crystalline state, with potential impact on their magnetic behaviour, the original H2LA ligand was systematically tuned at specific positions. Among the ligands tested, i.e. ca. 20, single crystals of Mn dimers were obtained only with two additional aroylhydrazones, namely the HLC and HLF ligands.
In 2012, we have reported on the synthesis of a MnIV dimer with the HLF ligands [28]. From a synthetic point of view, HLF ligand was reacted with [MnIII(OAc)3]·2H2O in MeOH at room temperature in the presence of potassium acetate (AcOK), followed by recrystallization under exposure to CHCl3. The elucidation of the dark-red crystals (obtained after two days, 11% yield based on Mn) using X-ray diffractometry revealed a Mn+IV dimer of structure [MnIV2(LF)4(μ-O)2]·2MeOH (C3). When the crystallization is carried out in a solution of THF with slow diffusion of pentane (C5H12), the solid-state structure includes water and THF molecules in the lattice instead of CHCl3 giving the general formula [MnIV2(LF)4(μ-O)2]·H2O·THF; the geometrical differences between the two solid-state structures of C3 driven by the inclusion of these different molecules in the lattice are negligible and thus only the best refined structure has been studied.
In 2014, a new dinuclear complex of MnIV bearing the HLC ligand, with a very similar structure, has been published by our group among other dinuclear complexes introducing ab initio CASPT2 calculations on unprecedented large systems whose interests will be discussed below [6]. The reaction of manganese(III) acetylacetonate with HLC ligand in THF followed by the diffusion of Et2O has allowed the crystallization of [MnIV2(LC)4(μ-O)2]·THF C2.
Complexes C2 and C3 are composed of two MnIV ions bridged by oxo anions that originate from residual water molecules in the solvent and/or hydrated metal sources. As reactions have been carried out under anaerobic conditions, the formation of MnIV from MnIII sources seems to arise from a dismutation reaction. Despite numerous efforts, we neither were able to isolate the MnII derivative(s) that should result from the dismutation reaction, nor succeeded to pinpoint the ligands and reaction conditions promoting the formation of MnIII complexes over MnIV dimers.
Last, the hypothesis that the similarity between the structures of ligand H2LA and the 2-salicyloyl-hydrazono-1,3-dithiane H2LE would allow H2LE to be a suitable ligand to stabilize a dinuclear μ-methoxo MnIII complex, which is expected to exhibit very strong intramolecular ferromagnetic interaction, prompted us to extend the coordination chemistry of ligand H2LE to MnIII. Based on the observation that the μ-methoxo dinuclear complex C1 is usually unstable in solution and probably dissociates into mononuclear entities, which in the solid state again tends to assemble into the well-organized μ-methoxo dinuclear structure, we initially focused our efforts on crystallization conditions that could promote the condensation of two mononuclear entities to form the dinuclear complex [7,29]. However, only single crystals of the monomer [MnIII(HLE)3]·CH2Cl2 were obtained by slow diffusion of pentane into a solution of CH2Cl2. Combinations of several solvents and non-solvents have been tested, i.e. CHCl3/MeOH, CH2Cl2/EtOH, 1,2-dichloroethane/iPrOH, DMF/Et2O, THF/H2O, on different proportions using slow liquid or vapour diffusion, but all failed to give a crystal of the desired dinuclear complex.
2.3.2 Iron dimers: [FeIII2(HLA)4(μ-OCH3)2], [FeIII2(HLE)4(μ-OMe)2]·1/2MeOH, [FeIII2(HLE)4(μ-O)2]·3CH2Cl2, [FeIII2(Lc)4(μ-OCH3)2], [FeIII2(LF)4(μ-OCH3)2], and [FeIII2(LB)4(μ-OCH3)2]
In view of exploring the roles played by the organic periphery on the exchange coupling constants between the two metal centres, we have first prepared and reported in 2008 an isostructural ferric analogue of the original [Mn2] system C1 (see Fig. 4) [26]. Likewise dimer C1, this FeIII complex of the general formula [FeIII2(HLA)4(μ-OCH3)2] C4 only exists in the solid state, and disintegrates to a monomer in solution. Dinuclear complex C4 can be synthesized in two ways, starting from commercial Fe(acac)3 or FeCl3 as metal sources. The first way consists in a stepwise synthesis with a good yield of 73% via the isolation of the monomer obtained by reacting in DMF (N,N-dimethylformamide) the salt with two equivalents of H2LA ligand at room temperature and further crystallisation by slow diffusion of MeOH to the THF organic phase. In the second route, a one-pot preparation from a stoichiometric amount of HLA ligand with the chloride salt in MeOH, using the weak base sodium acetate (NaOAc), where the recrystallization from THF with slow diffusion of MeOH yielded well-formed crystals of dimer C4.
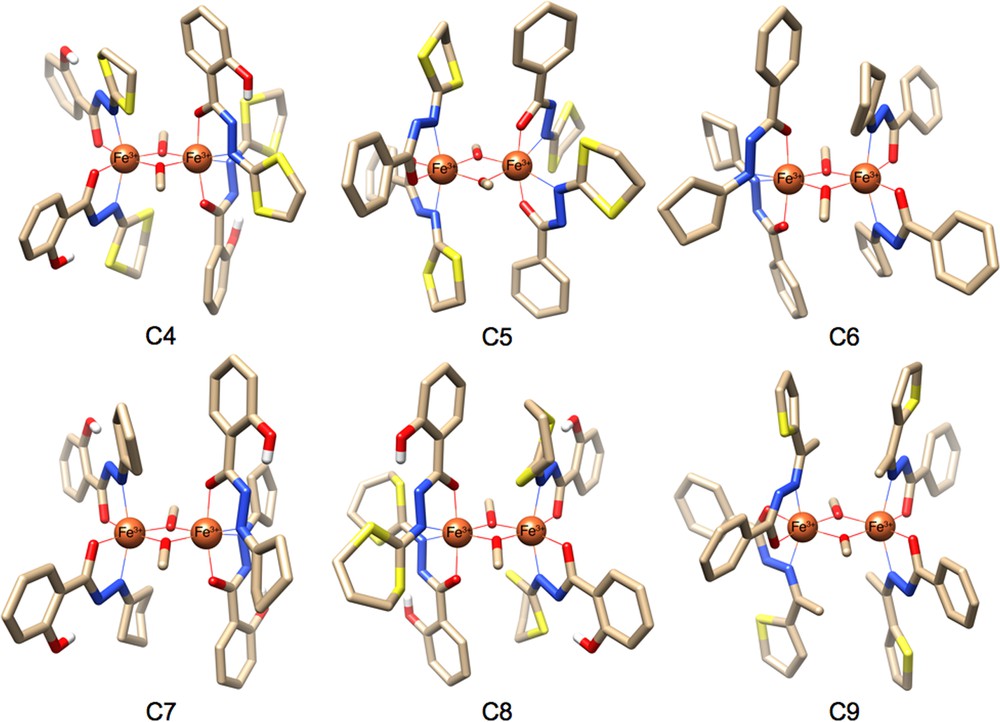
(Colour online.) Molecular structures of the six dinuclear ferric complexes C4, C5, C6, C7, C8 and C9, using ligands H2LA, HLB, HLC, H2LD, H2LE and HLF, respectively. Solvent molecules and hydrogen atoms (except those of phenol groups) have been omitted for clarity; O, red, C, brown.
