

1 Introduction
The selective hydrogenation of α,β-unsaturated aldehydes assisted by supported metal catalysts is a key stage in the preparation of pharmaceutical products, flavors and fragrances[1–3]. Cinnamaldehyde (CAL) selective hydrogenation is a good example of this commercially important process that allows obtaining cinnamic alcohol (CA), hydrocinnamaldehyde (HCAL) and hydrocinnamic alcohol or 3-phenylpropanol (PP). Cinnamic alcohol is a valuable product in the perfume industry for its aroma as well as for its fixation properties. Also, it is used in the pharmaceutical industry in the synthesis of chloromycetin antibiotic [4,5]. More recently, important applications were found for HCAL such as its use as an intermediate in the synthesis of medicines for HIV treatment [6–8].
Although extensive research has been carried out on the preparation of catalysts for the selective hydrogenation of cinnamaldehyde to CA or HCAL, there is no single study that adequately covers the origin of the selectivity [9–11]. In particular, several attempts have been made to synthesize catalysts based on selective noble metals toward HCAL. Bimetallic catalysts have been found to exhibit an important synergetic effect that promotes the selectivity to HCAL. Also, catalysts based on Rh are some of the most effective catalysts for selective hydrogenation [12].
In general, species based on heteropolyacids or polyoxometalates (HPOMs), both as bulk and supported systems, have started to replace a large number of conventional oxide precursors, improving the efficiency and environmental conditions of several catalytic processes [13–17].
Recent studies have shown that the Rh(III)-heteropolymolybdate with Anderson-type structure supported on gamma alumina is an interesting precursor in heterogeneous catalysis for the selective hydrogenation process of cinnamaldehyde toward HCAL[18]. Such studies have shown that the planar structure of the heteropolyoxoanion as well as its redox and solubility properties are key factors in the heteropolyanion-γ-alumina interaction process, producing an active surface with an ordered surface and uniform deposition of Rh and Mo, which favors the synergetic effect allowing a high catalytic activity and high yield of HCAL.
In the present paper, a Rh(III)-containing Anderson phase has been synthesized and deeply characterized by physicochemical analysis. This phase was employed for the preparation of composites by equilibrium impregnation on natural aluminosilicates from Argentinean deposits (bentonite clay), previously modified and functionalized by different techniques so that they can be used as catalytic supports. Clays and their synthetic derivatives or those obtained by chemical modification are widely used in many applications as they exhibit specific features, such as acidity, swelling, ion-exchange properties, and a wide range of preparation variables, and they can be used in catalysis, are ease to workup, can be employed under mild experimental conditions with good yield and/or selectivity, low cost, etc. [19–21]. Several methods to obtain chemically modified clays have recently been reported in the literature. This chemical transformation gives rise to porous materials with high specific surface areas, commonly known as PILCs (pillared clays) and PCHs (porous clay heterostructures) [22–29]. These modified clays have micro- and meso-porosity and can then undergo further functionalization using another amine-siloxane compound as the neutral surfactant that provides a surface with a greater number of positive charges to adsorb heteropolymetalates.
In summary, the aim of the present work is the preparation and characterization of RhMo6 composites based on Anderson-type heteropolyoxomolybdate of formula (NH4)3[RhMo6O24H6].7H2O supported on Argentinean functionalized clays (PCH-F and PILC-F) to be used as catalysts in the liquid-phase hydrogenation of cinnamaldehyde.
2 Materials and experimental procedure
2.1 Materials
The heteropolymolybdate (NH4)3[RhMo6O24H6]·7H2O (hereafter RhMo6) was synthesized from the aqueous solution reaction of ammonium heptamolybdate and RhCl3.6H2O in stoichiometric proportions as previously reported [14–17]. Its characterization was conducted by X-ray powder diffraction, FTIR and Raman Microprobe vibrational spectroscopies, following previously reported methods [14, 15].
The selected bentonite clay was extracted from an Argentine deposit and was provided by MINMARCO S.A. company as the original material enriched in the montmorillonite fraction by Stokes' method. This mineral has the molecular formula Mx(Al2−xMgx)(Si4)O10(OH)2·nH2O, where Mx are interlayer cations (Na+, K+, etc.). Chemical modification of this material by conventional techniques allowed PILCs (pillared clays) and PCHs (porous clay heterostructures) to be obtained [29].
