

1 Introduction
Alcohols are widely used as solvents and intermediate products in the petrochemical synthesis. Alcohols have extensive applications as precursors of surfactants, fragrance compounds, plasticizers, extragents, etc. in various industries. They have rather low production cost and are environmentally friendly additives for high-octane fuels and could be alternatives to hydrocarbon fuels [1–11].
Alcohol-containing fuels reduce the emission of greenhouse and toxic gases (CO and NOx) and generation of soot particles [5,8,10,12–14]. Fuels with high ethanol concentrations significantly build up engine power though they require modification of the engine.
Alcohols act as octane number-increasing additives in fuel production. Methyl tert-butyl ether (MTBE) used to be utilized as an anti-knock additive, but now it is no longer compliant with environmental standards [15,16]. Restriction of MTBE has initiated interest in alcohol-based additives. Methanol and ethanol have the highest octane numbers among alcohols, yet their use is limited due to low solubility in hydrocarbons. Higher alcohols are better as fuel additives because they are less volatile and more easily dissolve in hydrocarbons than methanol [8–10]. The C1-C4 alcohol mixture can be used as a high-octane additive and as an emulsifier.
A few groups of catalysts for alcohol production – Zn-, Cu- and Cr-oxide based systems, modified Fischer–Tropsch catalysts, platinum group metals (Rh, Ru and Pd), and transition metal sulphides are utilized in alcohol production [8]. Among them, only ZnO, CuO and Cr2O3 supported catalytic systems have extensive industrial applications. These types of catalysts have been used for methanol synthesis since 1923. Catalysts for C2+-alcohol synthesis, resistant to sulphuric compounds in the feed, are a R&D challenge today [17,18].
Synthesis gas is produced from coal, associated gas or biomass. After primary purification, it contains up to several dozen ppm of sulphuric compounds such as hydrogen sulphide, carbon disulphide, mercaptanes, and thiophenes [19–24]. Oxide systems based on ZnCu or ZnCr are conventional catalysts for the synthesis of alcohols in the industry [8,25,26]; however they rapidly lose activity in the presence of sulphur. Oxide catalysts are deactivated by very small concentrations of sulphur compounds (0.1 ppm of H2S) [24,27–29]. Catalysts based on Zn, Cr, and Cu oxides or reduced Co noticeably lose activity during the first 10 h of the reaction with syngas that contains ∼100 ppm H2S [30]. A commercial CuZnO/Al2O3 catalyst completely loses its activity whereas hydrogen sulphide passed through it reaches one third of the metal amount in the active phase [31]. Therefore, additional purification is required to bring the sulphur content down to a few ppb, making the process more sophisticated and expensive. Catalysts based on transition metal sulphides would be a good solution here.
The design of molybdenum sulphide based catalysts for alcohol production from syngas attracts interest both of academic and industrial research. This paper discusses how different parameters such as the synthesis gas composition, modification of alkali metals, promotion with Co and Ni, active phase morphology, and the reaction mechanism impact MoS2 catalytic activity. Recently, several reviews have been published in this field [7,8,32–35]. This review is focused on the similarities found between the MoS2 catalytic behaviour in the synthesis gas conversion and the hydrodesulphurization reaction [36–43].
2 Sulphide catalysts for the alcohol synthesis from synthesis gas
It is well known that molybdenum disulphide can catalyse various types of chemical reactions: synthesis of hydrocarbons from CO and H2 [34], methyl mercaptan synthesis from CO/CO2 with H2 and H2S [44], amide conversion into thioamides [45], alcohol synthesis through hydroformylation of olefin or aldehyde hydrogenation [46–48], desulphurization [40,43,49], etc.
The first alkali-modified MoS2 catalysts for alcohol production from syngas were designed in the mid-1980s by Dow Chemical Company and Union Carbide Corporation [26,50,51]. A number of USA patents on the methods of the alcohol synthesis on sulphide catalysts and the manufacture of cobalt-molybdenum catalysts have been issued to date [33,52,53]. A notable feature of such catalysts is their ability to produce a mixture of methanol, primary C2+-alcohols and hydrocarbons during the syngas conversion. The resulting C2+ products generally have a linear carbon chain [54,55].
The feed does not require deep purification anymore, which reduces expenses on the installation and maintenance of hydrogen disulphide adsorption units. Unlike oxide catalysts, the sulphide ones require the presence of 50–100 ppm H2S in the feed to retain sulphidation and maintain activity [33,56,57]. These catalysts are more resistant to the СО2 exposure and carbonization than other catalysts [33,35,58]. However, production of C2+-alcohols from synthesis gas has not been commercialized at a large scale. The development of high performance catalysts for the synthesis of high alcohols from syngas is in the focus of the industry [7,59,60].