By applying our previously reported procedure [26], dimer [FeIII2(HLE)4(μ-OMe)2]·1/2MeOH C8 was synthesized in good yield (72%) by the reaction of H2LE with Fe(acac)3 in DMF, followed by the slow diffusion of MeOH into a THF solution of the product. Similarly, it was hypothesized that the formation of this complex occurs via a non-isolated mononuclear species [FeIII(LE)2L1L2], which possesses two deprotonated 2-salicyloylhydrazono-1,3-dithiane ligands and two additional coordination moieties, i.e. L1 and L2, which are probably remaining acetylacetonate ligand (acac) from the metal precursor or the methoxy group and a molecule of solvent coordinated. The reaction of two equivalents of the ligand with Fe(acac)3 in MeOH generated black precipitates, which can be crystallized in a mixture of CH2Cl2–Et2O to yield dark-green crystals of dimer [FeIII2(HLE)4(μ-O)2]·3CH2Cl2 in 20% yield (based on Fe). Dinuclear iron complexes with oxo bridges have been extensively studied mainly due to their peculiar physical properties and their relevance as intermediates in catalytic reactions, and a large number of μ-oxo bridged diiron complexes have been structurally characterized. Similar structures to dimer [FeIII2(HLE)4(μ-O)2]·3CH2Cl2, reported in the literature, mainly bear ligands of salen [30], salicylaldiminato [31] and N,N’-o-phenylenebis(oxamate) [32] type.
In view of preparing an isostructural dimer of C1 with HLB ligand, Fe(acac)3 was first used as our primary ferric sources. The reaction of two equivalents of HLB with Fe(acac)3 in methanol gives the mononuclear FeIII complex [FeIII(LB)2(acac)], which was isolated as air-stable dark-blue crystals in correct yield (53%, CHCl3/MeOH). The same method used with original H2LA ligand, which has led to the formation of the dinuclear FeIII complex [26], was applied. But all attempts to form the analogous FeIII dimer with HLB molecule starting from [FeIII(LB)2(acac)] have failed so far. Nonetheless, in 2010, we have reported the synthesis, molecular structure and magnetic properties (magnetic susceptibility measurements and EPR studies) of the new [FeIII2(LB)4(μ-OCH3)2] C5 compound [21] using FeCl3 as FeIII sources. From the synthetic point of view, the direct reaction of two equivalents of ligand HLB with FeCl3 in MeOH in the presence of two equivalents of sodium acetate has afforded a dark purple polycrystalline powder, which was further recrystallized (one-day-long diffusion of methanol into a chloroform solution of the crude product) to yield large and dark purple crystals of the targeted FeIII dimer C5 in excellent yield (95%).
Finally, in early 2014, we have reported on the synthesis and magnetostructural aspects of the new dinuclear complex [FeIII2(Lc)4(μ-OCH3)2] C6, using HLC ligand that features methoxo-bridged ferric metal ions [6]. The reaction involved iron(III) chloride salt with HLC ligand in alcoholic media using the weak base KOAc. The filtrate obtained was submitted to a 4-day standing in MeOH at room temperature, which has resulted in the formation of red single crystals of C6 (18% yield based on Fe).
Notably, we were also successful in preparing a new dinuclear FeIII-based complex [FeIII2(LF)4(μ-OCH3)2] C9 using ligand HLF. The detailed synthesis procedure, the crystal structure and the magnetic properties of this new dimer will be published elsewhere.
2.3.3 Chromium dimers: [CrIII2(HLA)4(μ-OCH3)2], [CrIII2(HLE)4(μ-OCH3)2], [CrIII2(Lc)4(μ-OCH3)2]·CHCl3·CH3OH, [CrIII2(LF)4(μ-OCH3)2]·CH2Cl2
Only two μ-methoxo dinuclear CrIII complexes with N,N,O,O coordination spheres have been reported to date [33]. This literature precedent along with our theoretical model prompted us towards the synthesis of Cr(III) analogues to further assess their magnetic properties in the solid state. Our efforts were successful, with the synthesis of four new Cr(III)-based dinuclear complexes, i.e. [CrIII2(HLA)4(μ-OCH3)2] C10, [CrIII2(HLE)4(μ-OCH3)2] C11, [CrIII2(Lc)4(μ-OCH3)2]·CHCl3·CH3OH C12 and CrIII2(LF)4(μ-OCH3)2]·2CH2Cl2 C13 (see Fig. 5).