To prepare PILC, a solution of [Al13] oligomer ([AlO4Al12(OH)24(H2O)12]7+) was used as the pillaring agent. The clay was treated and shaken with the pillaring solution at 25 °C for 2 h. The material obtained was filtered, washed, dried at 80 °C and calcined at 550 °C.
PCH was prepared by mixing the bentonite clay with the cetyl-trimethyl-ammonium-bromide (HDTMA-Br) surfactant, and then the mixture was continuously shaken for 24 h. Tetraethyl-orthosilicate (TEOS) was added as a source of silica and the mixture was shaken again at room temperature for 4 h. The product obtained was filtered and calcined at 550 °C [29] (See Scheme 1).
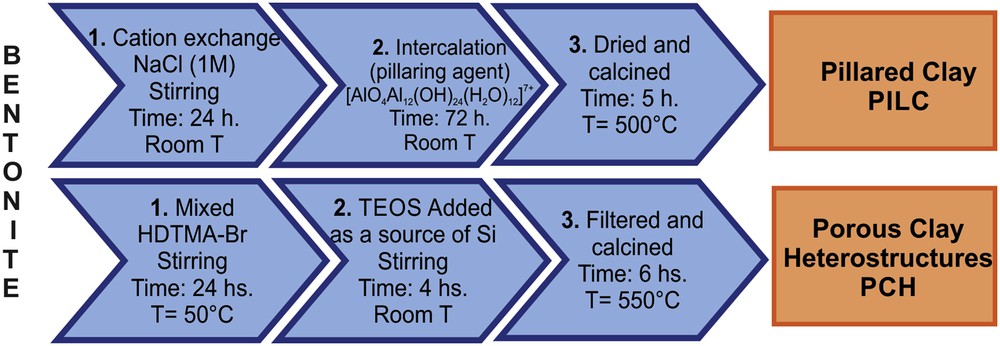
Preparation steps of bentonite-based systems, PILC and PCH.
2.2 PILC and PCH functionalization
PILC and PCH clay surfaces were further functionalized with 3-aminopropyl-trimethoxysilane (F) to obtain an adequate surface for the adsorption of the heteropolymetalate used as the active phase. Equal amounts of water and ethanol were mixed with the clay under stirring at room temperature for 24 h to activate the surface of the aluminosilicate by generating silanol groups (Si–OH).The mixture was then cooled centrifuged and dried in an oven at 70 °C for 24 h to ensure the complete removal of solvent and water. The functionalization of PILC and PCH was conducted in a shaker at 70 °C and 60 rpm for 12 h. Under these conditions, the functionalization agent (F) and toluene were used. Then, the materials were filtered and washed in toluene and acetone to prevent hydration. Finally, they were dried in an oven at 70 °C for 24 h (See Scheme 2).
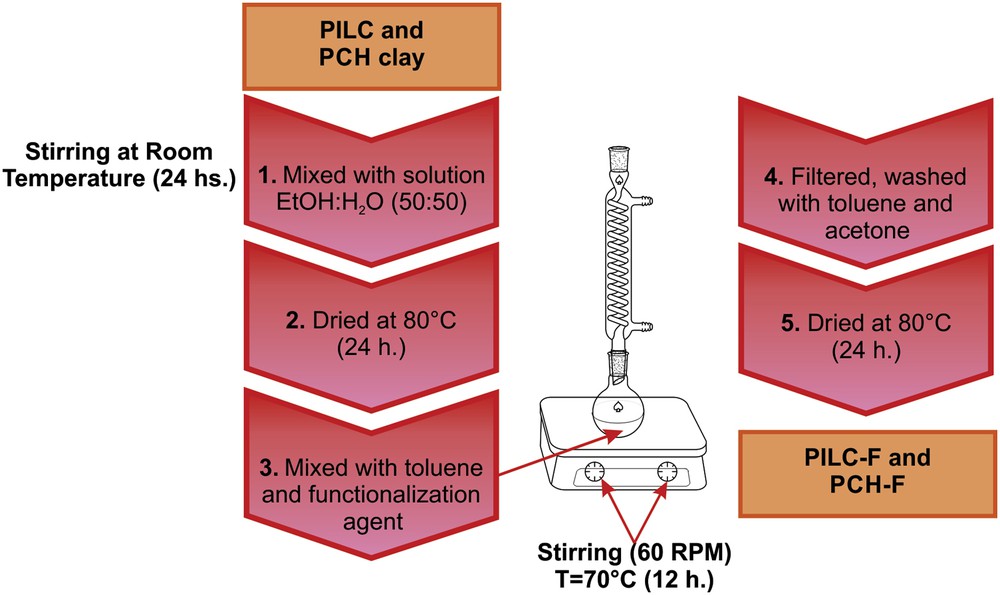
Stages of functionalization of PILC and PCH systems.