3 Molybdenum disulphide active phase morphology
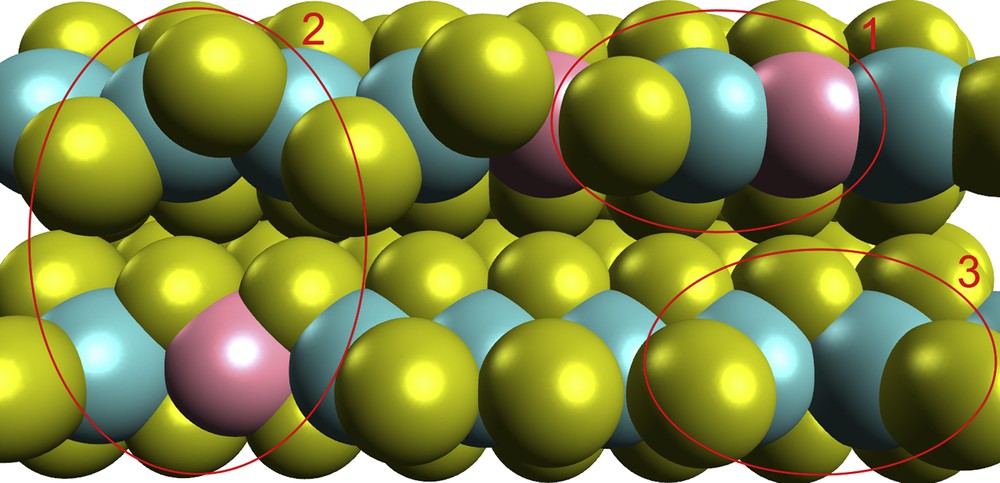
Molybdenum sulphides have been widely used since the thirties of the 20th century as a key component of the active phase of catalysts for hydrotreatment of oil fractions and crudes because of their high activity and resistance to sulphur poisoning [61]. Later on, several attempts were made to adapt them to oil fraction cracking and a water-gas shift reaction. Catalytic activity was found to be greatly dependent on the MoS2 crystallite structure, especially on the edge defects and intercalation [36,38,62–64]. Molybdenum disulphide crystallite can be of a hexagonal or triangular shape depending on the sulphidation conditions and the presence of a promoter [65–67]. Each crystallite layer consists of Mo4+ cations located between two levels of S2− anions. Each molybdenum atom coordinates with six sulphur atoms in the trigonal prismatic or octahedral structure with a strong interaction through S–Mo–S bonds. Adjacent layers are attracted to each other by van der Waals interactions. The distance between the layers measured by X-ray structure analysis is ∼6.2 Å [68]. The intercalation of various compounds can increase interlayer distance and initiate deformation [62]. It affects the crystallite reactivity. Molybdenum atoms located on a basal plane are fully saturated with sulphur atoms and inactive in catalytic reactions [69–71].
MoS2 layers are terminated by two types of edges, commonly referred to as the Mo-edge and S-edge (Fig. 1). Under HDS conditions most of the Mo atoms on the Mo-edge are surrounded by 6 sulphur atoms and are coordinatively saturated. Under reducing conditions the Mo-edge may lose some sulphur [67]. The result of this process is the formation of anion vacancies or, what is the same, coordinative unsaturated sites (CUS), clearly seen on Scanning Tunneling Microscopy (STM) images [72]. Concentration of these CUS is rather low. The S-edge is terminated by sulphur in the bridge position in a zig-zag pattern. The Mo atoms located on the S-edge are in incomplete prismatic coordination with two coordination vacancies per atom. The CUS on the S-edge are less reactive than sites formed on the Mo-edge [73].
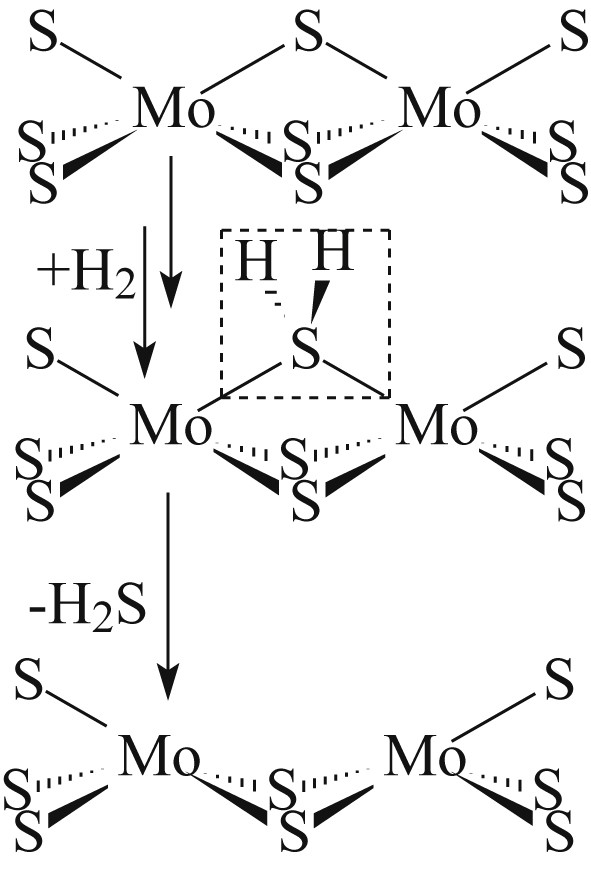
Crystallite edges are unstable in hydrogen atmosphere at high temperatures (>300 °C). Hydrogen reacts with the S-edge sulphur to generate an H2S and vacancy (Fig. 2) [36,74,75]. Formally the process of HDS is changing of the Mo oxidation degree from +4 to +6 (adsorption of the S-containing compound) and, further, to +4 (desorption of hydrogen sulphide). Under the reaction conditions, desorption of sulphur in the form of hydrogen sulphide followed by the vacancy formation on the crystallite edges determines the formation of active sites.