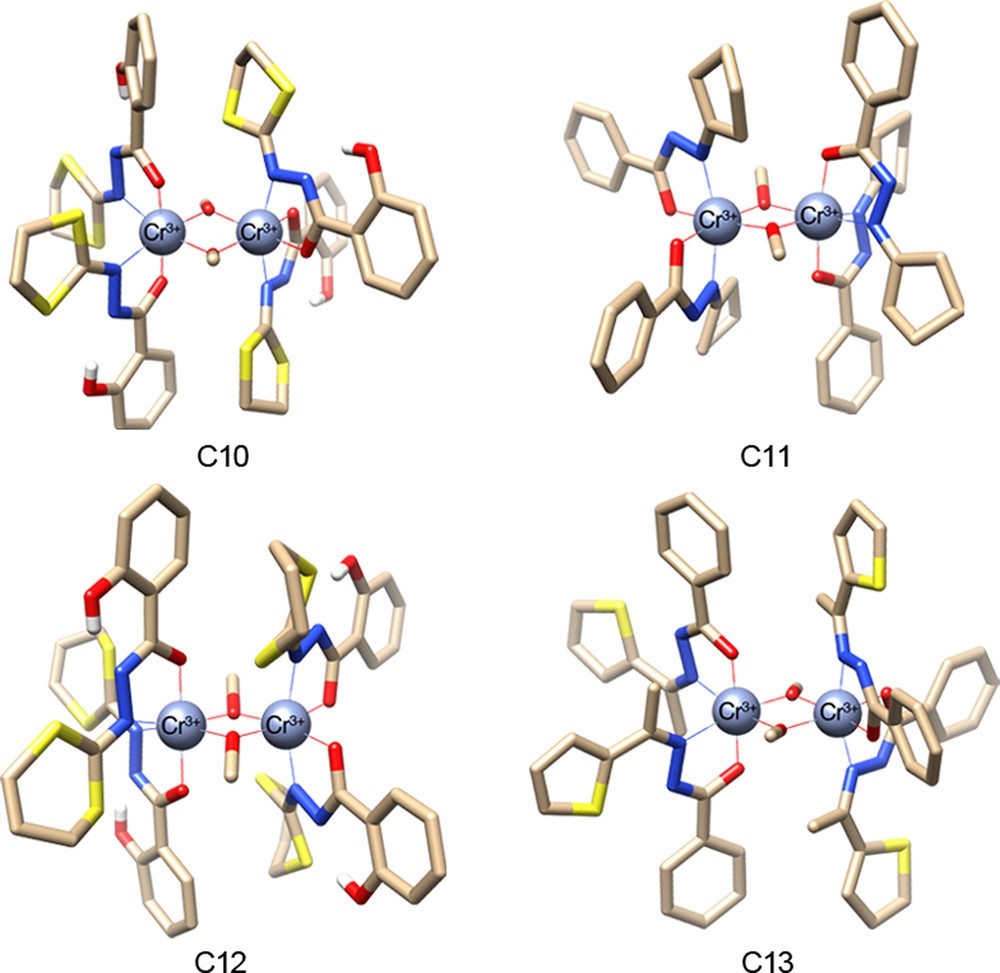
(Colour online.) Molecular structures of the four dinuclear chromium complexes C10, C11, C12 and C13, using ligands H2LA, HLC, H2LE and HLF, respectively. Solvent molecules and hydrogen atoms (except those of phenol groups) have been omitted for clarity; O, red, C, brown.
In 2010, we have reported the preparation of two new μ-methoxo dimeric species based on CrIII metal ions, using H2LA and ring-extended H2LE ligands [25]. The reaction of ligand H2LA (3 equiv) with anhydrous CrCl3 in MeOH afforded the mononuclear CrIII metal complex [CrIII(HLA)3] in a 53% yield. But, when excess zinc dust (Zn0, 4 equiv) was added to the ligand and CrCl3 in a 2:1 molar ratio, the reaction yielded the targeted dinuclear CrIII metal complex [CrIII2(HLA)4(μ-OCH3)2] C10 in a 35% yield. Dimer C10 could be isolated as single crystals via a classical solvent/non-solvent (CH2Cl2–MeOH) diffusion process, starting from a relatively diluted CH2Cl2 solution. The same procedure was used for the synthesis of the [CrIII2(HLC)4(μ-OCH3)2] C11 and [CrIII2(LE)4(μ-OCH3)2] C12 complexes, with respective 10% and 17% yields.
As mentioned previously, we were also successful in preparing the new dinuclear chromium complex [CrIII2(LF)4(μ-OCH3)2]·2CH2Cl2 C13. This was obtained by reacting in refluxing MeOH a suspension of ligand HLF, CrCl3, and Zn dust. Black crystals suitable for X-ray diffraction analysis were obtained (12% based on Cr) subsequently to the slow evaporation of a one-to-one MeOH-CH2Cl2 solution mixture. The detailed synthesis procedure, the crystal structure, and the magnetic properties will be published elsewhere.
2.3.4 Cobalt dimer: [CoIII2(LA)4(μ-OH)2]
The most common oxidation states of the Co metal are +II and +III. Cobalt(II) complexes are paramagnetic compounds, thus potentially molecular magnets considering the polynuclear species. On the contrary, CoIII complexes are almost exclusively octahedral and diamagnetic. This is due to the weak ligand field necessary to cause electron pairing; exceptions are [CoF6]3–and [CoF3(H2O)3], which are paramagnetic [34].
We thus decided to explore the chemistry of Co complexes with the supported ligands H2LA and ring-extended H2LE with, expectedly, a comparison that could be made with the MnIII and FeIII complexes previously obtained during our systematic approach [22]. The reaction of three equivalents of the ligands with either CoCl2 in MeOH at RT, followed by air oxidation in the presence of KOH, or Co(OH)3 in refluxing ethanol, yielded mononuclear CoIII complexes [CoL3]. The complex [CoIII2(LA)4(μ-OH)2] C14 can be obtained, however, by the reaction of two equivalents of H2LA with Co(OH)3 in refluxing ethanol with 21% yield (See Fig. 6). Slow diffusion of pentane into a CH2Cl2 solution of the crude product allows its isolation as black crystals. As expected, the octahedral CoIII metal ions of C14 are diamagnetic.

(Colour online.) Left: Molecular structure of the dinuclear cobalt complex C14, using ligand H2LA. Solvent molecules and hydrogen atoms (except those of phenol groups) have been omitted for clarity; O red, H white. Right: corresponding ChemDraw representation of C14.
2.4 Remarks about molecular structures of dinuclear complexes
It is first important to note that all molecular structures discussed in this article were obtained from single-crystal X-ray diffraction results, which have allowed a first effective molecular characterization of these objects in the solid state. Most of these complexes are paramagnetic; therefore, their fine structures could not be determined by conventional techniques. Structural data and crystal chemistry details for each one of these dinuclear complexes are reported in our articles published between 2005 and 2014. This technique allows a fine comparison of the magnetic properties of the complexes regarding their solid-state structures (Section 3).
2.4.1 Unsymmetrical coordination around both metal ions
The peculiarity of most of these dinuclear complexes is the unsymmetrical arrangement of the ligands observed in the solid state. In the case of C1, this unsymmetrical situation (see Scheme 2) leads to a noncollinearity between the distortion axis of the two MnIII centres, establishing an efficient ferromagnetic interaction pathway (see also Scheme 3). This arrangement of the metal orbitals are seen thanks to the occurrence of a clear Jahn–Teller deformation with Mn(III) ions, as discussed by Beghidja et al. [9]. Moreover, specific intramolecular non-classical H-bonds (about 10 kcal·mol−1 each) occurring between hydrogen atoms of the five or six-membered saturated rings and the centroids of the phenol groups could explain this unexpected geometrical conformation. We observed in some complexes with six-membered saturated rings two CH-π hydrogen bonds per ring, for instance in complexes C8 or C12 [27,32]. Although the unsymmetrical arrangement seems to be favoured by this type of aroylhydrazone ligands, we observed symmetrical arrangements in five specific cases, as discussed hereafter.