2.3 Composite preparation
The following composites: RhMo6/PCH, RhMo6/PCH-F (on functionalized PCH), RhMo6/PILC and RhMo6/PILC-F (on functionalized PILC) were prepared by equilibrium adsorption impregnation in excess of pore volume on the different supports using aqueous solutions of heteropolymolybdates (10 mg/mL of Mo) which were prepared following previous studies of Mo adsorption isotherms on alumina [15].
2.4 Characterization of modified clays and their RhMo6 composites
The specific surface area, pore volume and pore diameter were determined by the BET method, using Micromeritics ASAP 2020 equipment. The samples were characterized by X-ray powder diffraction with a Philips PW 1714 diffractometer (Cu Kα radiation and Ni filter).
Thermogravimetric and differential thermal analysis (TG-DSC) measurements were carried out using a Shimadzu thermoanalyzer (TGA-50 and DTA-50), at a heating rate of 10 °C/min, from room temperature up to 500 °C in air atmosphere. TG-DSC assays were performed using 10 mg of each sample.
Mo contents of the initial and final solutions were determined by atomic absorption spectrometry (AAS), using an IL-457 spectrometer. The concentration of Mo adsorbed was then determined by mass balance [15]. Complementary chemical composition measurements were made using scanning electron microscopy and energy-dispersive X-ray spectroscopy (SEM–EDS) by using a Philips SEM 505 microscope with a dispersive energy system for microanalysis by using an X-ray, EDAX 9100.
Raman spectra were recorded at room temperature with a Raman microprobe (Infinity from Jobin-Yvon) equipped with a photodiode array detector. The exciting light source was the 532 nm line of an Nd YAG laser and the resolution of spectra was 2 cm−1.
XPS spectra were registered on a SPECS Multi-technique analysis instrument spectrometer equipped with a dual X-ray Mg/Al source and a PHOIBOS 150 hemispherical analyzer in the mode of fixed analyzer transmission (FAT). The spectra were obtained with a pass energy of 30 eV and an Mg anode operated at 200 W. The pressure during the measurement was lower than 2.10−8 mbar.
The sample was calcined in an oven at 150 °C for 30 min, then was pressed, placed on the sample holder of the instrument and evacuated to ultra-high vacuum for at least 2 h before the measurement. It was later reduced to 350 °C for 10 min in a H2-5%/Ar flow and evacuated to ultra-high vacuum for at least 2 h before the second measurement. The Rh3d, Mo3d, O1s, Si2p and Al2p regions were processed by a computer. Electron binding energies were referenced to the C1s peak at 284.6 eV. The intensity ratios (for Rh or Mo) were obtained from peak area determination by integration of the appropriate peak. To assess a quantitative relationship between the XPS peak intensity ratio and surface composition, the experimental results were compared with the values predicted by the sensitivity factor of Scofield [30].
Temperature-programmed reduction (TPR) [15] used to analyze the precursor–support interaction was conducted with Quantachrome equipment, Quantasorb Jr. model. The reactor was fed with a10% H2/N2 mixture (flow = 20 cm3/min) and the temperature was raised from room temperature to 850 °C at a heating rate of 5 °C/min.