In 1980s, H. Topsøe and co-workers studied a Co-promoted MoS-catalyst using Mössbauer spectroscopy and found the separate CoMoS phase. They attributed it as an active phase of the catalysts [76–79]. The Co(Ni)MoS-phase consists of molybdenum disulphide crystallites with edges decorated with promoter atoms. Incorporation of the Co atoms into the edges of the MoS2 proceeds in the course of replacing the Mo atoms by Co atoms under sulphidation conditions. This has been supported by STM methods [72,80,81] and by recent computational studies [82]. The presence of Co/Ni in the active phase facilitates the CUS formation [83,84]. Catalytic tests have shown that hydrodesulphurisation activity depends on the Co(Ni)MoS-phase concentration. The Co(Ni)MoS crystallites can be single- (Type I) or multilayered (Type II) (Fig. 1) [72,79,81,85]. Catalysts with a higher content of the Type II Co(Ni)MoS phase are more active than those with the Type I Co(Ni)MoS phase. This can be explained by weaker interaction of multilayered crystallites with the support. According to the rim-edge model, two types of active sites exist on the surface of molybdenum disulphide crystallite [86]. These sites are differentiated by their location. Active sites located on the crystallite edges are responsible for hydrogenolysis and active sites located on the rims – for hydrogenation. Catalyst activity directly depends on the linear size and the number of layers of MoS2 crystallites.
Using 35S tracer methods, it was found that promoted molybdenum disulphide contained two different types of active sites – more active (“rapid”), and less active (“slow”) [87–90]. Later, the site types were specified by a dynamic model [91,92]. According to the model the “rapid” sites consist of a combination of two single Mo-sulphide clusters – promoted by Co and unpromoted. “Slow” sites are comprised of single unpromoted Mo-sulphide clusters. The mutual transformation of the two site types is postulated by the model. Terminal sulphur and promoter atoms can migrate from one layer of MoS2 crystallites to another under the reaction conditions thereby changing the distribution of “rapid” and “slow” sites. Their transformation frequency determines catalytic activity [90,92].
Modification of CoMoS catalysts by potassium additives makes them active in alcohol synthesis from CO and H2 because of formation of the KCoMoS phase [93,94]. However, the potassium position in the active phase and its role in alcohol synthesis have not been clarified so far.
4 Change in the catalyst sulphidation degree during the synthesis gas conversion
One of the most important factors affecting the KCoMoS catalytic activity and stability in alcohol formation is sulphur removal from the catalyst surface during the reaction. To keep its activity constant for a long time it is necessary to add some amount of sulphur continuously to the ongoing reaction. It was found that the selectivity of the KMoS catalyst oxidized in air changed from the formation of alcohols to hydrocarbons [95–98]. To sum up, the oxysulphides are formed as intermediates in the course of the molybdenum disulphide interaction with syngas. The authors of [97,99] supposed that the following factors should affect catalytic activity. Continuous reaction in the absence of a sulphiding agent causes the Mo-OH group formation that leads to intermediate decomposition. In the course of conversion of S-free synthesis gas Mo-carbide formation takes place. When the sulphidizing agent is in the crude Mo-carbide particles are not formed. These observations were made using XPS by Hensley et al. in [97]. The authors mentioned the opposite slope trend between catalytic activity and Mo-carbide formation. It could be explained by coke formation accompanying the process of carbide formation and blocking the active sites of the sulphide catalyst. There is a likelihood of the promoter separation and formation of the Co9S8 phase. The sulphur-containing feed can be used to avoid catalyst deactivation. The H2S present in syngas maintains the activity of the catalyst and extends its service life [97]. A rise of the H2S concentration in the feed over 100 ppm enables reaching the stable state of the KCoMoS/C catalyst faster than in purified syngas [56,57]. The presence of H2S increases C2+-alcohol selectivity.
5 Effect of support nature on MoS2 catalyst activity in alcohol synthesis
Materials such as metal oxides (Al2O3, MgO, ZrO2, and SiO2) [56,100–104], carbon materials (activated carbon and nanotubes) [56,102,103,105–110], and clay [102,111] were tested as possible carriers for MoS2-based catalysts. Studies on the effect of support surface pH on catalyst activity show that high acidity suppresses the formation of alcohols and accelerates hydrogenation [101,112]. Inert carbon materials are free from this disadvantage. A series of studies show that carbon-supported catalysts exhibit higher activity than those supported on metal oxides [56,103]. It is true both for alcohol synthesis and for hydrodesulphurisation reactions.
Support morphology plays an essential role in the active phase formation. A series of KCoMoS catalysts supported on different types of activated carbon and nanotubes were tested by Surisetty and co-workers [113]. According to the test results, the dispersion of the active phase and yield of alcohols rise with an increase in pore size. Carbon-based materials provide the catalysts with a few advantages as compared to conventional alumina: a weak interaction between the support and active phase, resistance to acidic and basic compounds, and stability at high temperature and pressure. However, due to certain properties, alumina retains its position as the main support for commercial catalysts. It has a developed surface, high packed density, thermal stability, and durability. So, it was considered reasonable to combine positive properties of both supports. To this effect, carbon-coated alumina was synthesized as a carrier for sulphide catalysts [114–116]. The carbon content and the method of carbon deposition over alumina were found to be the key factors impacting catalytic activity.