(Colour online.) Schematic representation of the two possible configurations observed for the dinuclear complexes.

(Colour online.) Simplified scheme of dominant ferromagnetic exchange pathways in MnIII dimer C1.
2.4.2 Occurrence of some symmetric complexes
The first symmetric complex that we observed is the complex C10’ (Fig. 7). This complex was obtained during the test synthesis of the Cr3+ complex C10 (which is unsymmetrical) [32]. Only very few single crystals of complex C10’ have been obtained and analysed by X-ray diffraction, although its synthesis could not be rationalized. Therefore, no magnetic measurement could be performed on this sample.

(Colour online.) Dinuclear complexes C2, C3, C9, C14 and C10’ [27] with symmetric configuration around the metal ions. Framing: Complex C1 with asymmetric configuration for comparison. The black arrows represent CH-π hydrogen bonds; C, brown; O, red; C, brown; H, white.
We were therefore surprised that the Co3+ complex C14 [24] is only found with symmetrical configuration and no observation of an unsymmetrical complex has been, to the best of our knowledge, reported to date. We then attempted to correlate this symmetric configuration with the non-magnetic (diamagnetic) character of this compound.
Despite numerous attempts to synthesize an isostructural Mn3+ complex to complex C1, only two symmetrical complexes of Mn4+ have been obtained, the latter were formed by disproportionation of Mn3+ to Mn4+ and Mn2+. Both complexes C2 and C3 are strongly antiferromagnetic (see below chapter 3). And, despite our several attempts and strategies, no additional dinuclear Mn3+ (potentially ferromagnetic) could be stabilized, which is very surprising… and of course frustrating for the chemist. No satisfactory explanation has been brought to date to this crucial point.
Finally, we very recently synthesized a new symmetric Fe3+ complex with the ligand HLF. These observations make more complex the effort to explain the relative stabilities of these different molecular entities. To better understand these subtle geometric conformations, we believe that other complexes must be characterized along electronic density maps (from high-resolution single-crystal diffraction) with and without a magnetic field to provide valuable information about the potential relationship in these systems between the magnetism of the transition metal ions and the molecular geometry of the dinuclear entities.
3 Magnetic properties
3.1 Preamble
As mentioned earlier, the dependence of coupling pathways on the structure and symmetry of the organic ligands makes dinuclear complexes valuable systems to tune metal-to-metal interactions by subtle structural changes within the organic periphery. Before considering possible structure–magnetism correlations, we will recall the main points of our magnetic investigations as detailed in the associated articles.
The magnetic properties of complexes C1–13 were investigated from magnetic measurements obtained with SQUID magnetometer in the solid-state typically in the 300–1.8 K temperature range with an applied field of 5 KOe except for complexes C3, C9, C11, C13 (10 KOe) and C6 (0.5 KOe). The magnetic susceptibility data were fitted using the following spin Hamiltonian where all parameters have their usual meaning and the spin operator S is defined as :
To reproduce the data satisfactorily, we sometimes had to consider a certain amount ρ of paramagnetic impurity supposed to follow the Curie law and being of the same spin states than the isolated metal ion considered. Intermolecular magnetic interactions are neglected given the large distances between metal ions of different molecules (>8 Å). Magnetic coupling constant J and selected structural properties of some of the complexes can be found in Table 1.
Magnetic coupling constant J and selected structural properties of complexes C4–13.
| FeIII complexes | J (cm−1) | M–M (Å) | M–O–M (°) | P (Å) | μ-OR tilt angle (°) |
| C4 | –27.4 | 3.085 | 102.75 | 1.975 | 19.68 |
| C5 | –27.2 | 3.097 | 103.31 | 1.977 | 17.33 |
| C6 | –23.0 | 3.107 | 102.30 | 1.995 | 25.67 |
| C7 | –17.5 | 3.057 | 101.71 | 1.971 | 18.86 |
| C8 | –21.1 | 3.094 | 102.41 | 1.984 | 25.18 |
| C9 | –32.3 | 3.198 | 105.75 | 2.006 | 2.43 |
| CrIII complexes | |||||
| C10 | –21.5 | 3.024 | 100.91 | 1.961 | 27.28 |
| C11 | –18.6 | 3.060 | 101.60 | 1.973 | 20.55 |
| C12 | –28.1 | 3.021 | 100.97 | 1.958 | 29.22 |
| C13 | –12.6 | 3.060 | 102.38 | 1.964 | 18.99 |
3.2 Manganese(III/IV) complexes
MnIII2(HLA)4(μ-OMe)2 C1 is a neutral asymmetric complex where each manganese ion is chelated by two HLA–bidentate ligands and bridged by two methoxy anions. The magnetic susceptibility is characteristic of a dominating ferromagnetic exchange between the metal centres resulting in an S = 4 ground state. Axial zero field splitting was introduced only in the ground state using the following Hamiltonian:
The fit leads to a J value of +19.7 cm−1, g = 1.90 and DS = 4 = –0.83 cm−1, which is, to our knowledge, one of the strongest ferromagnetic interactions ever found in binuclear MnIII complexes. Field-dependent magnetization measurements at low temperatures were performed to characterize more precisely the magnetic anisotropy of the ground state, the best-fit yielded DS = 4 = –0.85 cm−1 and ES = 4/|DS = 4| = 0.21. The amplitude of the anisotropy is considerably reduced compared to what is usually observed for monomeric MnIII complexes (|D| ∼ 4 cm−1) although it remains considerably larger than typical MnIII dinuclear complexes. Modelling of the full molecule by first-principles density functional theory (DFT) calculations found the ferromagnetic configuration to be lower in energy by 182 cm−1 than the antiferromagnetic one. Considering the 10 J energy difference between the S = 4 and S = 0 states, we obtain J = –18.2 cm−1, which in very close agreement with the experimental data.
Both the reduced amplitude of anisotropy and the peculiar strength of the ferromagnetic coupling that are proposed originate from the noncollinearity between the distortion axis of the two MnIII centres, establishing ferromagnetic interaction pathways which arises from the efficient “crossed interaction” between the singly occupied dxy orbital on a metal centre and the empty on the other metal centre (Scheme 3).