To analyze the type of active site present on the surface of the prepared materials, fresh composites were treated by isopropanol adsorption followed by the temperature-programmed surface reaction (TPSR). Thus, possible acidic, basic and redox sites were determined [31–33]. Before being submitted to the test, the supports and composites were heated at 350 °C in 99.999% purity He for 30 min, and then, they were cooled to 40 °C and 5 pulses of isopropanol were injected. The solids were then heated to 350 °C at a heating rate of 10 °C/min and finally, the desorbed species were measured with a Balzers QMG 112 mass spectrometer. The following masses were assigned: 44 (CO2), 41 (propene) and 43 (acetone).
2.5 Hydrogenation of cinnamaldehyde
Liquid-phase hydrogenation of cinnamaldehyde was conducted in an Autoclave Engineers reactor at 10 atm H2 pressure and 85 °C. A 0.20 g mass of the catalyst previously reduced in flowing H2 at 350 °C for 2 h, 0.8 mL of cinnamaldehyde and 60 mL of toluene as solvent were used in each test. The reaction progress was monitored by gas chromatography using a Varian CP-3800 chromatograph equipped with a capillary column CP wax 52 CB (30 m × 0.53 mm) and a FID detector. Identification of the reaction products was carried out in a GC/MS Shimadzu QP5050 mass spectrometer equipped with a capillary column SUPELCO SPBTM-5 (30 m, 0.25 mm i.d.).
3 Results and discussion
3.1 Physicochemical characterization of modified clays and composites
Pillared clays are formed through a process that involves the intercalation of inorganic cations ([Al13]), while to obtain heterostructured clays a cationic surfactant and neutral amine are intercalated between the clay layers. Thus, a structural micelle with pillars of nanoscopic oxides of Al(III) is produced in the PILC system. Regarding the PCH, the subsequent addition of a siliceous (TEOS) precursor causes an in situ polymerization of the silica pillars around the micelle formed in the previous stage. Finally, these inorganic or organic “templates” are removed by calcination at 550 °C, producing materials with high specific surface [23]. Thus, PILC and PCH materials of high surface area are obtained (Table 1).
Textural parameters and EDS chemical dataa of modified and functionalized clays.
| Type of clay | SBET (m2/g) | Pore volume (cm3/g) | Pore size BJH (Å) | Si % | Al % | Si/Al |
| Original | 33 | 0.07 | 84 | 80.11 | 19.89 | 4.00 |
| PILC | 128 | 0.11 | 39 | 72.00 | 28.00 | 2.60 |
| PILC-F | 59 | 0.04 | 47 | 74.50 | 25.50 | 3.00 |
| PCH | 579 | 0.58 | 56 | 89.46 | 10.54 | 8.50 |
| PCH-F | 105 | 0.20 | 82 | 94.75 | 5.25 | 18.00 |
a Data calculated from a content of Si + Al = 100%.
Additional functionalization of the materials (PILC and PCH) was carried out with a unit of 3-amine-propyl-trimethoxysilane (F) to obtain a surface with a higher positive charge to allow the adsorption of heteropolyanions. The functionalization mechanism implies the interaction of a unit of organosilane group with two silanols of the modified clay surface which produces a terminal amino group on the surface according to the following equation [34]:
| 2Si–OH + Si(OMe)3(CH2)3NH2→(SiO)2Si(OMe)(CH2)3NH2 + 2 Me–OH |
Once anchored to the channel walls, the organosilane groups that possess donor amines can be used to prepare polymetalates grafted to the clay modified surface.
Table 1 shows that the BET surface area is reduced practically to 50% (for the PILC system) and to 20% (for the PCH system) of the previous value, respectively. This fact is probably due to the condensation reaction that is caused by the functionalization process, where the amine groups would produce electrostatic repulsions and the subsequent reduction of the specific area. The functionalization cannot completely eliminate the surface silanol groups, which imparts a degree of hydrophilicity to the surface. This effect is explained by the steric effect of the functionalizing agent. This would act like an umbrella, protecting surface hydrated silanol groups and reducing the surface area [35,36].
The average pore diameter was 56 and 82 Å for PCH and PCH-F, respectively, which, according to the classification by Dubinin, correspond to mesoporous materials [37]. However, the proportion of super-micropores for PCH was also high [38,39].