To determine the optimal carbon content on the γ-Al2O3 surface we prepared and tested in the alcohol synthesis a set of KCoMoS catalysts with 0–4.8 wt.% of carbon [117]. Textural characteristics of γ-Al2O3 remained unaffected by carbon coating. The catalytic test results indicated that the carbon interlayer increases the CO conversion and alcohol selectivity (Fig. 3). The plotted ‘alcohols to hydrocarbons’ ratio has its maximum at 0.8 wt.% of the carbon content. The sample with 0.8 wt.% carbon has up to a 16-fold higher ‘alcohols to hydrocarbons’ ratio than that on pure γ-Al2O3.
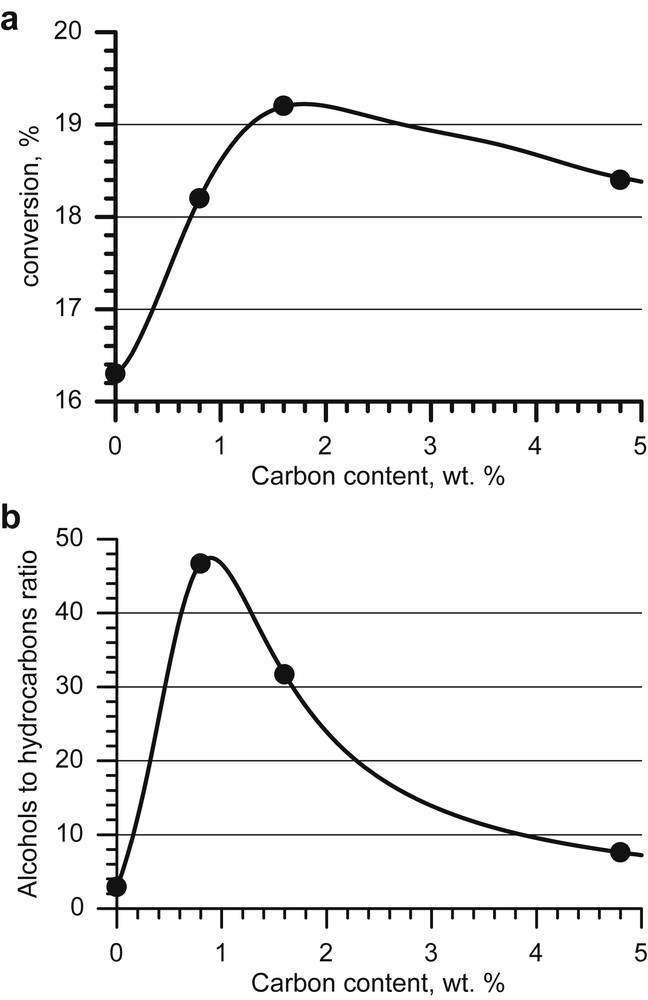
Similar dependences of catalytic activity from the carbon content were observed earlier for Co(Ni)MoS catalysts in the hydrodesulphurisation reaction. This is indicative of the uniformity of carbon effects on the formation of active phases for different sulphide catalysts. These calculations [118] suggest that carbon does not deposit on the alumina surface uniformly when its content is below 5 wt.%. According to the acidity measurements [118], in the course of the carrier impregnation by organic precursors of the carbon interlayer, they adsorb preferably on Lewis sites and block them. A drop in carrier's acidity reduces the interaction between the carrier and the active phase. Carbon deposits localize oxide precursor particles on the alumina surface that is free of carbon “islands” and promote building crystallites with a higher stacking number and, thus, the formation of Type II sites.
6 Effect of alkali metal addition on the activity of MoS2 catalysts
Alkali metals are widely employed in heterogeneous catalysis to control the activity and selectivity of different reactions [119]. The syngas conversion on unmodified MoS2 catalysts produces basically hydrocarbons. After the modification by an alkali metal, alcohols become the main product [56,98,120]. Therefore alkali metal functions are inhibition of hydrogenation activity and promotion of active sites responsible for the alcohol formation [33,54].
Quantum chemical calculations of CO [121] and methanol [122,123] hydrogenation pathways on the unpromoted molybdenum disulphide surface show that a C–O bond cleavage accompanied by the formation of methane and water is thermodynamically more preferable than carbon chain growth. It is likely that the active site modification with potassium decreases the energy barrier of the alcohol formation. Potassium ions can serve as CO coordination sites and facilitate the formation of an acyl intermediate on the surface. It was reported that an alkali metal affects the dispersity of the active phase and thereby brings about a structure-formation property [32,33,124].