This noncollinearity is the main feature of the unexpectedly asymmetric structure (see also Scheme 2) of MnLA, with an angle of approximately 100° between the two distortion axes, and seems to find its origin in two intramolecular nonclassical H-bonds. Given this very peculiar metal orbitals arrangement and magnitude of ferromagnetic coupling, it appears difficult to link C1 to any magnetostructural studies of dinuclear MnIII.
The manganese oxo dimers [MnIV2(HLC)4(μ-O)2] C2 and [MnIV2(HLF)4(μ-O)2] C3 are closely related neutral symmetric complexes where each MnIV ion is chelated by two HL- bidentate ligands and bridged by two oxo di-anions. Their magnetic susceptibility is indicative of strong antiferromagnetic exchanges resulting in S = 0 ground states; best fits for C2 and C3 lead to J = –384 and–298 cm−1 respectively, with a fixed g = 2.
These two complexes share the typical magnetic properties of complexes with MnIV2(μ-O)2 core. Although this class of complexes has been studied extensively for their model role to the oxygen-evolving complex of photosystem II, comparatively little has been done to establish a magnetostructural correlation. Law et al. determined a direct relation between the MOM angle and the coupling constant while the data did not show a dependency as strong upon M–M and M–O distances [35]. In this regard, the high J coupling values of the two complexes are in line with their fairly large MOM angles of 99.47 and 99.73°, respectively (Fig. 8). Using Hückel calculation, Holtzeman et al. [48] rationalize the angular dependence of the coupling constant J through the variations of the xz–z2, xz–xz, and yz–yz interactions.
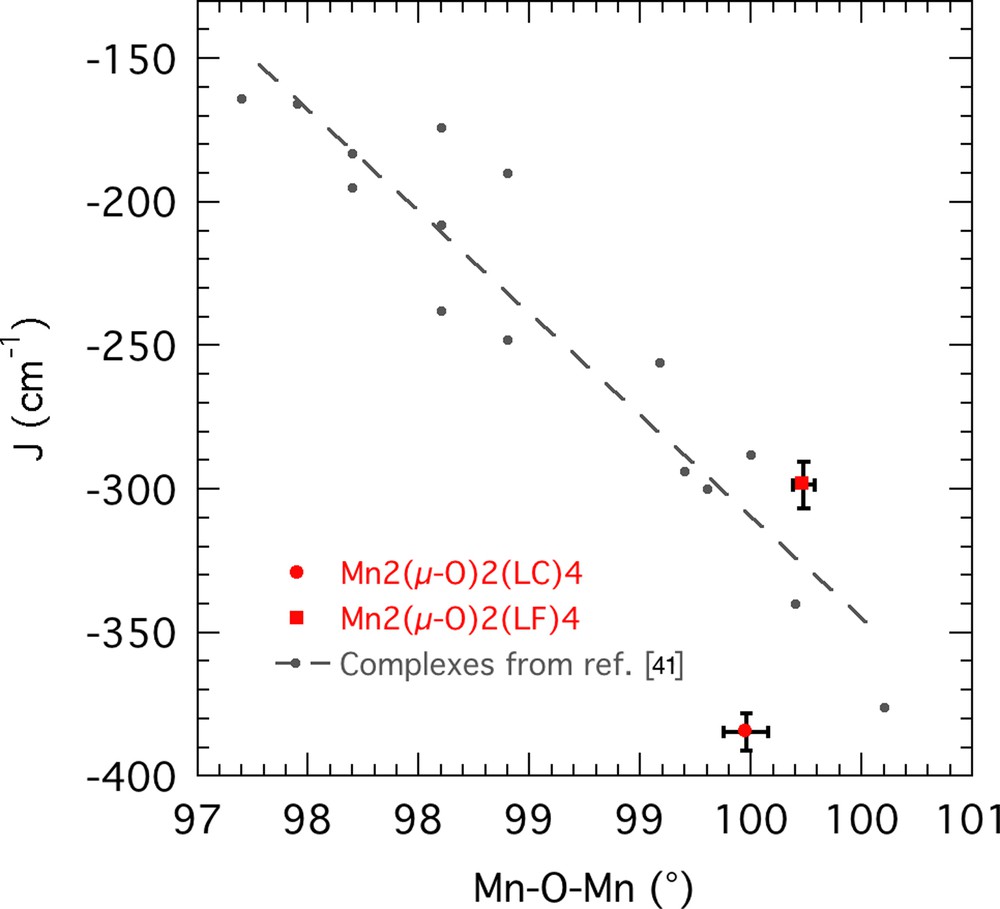
(Colour online.) Plot of coupling constants J (cm−1) vs. Mn–Oxo–Mn angles (°) for C2 and C3 (red) compared with data found in ref [41] and ref. therein (black).
Although the correlation has proved to be valid for a wide range of chelating ligand coordinated through a neutral nitrogen and an anionic oxygen, several outliers complexes in which C2 could be included do exist. Indeed, it presents a larger J coupling, but a narrower Mn–O–Mn angle than C3, indicating that other parameters, however of less significance, might be taken into account. The solid-state structure of C2 shows an inversion centre at the MnIV2(μ-O)2 core, in contrary with C3, as well as N–Mn bonds closer to a perfect octahedral geometry than C3, which may result in better orbital overlaps along the superexchange pathway and thus explain the higher J value of C2, though no other structural parameters could help to understand this difference. Fitting the magnetic susceptibility of both complexes has also shown to be difficult due to important corrections for paramagnetic impurities (ca. 8%) and degradation of the complexes at relatively low temperatures, preventing to collect data “where it matters” for such high antiferromagnetic coupling.
3.3 Iron complexes
The iron dimers [FeIII2(HLA)4(μ-OMe)2] C4, [FeIII2(HLB)4(μ-OMe)2] C5, [FeIII2(HLC)4(μ-OMe)2] C6, [FeIII2(LD)4(μ-OCH3)2] C7, [FeIII2(LE)4(μ-OCH3)2] C8, and [FeIII2(LF)4(μ-OCH3)2] C9 are similar neutral complexes where each FeIII ion is chelated by two HL–bidentate ligands and bridged by two methoxy anions. Their magnetic susceptibility is indicative of antiferromagnetic exchanges resulting in S = 0 ground states. Best fits for each complex are gathered in Table 2.