Fig. 1(a) and 1(b) show the XRD diagrams of (a) PILC and (b) PCH and their functionalized system, and PCH-F sample treated at 300 °C for 3 h with no noticeable structural changes.
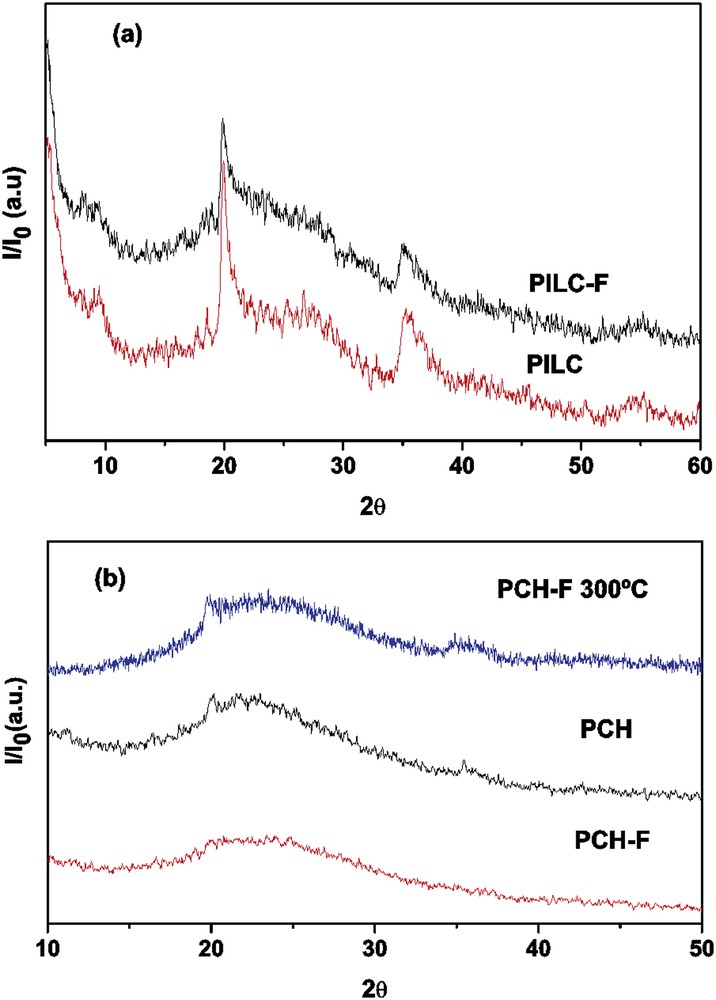
(a) Comparative XRD of PILC (as prepared) and PILC-F (functionalized sample) and (b) PCH (as prepared); PCH-F (functionalized sample) and PCH-F treated at 300 °C.
Figs. 2 and 3 show the results of the TGA profiles for the PILC and PCH and TG-DSC for PILC-F and PCH-F systems. Both PILC-F and PCH-F functionalized samples treated at 300 °C undergo at least 5% more weight loss compared to PILC and PCH, respectively. After functionalization, the silicon content increases to at least 4 wt% for PILC-F and 6 wt% for PCH-F, and the presence of new molecular units on the surface also causes a large increase in weight and additional adsorption of water molecules resulting in a greater weight loss with increasing temperature. DSC profiles show an endothermic signal around 100 °C, corresponding to the loss of hydration water, then the curve continues with a negative slope without signals, suggesting slow loss of molecular unities such as NH3 and CO2 and the subsequent dehydroxylation at higher temperature.