Effect of alkali metals (K, Na, and Cs) on MoS2 catalyst activity has been in the focus of research over the past decades [55,125]. According to Youchang and co-workers [126] the catalyst modified with Na exhibits lower activity in comparison with other alkali metals. Lithium intercalants are applied for batteries [62], but not as components of the active phase for MoS2 catalysts in the syngas conversion [127]. A comparison study of the effect of K and Cs additives on the morphology by methods of scanning electron microscopy and X-ray photoelectron analysis was performed for modified CoMoS catalysts [54,111,124]. The effect of these additives on catalytic activity was also studied. The distribution of potassium changes in the course of the alcohol synthesis. Iranmahboob and co-workers [111] showed that initially non-uniform potassium distribution became uniform after the reaction. This is not the case for Cs that does not change its non-uniform distribution during the reaction because of the cluster formation. Potassium is supposed to be an optimal modifier. An induction period in the alcohol synthesis was observed in [57,96,128,129]. This phenomenon may be explained by a period of time needed for the potassium distribution along and under the MoS2 slab layer.
Potassium modification impact strongly depends on precursors used for the catalyst preparation. The most suitable salts [96,101,124,128,129] for the active phase modification are those that easily dissociate on counter-ions and can be uniformly distributed on the surface of supports. Depending on a potassium precursor, the catalyst can intensify either the formation of alcohol (K2CO3, KOH, KNO3, CH3COOK, K2S, and K2O2) or С2+-hydrocarbons (K2SO4, KCl, KBr, and KI). The CO conversion decreases irrespective of the nature of K precursors. For the MoS2 catalysts modified with К2СО3, К2О2 and К2S, Woo H.C. and co-workers [96] observed signals that corresponded to metalorganic substances in the IR spectrum of adsorbed CO. The signals were associated with bond oscillations in the surface intermediate formed during the reaction. No signals were recorded for sulphide catalysts modified with K2SO4 or KCl, or exposed to air (oxidized). The mechanism of carbon chain growth and alcohol formation including intermediate configuration Mo(Co)–C–O–K was suggested [96].
Recently, we have conducted a comparative study of the potassium modification of CoMoS catalysts in the syngas conversion and thiophene hydrodesulphurisation [120]. We tested the catalysts with 0–15 wt.% potassium (Fig. 4). Potassium addition to CoMoS active phases leads to a decrease of n-hexene and thiophene conversions and promotes alcohol synthesis. As reported in [120], the hydrogenation sites are more sensitive to potassium than the hydrodesulphurization sites. According to the rim-edge model [86] it is possible to assume that initially potassium modifies the active sites placed on the MoS2 slab rims and then the sites placed on the slab edges.
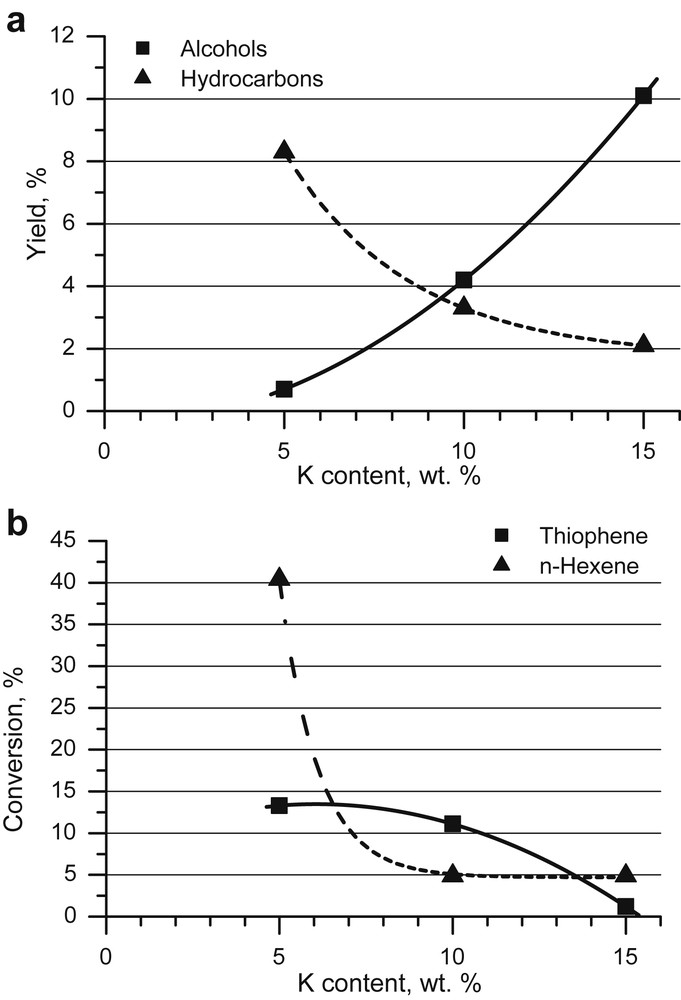
A comparative study of HDS and syngas conversion over alkali-modified CoMo-sulphides together with investigation into the MoS2 crystallites morphology led us to conclude that the sites responsible for alcohol synthesis are the potassium modified sites that provide C–S bond hydrogenolysis in the course of HDS. The sites responsible for hydrocarbon formation in the course of syngas conversion are the same as the sites providing hydrogenation during HDS [120].
Thus, the alkali metal incorporating into active sites reduces acidity of the support, influences the active phase morphology, increases the MoS2 crystallite size and layer number. However, potassium localization remains unclear and no reliable information on the mechanism of potassium influence on catalytic activity is currently available. The change in the potassium distribution on the catalyst surface in the ongoing interaction with synthesis gas and diffusion of potassium atoms under the MoS2 [96,128,129] can be explained by the potassium intercalation into the molybdenum disulphide structure. The formation of intercalating compounds of alkali metals is well known in MoS2 crystallites and has been described elsewhere [62,130–132].