Best fit parameters for complexes C4–9. The two dashed lines are both complexes whose magnetic properties are far out of the range of expected magnetostructural correlations.
| Complexes | J (cm−1) | g |
| C4 | –27.4 | 2.05 |
| C5 | –27.2 | 2.03 |
| C6 | –23.0 | 2.01 |
| C7 | –17.5 | 2.2 |
| C8 | –21.1 | 2.06 |
| C9 | –32.3 | 1.97 |
Several attempts have been done so far to propose accurate magnetostructural correlations for weakly coupled FeIII binuclear complexes with a general agreement that some parameters play a key role, e.g., Fe–Fe distance or Fe–O–Fe angle. But, none of them allows us to establish their relative contribution to the coupling constant J applicable to a wider range of examples than complexes of specific families of ligands. As a representative example, Le Gall et al. established a correlation between J and the Fe–O–Fe angle for a family of binuclear FeIII complexes with β-diketonate-alkoxide peripheral ligands [36], which does not apply to all the other complexes reviewed by Werner et al. [37] in their refinement of the semi-empirical relation established by Gorun and Lippard [38]. They observed that J could be expressed as a function of the structural parameter P, defined as half the averaged shortest superexchange pathway between the two FeIII, under the form: . The refinement of the A and B parameters based on an enhanced number of examples gives cm−1 and B = –13.9Å–1. A plot of the coupling constant vs. P using this expression can be seen in Fig. 9 along the report of the parameters of complexes C4–9.
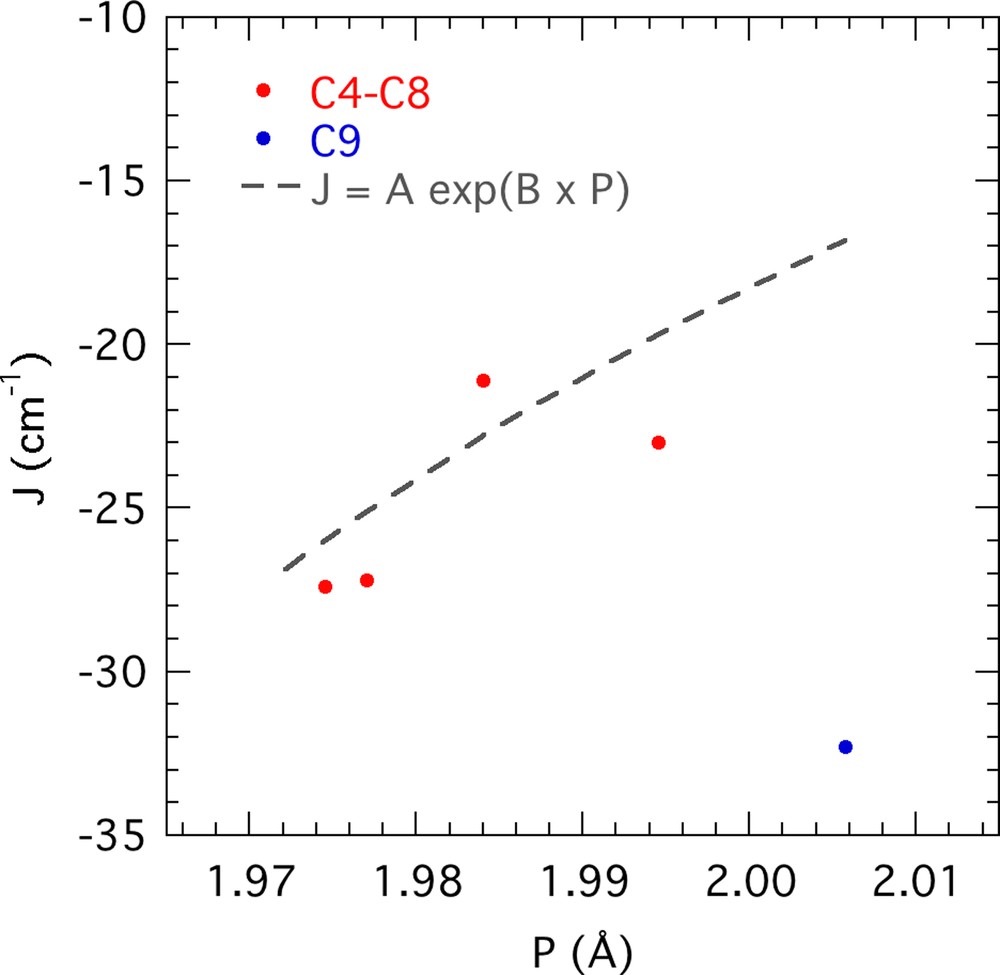
(Colour online.) Representation of the Gorun and Lippard semi-empirical relationship with Werner et al.’s refined parameters (grey line) with the reported values of complexes C4–9, except C7, for which the quality of magnetic susceptibility data does not allow us to discuss any magnetostructural correlation. Depicted in blue is complex C9 whose coupling constants falls far off expected values.
Complexes C4–6 and C8 are in very good agreement with the semi-empirical relationship, which is not the case of complex C9. Complex C7 best-fit parameters are given with great caution and had not been included in Fig. 9 since the measurements had to be taken on the crude polycrystalline sample, the single-crystal sample having shown a propensity to rapidly decompose, thus resulting in imprecise fitting. Although complex C10 shows the greatest Fe–Fe distance and the widest Fe–O–Fe angle, these differences offer little insight into the coupling constant value: the large Fe–Fe distances (>3 Å) prevent any significant impact of the direct interactions of the metal orbitals on the coupling constant. Moreover, Werner et al. tried to include the Fe–O–Fe angle in their correlation, following the route of Weihe and Güdel [39], but they did not observe any improvement of the correlation. This can be rationalized by the high number of superexchange pathways existing between two S = 5/2, each presenting different angle dependencies of their overlap integrals S. In the case of the high-spin d5 ions, the spherical electron distribution results in a compensation of these different types of angular dependencies. As observed in the case of bis(μ-hydroxo)dichromium(III) complexes, the hybridization of the bridging oxygen atom, reflected in the variation of the angle between the O–R vector and the Fe2(OR)2 plane, could play a role as C9 presents the bridges closest to collinearity with Fe2(OR)2 plane. However, the symmetrical organization of the ligand in C9 is probably the main factor governing the differentiation from C4–8, which show a noncollinearity of the distortion axis of the two metals in a manner similar to that of C1; this organization imposes several changes in the superexchange pathways that may favour antiferromagnetic exchanges. This interesting differentiation, supposed to be related to intramolecular interaction between the ligands, opens up the perspective to new prospects for theoretical calculations.