7 Effect of transition metal promoters on the activity of MoS2 catalysts
Co and Ni are the main promoters for sulphide catalysts used both for HDS, and for the catalysts used in alcohol synthesis [56,103,133]. Ni or Co addition to the active phase of an alkali-modified sulphide catalyst promotes the CO conversion and carbon chain growth. The Co added to the catalyst affects hydrogenation activity and increases the ratio of alcohol to hydrocarbon yields [103,134]. Ni addition mostly improves the selectivity of the hydrocarbon formation [103,135].
Studies of Co and Ni content influence on KCo(Ni)MoS catalyst activity in the alcohol synthesis show that the maximal C2+-alcohol yield is at Co(Ni)/Mo = 0.5 atomic ratio [93,133,134,136,137]. The Mo:Co = 1:2 ratio was found most efficient in the alcohol synthesis and hydrodesulphurization [138]; this fact suggests similar roles of the promoter in both reactions. At a low ratio, Co(Ni)/Mo <1/3, promoter atoms incorporated into the molybdenum disulphide active phase increase its dispersity. With the promoter content increase, vacancies on the MoS2 crystallite edges become saturated and form separate Co9S8 and NiSx phases [134,138].
The authors of [32,136,139] assume that the promoter participates in the formation of two phases – a separate Co/Ni sulphide phase responsible for hydrogenation and a mixed “KCoMoS” or “KNiMoS” phase that promotes carbon chain growth.
Alternatively, the authors of [91,93,120,137] believe that the formation of alcohols and hydrodesulphurization proceed rather on the CoMoS phase sites than on the sites of the two separate phases. Cobalt content facilitates the growth of the MoS2 crystallite linear size and the number of layers. The cobalt excess leads to the formation of a low-active Со9S8 phase that blocks CoMoS active sites. At the same time the Co9S8 phase could play the role of an H2 activator and thus increase catalytic activity in hydrodesulphurization [140].
8 Suggested schemes for the mechanism of the alcohol formation from СО and Н2 on K(Co)MoS catalysts
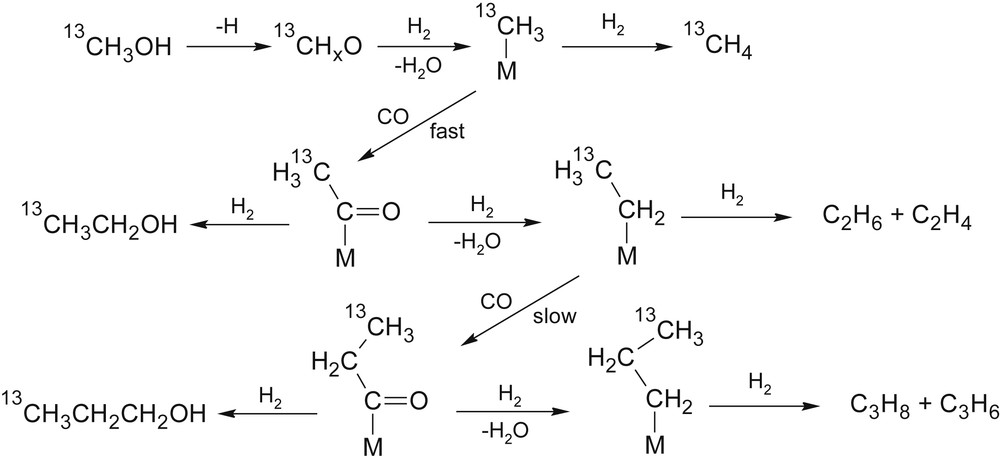
In the past decades, several hypothetic mechanisms on the CO conversion over MoS2 were considered [26,32–34,54,96,141,142]. Studying the mechanism of carbon chain growth Santiesteban and co-workers [54,125] performed a series of tests of K2CO3/Co/MoS2 with syngas-13CH3OH co-feeding. An NMR analysis of the reaction products revealed that only the terminal C atom of the ethanol formed was isotopically labelled. Propanol contained isotopic labels in two alternative positions (13CH3CH2CH2OH and CH313CH2CH2OH) but not in both (13CH313CH2CH2OH). The authors suggested a scheme of carbon chain growth (Fig. 5) where the key step was CO insertion in α- or β-positions of an adsorbed alkyl intermediate.
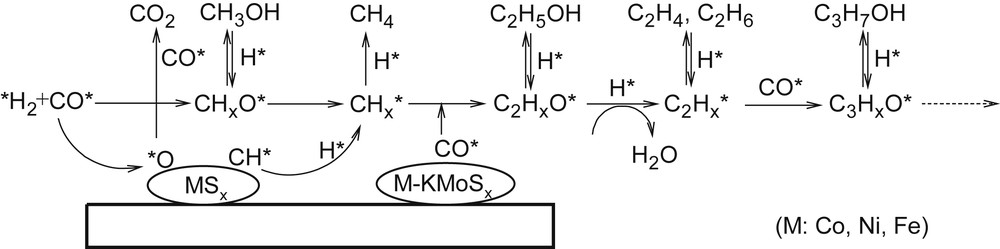
According to [101,136], the first step of the reaction mechanism is fast dissociation of CO and the formation of a CHx* particle on the surface of a separate MSx phase (M = Fe, Co or Ni) (Fig. 6). The methane is formed by direct hydrogenation of CHx*. Non-dissociatively adsorbed CO* is inserted in the metal-carbon bond with the formation of an acyl intermediate on the active site of the mixed M-KMoS phase. Further hydrogenation of the acyl intermediate can lead to alcohol or hydrocarbon molecule desorption.
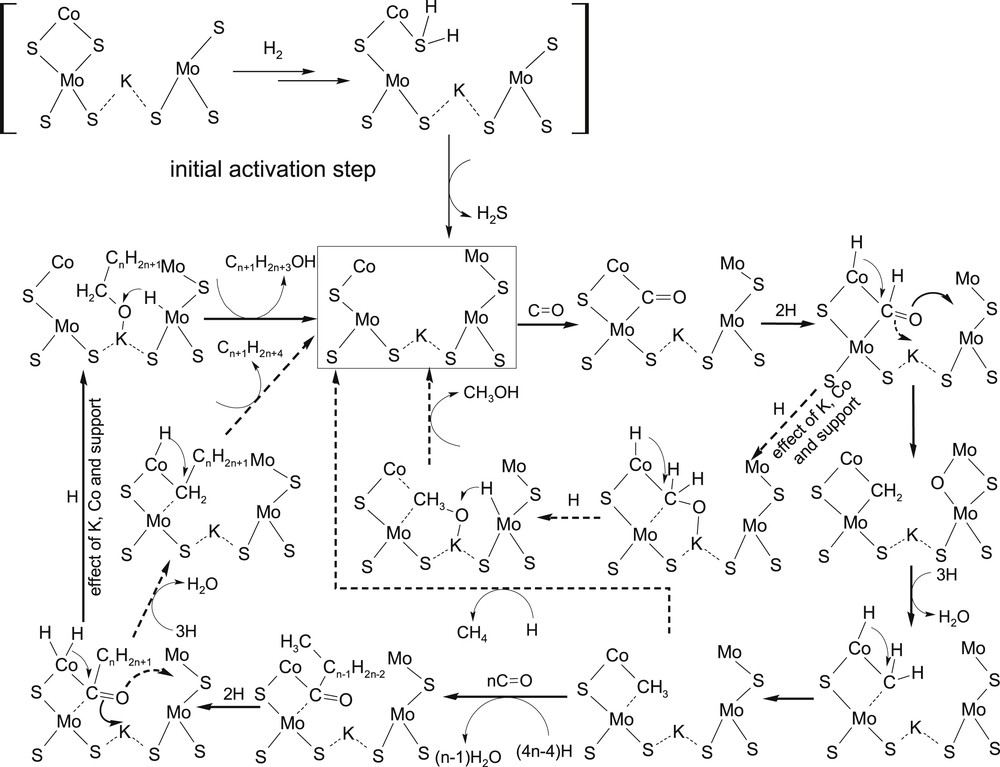
In [142,143] we proposed a scheme of CO conversion on KCoMoS catalyst's active sites (Fig. 7). An initial reaction step is CO molecule adsorption on a vacancy. After partial hydrogenation of CO, its bond length extends and oxygen can coordinate on transition metal atoms or potassium ions intercalated between MoS2 layers depending on the catalyst structure and reaction conditions. It means that the CO coordination is a key factor determining the formation of either an alcohol or hydrocarbon. The coordination of oxygen on the alkali metal ion stabilizes the alcoxyl intermediate and prevents C–O bond breaking, which favours the alcohol formation. The coordination of oxygen on the Mo or Co atom results in the hydrogenolysis of the C–O bond. The formed alkyl fragment can be reduced to hydrocarbon or undergo a chain-growth reaction. Chain growth occurs through the insertion of a new CO molecule between carbon and transition metal atoms.
Alkyl and alkoxide intermediates play a significant role in all earlier suggested mechanisms of the alcohol synthesis [54,56,101,136,141,142]. The KCoMoS catalyst tests where ethanol or ethylene was co-fed with synthesis gas confirm this [143]. A simultaneous conversion of synthesis gas with additives led to profound changes in the reaction rate and product composition. Probably, ethanol or ethylene involved CO in a catalytic reaction significantly increasing the CO conversion. Using GC–MS analysis of the reaction products the following compounds were identified: esters, ketones and branched alcohols. We found various acetals and tracer amounts of acetic and lactic acids in the products of the alcohol synthesis in an ethanol excess. Probably, compounds of different classes may be manufactured by varying both additives to syngas and reaction conditions in scaled-up processes.