3.4 Chromium complexes
The chromium methoxo dimers [CrIII2(HLA)4(μ-OMe)2] C10, [CrIII2(HLC)4(μ-OMe)2] C11, [CrIII2(HLE)4(μ-OMe)2] C12 and [CrIII2(HLF)4(μ-OMe)2] C13 are closely related neutral symmetric complexes where each CrIII ion is chelated by two HL–bidentate ligands and bridged by two oxo di-anions. Their magnetic susceptibility is indicative of weak antiferromagnetic exchanges resulting in S = 0 ground state. Best fits for C10–C13 are given in Table 3 given below.
Best fit parameters for dinuclear complexes C10-13.
| Complexes | J (cm−1) | g |
| C10 | –21.5 | 1.99 |
| C11 | –18.6 | 1.97 |
| C12 | –28.1 | 1.99 |
| C13 | –12.6 | 1.94 |
The magnetic interaction properties of the complexes are perfectly in line with previously published magnetostructural correlations of alkoxo-bridged Cr(III) binuclear complexes [40]. No magnetostructural correlation including a dominant single structural parameter has been observed. As an example, the narrowing of the Cr–O–Cr angle induces an increase in the ferromagnetic contribution to the exchange, but it also tends to shorten the Cr–Cr distance allowing for an antiferromagnetic direct interaction of the metal orbitals. Concerning the complexes C10–C13, an inverse trend is observed with a decrease in the antiferromagnetic contribution to the coupling with the widening of the Cr–O–Cr angle and the subsequent elongation of the Cr–Cr distance, although there is no clear correlation between these parameters and the magnitude of the coupling. The most relevant parameters seem to be the tilt angle of the OMe bridges with respect to the Cr2O2 plane: the collinearity of the bridging ligand with the plane either disfavours the antiferromagnetic contribution or favours the ferromagnetic one. However, in order to define such correlation, more examples of similar structures are needed as well as theoretical studies.
4 Some data of computational analysis
The first dinuclear complex synthesized and structurally characterized is the manganese-based molecule [Mn2(HLA)4(μ-OCH3)2] C1, which interestingly displays the largest ferromagnetic interaction observed so far between two MnIII (J = 19.7 cm−1). To get better insight into the structural factors governing this strong exchange coupling between the two trivalent manganese centres, our collaborators J. Kortus and G. Rogez have carried out first-principles density functional theory (DFT) calculations (Fig. 10) [7]. Starting from the X-ray structure, a single molecule was isolated for the calculation. In order to estimate the exchange coupling between the two MnIII ions, we considered magnetic configurations corresponding to ferromagnetic and antiferromagnetic coupled MnIII ions. All electronic degrees of freedom were allowed to relax during the self-consistent calculation. After reaching self-consistency, it has been checked whether the solution converges to the desired spin configuration by calculating the spin density in a sphere around the Mn metal centres. The ferromagnetic configuration was found to be lower in energy by 182 cm−1 than the antiferromagnetic one. Mapping this energy difference to the energy difference between the S = 4 and S = 0 states, which equals 10 J, we obtained J = +18.2 cm−1, in a very close agreement with the result from the fit, i.e. J = +19.7 cm−1. The fact that the ferromagnetic contribution competes more or less successfully with the antiferromagnetic one obviously depends on subtle chemical and structural features, as stated by Hotzelmann et al. [41]. But, as others however, we still do not understand which precise factors do govern this competition.

(Colour online.) Magnetic orbitals ordered with increasing energy from left to right for the left fragment. The z-axis is defined by the two nitrogen (grey) atoms above and below the plane given by the oxygen (red) atoms.
The [Mn2] system provides a new example of such a contribution of an interaction between one singly occupied orbital and one empty orbital to the overall positive exchange constant. The fact that in the present case the connection between the magnetic centres consists in two methoxo bridges, and not simply one oxo bridge as found in the reported work of Hotzelmann et al. [41], may play a role. To that, the fact that the distortion axes are no longer parallel, but almost perpendicular, which leads to a different and somehow more complicated scenario for the interactions between the magnetic orbitals (supporting information in [9]).
In a concerted effort to gain better evidence of the structural factors governing exchange coupling between paramagnetic centres, the above-mentioned DFT computational analysis were validated for the [Mn2] system, using the ab initio calculations developed by our collaborators V. Robert and A. Domingo, and further extended to four additional dimers of manganese, iron and chromium metal ions supported by H2LA and HLC ligands, i.e. [FeIII2(HLA)4(μ-OCH3)2] C4, [MnIV2(Lc)4(μ-O)2]·THF C2, [FeIII2(Lc)4(μ-OCH3)2] C6, and [CrIII2(Lc)4(μ-OCH3)2]·CHCl3·CH3OH C11 [6].
This technique is the result of the combination of the complete active space second-order perturbation theory (CASPT2) method [42], that has proved to be credible for the treatment of electron correlation effects on transition-metal compounds [43], with localized molecular orbitals (MOs), since it offers the benefit to enable or disable arbitrarily defined regions of the molecule, the possibility of unravelling the influence of different regions of the molecule on the overall magnetism of the compound.
We first validated the proposed theoretical method with complex [Mn2(HLA)4(μ-OCH3)2] C1, which exhibits a strong ferromagnetic coupling scenario, and then extended it to its iron analogue [FeIII2(HLA)4(μ-OCH3)2] C4, which shows an antiferromagnetic character. Subsequently, this robust theoretical strategy was applied to a new series of three dinuclear complexes based on parent HLC ligand. The simpler chemical structure of the HLC ligand in comparison to that of H2LA molecule was supposed to be of help to assess the influence of some remote substituents of the ligand and the role of the basic aroylhydrazone structure as well. The three HLC-based complexes are the discussed [MnIV2(Lc)4(μ-O)2]·THF C2, [FeIII2(Lc)4(μ-OCH3)2] C6, and [CrIII2(Lc)4(μ-OCH3)2]·CHCl3.CH3OH C11. The assessment of the magnetic properties of these five new dinuclear compounds offers estimates of their J values together with detailed information about the contributions of different parts of the molecule to the overall magnetism. The partitioning scheme for both HLC and H2LA ligands and the bridges, which was used to evaluate their influence on the molecular magnetism, is illustrated in Reference [8]. The various regions defined in the computational model incorporate the π system and lone pairs localized in these zones.