Gunturu and co-workers reported that no higher alcohols resulted from the methanol conversion in helium [144]. We compared the KCoMoS catalyst behaviour in ethanol conversion under nitrogen and hydrogen (Fig. 8) [117] and found an essential difference of ethanol conversion and product distribution in nitrogen and hydrogen media. Ethanol conversion under N2 was significantly higher than under H2. Moreover, conversion – gas pressure dependencies were found to be different for the two gases. Ethanol conversion products under either gas were similar and included acetaldehyde, acetic acid, ethyl acetate, butyl acetate, butyric acid, ethyl butyrate, diethoxyethane, methane, ethane, CO, and CO2. The oxygenate content was significantly higher in nitrogen atmosphere than in hydrogen atmosphere. It confirms that alcohols are able to disproportionate on the KCoMoS catalyst. Products of their disproportionation undergo side-reactions of condensation, etherification and decarboxylation. Hydrogen is likely to suppress ethanol disproportionation and to shift the reaction equilibrium toward reduction.

A presence of butyl acetate, butyric acid, and ethyl butyrate among the ethanol conversion products under inert atmosphere suggests a possibility of condensation mechanism along with the CO insertion. One of these mechanisms dominates depending on the reaction conditions and feed additives. Recently, based on our results [142] Wu and co-workers [145] suggested a possibility of two competing mechanisms for carbon chain growth over Cu-based catalysts.
9 Industrial horizons
Development of a poisoning-resistant catalyst for the alcohol synthesis has been a challenge in the past decades. Studies on the effect of various support materials, modifiers, promoters and reaction conditions on catalytic activity have greatly contributed to industrial utilisation of higher alcohol synthesis (Table 1). Sulphide-based catalyst applications change the syngas purification standards. Continuous tests of KCoMoS catalysts show that a presence of sulphur compounds in syngas significantly extends catalyst lifetime [97].
Comparison of sample compositions, reaction conditions and catalytic properties in alcohol synthesis from CO and H2 of alkali-doped transition metal sulphide catalysts taken from literature.
| Catalyst | Reaction conditions | Conversion of CO, % | Selectivity, % | Reference | ||||
| P, MPa | T, °C | GHSV (h−1) | H2/CO | HC | Alcohols | |||
| KCoMoS | 10.5 | 265 | – | – | 33 | 17 | 51 | [50] |
| KCoMoS | 10.5 | 305 | – | – | 39 | 12 | 50 | [51] |
| KCoMoS | 5.0 | 300 | 2400 | 2 | 47 | 58 (CO2-free) | 42 (CO2-free) | [103] |
| KCoMoS/C/Al2O3 | 5.0 | 360 | 600 | 1 | 18 | 1 | 65 | [112] |
| KCoMoS/MWCNT | 8.3 | 340 | 3600 | 1 | 47 | 27 | 32 | [129] |
| La/Ni/KMoS | 8.0 | 320 | 2500 | 1 | 33 | 34 (CO2-free) | 66 (CO2-free) | [130] |
MoS2-based catalysts are expected to be suitable for large scale production of alcohols from biomass-derived synthesis gas. In 2011 the first pilot plant for the alcohol synthesis from biomass-derived syngas over sulphide catalysts was put into operation [146]. The project aimed at varying parameters to investigate the optimal operation point of the plant and at using these parameters to perform trials over several days. Parameter variations showed the impact of reaction temperature, pressure, and space velocity on the product yield and distribution. The influence of the gas composition on the product distribution and side products was also investigated. The study focused on the influence of the ethylene content in syngas on the formation of propanol and ethyl mercaptan (C2H6S). The first experiments indicate that the ethylene content in syngas has a high impact on the product distribution and is responsible for side reactions to yield sulphur components. The alcohol mixtures can be distilled to separate alcohols. Ethanol can be separated from residual alcohols for blending with gasoline. Also, there is a possibility of producing olefins by dehydration. After dehydration, an oligomerisation process can be used to synthesize gasoline. Alcohol mixture thus produced is promising in a broad variety of applications.
10 Conclusions
Alkali-modified molybdenum disulphide is a promising catalytic system for the synthesis of higher alcohols from synthesis gas. The first stage of the pilot tests confirms a high industrial potential of the catalytic systems. This review discusses effects of various supports, modifiers and promoters on the activity of sulphide catalysts. The use of carbon materials with the developed surface as a catalyst support facilitates the alcohol synthesis. Alkali metals incorporated into active sites and operating in them reduce support acidity, affect the active phase morphology, and favour growth of the MoS2 crystallite size and layer number. Promotion of the catalyst with Co and Ni is useful for the C2+-alcohol formation.
The edge-rim and interlayer dynamic models can be applied for the explanation of KCoMoS catalyst activity in the alcohol synthesis. The active sites responsible for the alcohol formation were attributed to be alkali-modified hydrodesulphurization active sites.
Methanol, ethanol and ethylene addition dramatically increases catalytic activity and the number of reaction products. A difference between methanol and ethanol conversions in inert gases was evaluated: methanol disproportionates with the formation of methane and carbon oxides; ethanol disproportionates and condensates to yield higher molecular weight products. Ethanol conversion products in inert gas indicate a possibility of a condensation mechanism.
The alcohol formation may be a combination of two mechanisms: (i) carbon chain growth proceeding via the CO molecule insertion in the carbon-metal bond of the surface intermediate, and (ii) aldol condensation. Which of the two dominates depends on the reaction conditions.
Acknowledgements
The authors congratulate Professor Edmond Payen on his anniversary and wish him good health and new achievements in catalytic science.
The authors extend thanks to the Russian Foundation for Basic Research (Project No. 14 03 31769 mol_a).