The work has consisted in measuring the J values by activating other regions of the dimetallic core. Notably, the J value obtained for complex [FeIII2(HLA)4(μ-OCH3)2] C4 with the interactions included for all the regions is–22 cm−1, in good agreement with the reported experimental value, i.e. –27 cm−1, which confirms the smaller impact of the ligand σ orbital and the diffuse MOs on the J value [28]. The exception is the dithiolane group in H2LA ligand, which significantly favours the ferromagnetic coupling, while the other two parts, i.e. N,N,O-core and phenol group, do not seem to have any significant direct effect on the magnetic properties. In general, the largest antiferromagnetic contributions come from the methoxo bridges and the more diffuse virtual orbitals located at both the metal centres and first coordination sphere. Since the bridges open an indirect channel between the two metal centres that favours the antiparallel arrangement of their unpaired electrons, i.e. superexchange, we were not surprised by such phenomenon. Likewise, the virtual orbitals allow direct through-space interactions between the unpaired electrons of each metal centre. On the other hand, the behaviour of [Mn2(HLA)4(μ-OCH3)2] C1 is insensitive to its environment, with an overall J value stable and ferromagnetic with a final value of 4 cm−1, in qualitatively good agreement with the reported ferromagnetic character of this compound, i.e. ca. 20 cm−1. Reaching quantitative agreement for ferromagnetic systems is usually complex because of specific coupling mechanisms [44] or spin polarization effects [45], which are not included in the applied CASPT2 method. Though the methoxo bridges still seem to have a significant antiferromagnetic contribution, similar to that for [FeIII2(HLA)4(μ-OCH3)2] C4, however, neither the ferromagnetic effect of the dithiolane (S) nor the antiferromagnetic contribution of the virtual orbitals (M+) do appear in [Mn2(HLA)4(μ-OCH3)2] C1. These results indicate that the ferromagnetism of [Mn2(HLA)4(μ-OCH3)2] C1 is inherent to the dimetallic MnIII–MnIII core, and the factors that determine the J value are both the relative orientation between the metal centres and the intermetallic distance. Once strong ferromagnetic coupling is achieved between the metal centres, the influence of the ligands becomes insignificant.
The proportionality of the antiferromagnetic character with the coupling of the dimetallic core (reference J value) is observed for these complexes and better evidenced for the three complexes supported by aroylhydrazone ligand HLC. The diffuse virtual orbitals located at the metal centres (M) and the first coordination sphere (M+) also strengthen the antiferromagnetism of the three compounds. As observed for the bridges, their impact on the J value is also proportional to the reference J value, but to a lesser extent. The HLC ligand causes a weak increase in the antiferromagnetic component, which exclusively originates from the central ring (C region) directly coordinated to the metal cation. The aromatic system of the phenyl group (P region) is inert with respect to the exchange coupling of the molecule. Since the HLC molecule does not bear the dithiolane group (S) that induces a ferromagnetic contribution for the H2LA complexes, it could be expected that HLC would exhibit a stronger antiferromagnetic contribution in comparison. The clearest example is the [MnIV2(Lc)4(μ-O)2]·THF C2 complex, the J value of which becomes twice as antiferromagnetic with the C and P regions activated, i.e.–32 vs.–15 cm−1. Unfortunately, the J values of the other compounds are too small to allow any concluding remarks. Furthermore, the alterations of J induced by the different regions are additive for [MnIV2(Lc)4(μ-O)2]·THF C2 complex, but not for [FeIII2(Lc)4(μ-OCH3)2] C6 and [CrIII2(Lc)4(μ-OCH3)2]·CHCl3·CH3OH C11. The combined interactions between the individual regions are definitely not equivalent between the MnIV system and the FeIII and CrIII compounds. This distinct behaviour could be related to their bridging ligands, which are oxo for MnIVL and methoxo for FeIIIL and CrIIIL. Nevertheless, it is difficult to affirm whether this change is due solely to the different interaction of the bridges or also to the character of the involved metal centres.
All in all, the inspections along the series of compounds with the HLC ligand in combination with the data obtained from the two complexes with H2LA reveal clear common features between them. Precisely, the role of the dimetallic core and the bridges dominates the intermetallic exchange coupling constant (J). On the other hand, the HLC and H2LA ligands have a pretty minor direct effect over J. But, these have a structural role that is determinant in the control of the geometry of the dimetallic core and, thus, the magnetic interactions. Notably, we proved that the overall magnetism the ligand imposes by determining the structure of the dimetallic core is more effective than any external electronic interaction. Detailed information on the computational strategy developed and the results obtained can be found in our recently published work [6].
5 Conclusions
By reviewing our research effort in the very active and promising field of magnetic materials, we hope to have provided here an interesting example in the synthesis and characterization of small metallorganic-based molecular systems using aroylhydrazone ligands. Our synthetic strategy is to design compartmentalized ligands providing coordination pockets capable to strongly coordinate the metal ion centres. We were able to isolate and characterize a robust homometallic dimer of manganese with the largest coupling constant obtained so far for a MnIII–MnIII interaction, i.e. J = 19.7 cm−1. The possibility of obtaining isostructural systems of the [Mn2] system with FeIII, CrIII, and CoIII ions has contributed to elucidate the mechanism of exchange interaction in these systems. The DFT and ab initio CASPT2 calculations techniques applied to five representative binuclear transition metal compounds allowed us to explore the specific influence of the organic periphery, the bridging ligands, and the remote substituents on the exchange coupling constants in these systems. Valuable synthetic strategies are now in our hands and, from this review, de facto in the hands of the coordination chemists to broaden the area of coordination chemistry in general and molecular magnetism in particular. Our efforts are still dedicated to the preparation of novel ligands together with the self-assembly reactions using other bridging ligands, and single-molecule growth, expecting ultimately a pure 3d dinuclear compound with a SMM behaviour and/or, from a fundamental point of view, with an exchange coupling scenario that could cross beyond the horizon in which our [MnIII–MnIII] system stands since 2006!
Acknowledgments
The financial support of the “Centre national de la recherche scientifique” (CNRS) and the French “Ministère de la Recherche” has been key for the development of this research program. We also thank the “Fundação para a Ciência e a Tecnologia” (FCT) for funding. The results reviewed here have of course been obtained thanks to the outstanding contribution of other scientists. We thank Chahrazed Beghidja, Nicolas Clément, Nouri Bouslimani, Clément Toussaint, Khaled Cheaib, Mohamedally Kurmoo, Maxime Bernard, Marta M. Andrade, Clarisse Tourbillon, Weiwei Zuo, Vitor Rosa, Sylvie Choua, Guillaume Rogez, Jens Kortus, Marcel Wesolek, Philippe Turek, Lydia Brelot, David Martel, Alex Domingo, and Vincent Robert.