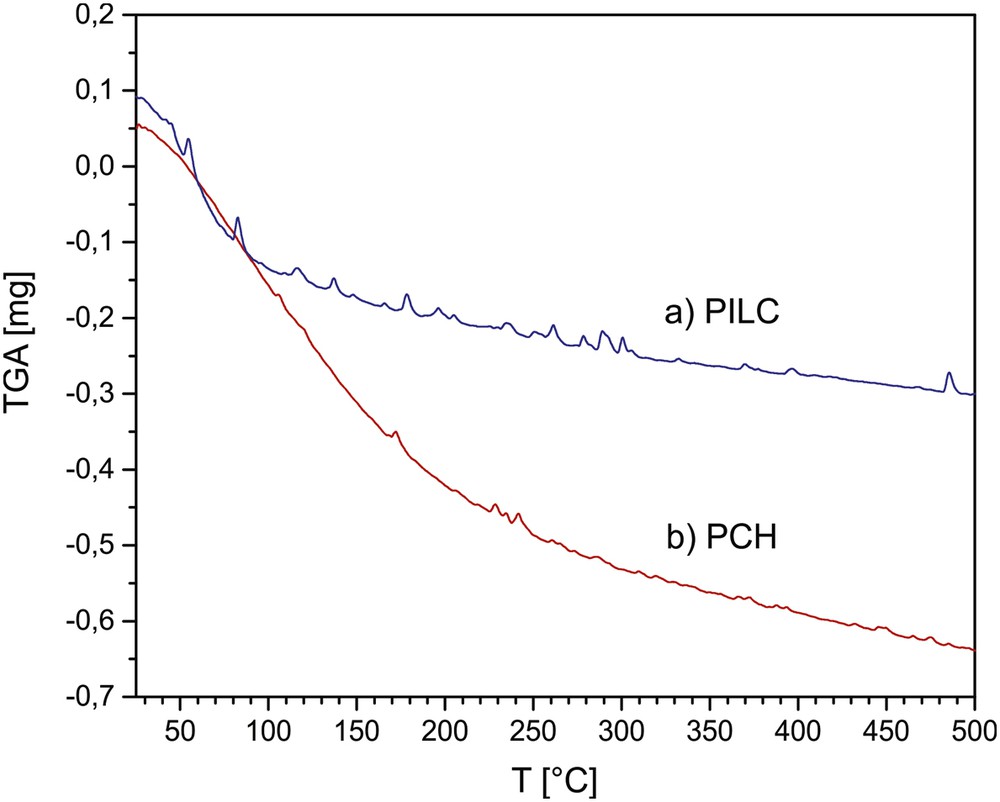
TGA profile of PILC and PCH between room temperature and 500 °C.
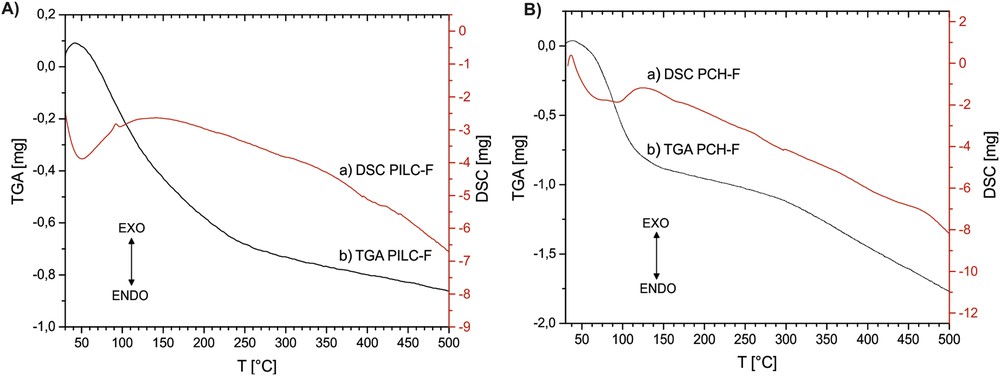
A) TG-DSC of PILC-F and B) TG-DSC of PCH-F between room temperature and 500 °C.
These results are related to the data from SEM-EDS reported in Table 1. Variations in the amounts of surface Al and Si confirm the effect achieved by functionalization.
The equilibrium impregnation process of PILC and PCH clays (untreated and functionalized) with RhMo6 solution produced four composites. In these processes the heteropolyanion is adsorbed by ionic-par formation according to the following surface reactions:
| PILC or PCH surface/3[≡Si–OH2]+:[RhMo6O24H6]3−+ 3NH4(OH) |
| PILC-F or PCH-F surface/[(≡Si–O)2Si(OMe)2(CH2)3NH3]+:[RhMo6O24H6]3−+ 3NH4(OH) |
When PILC or PCH is immersed in RhMo6 aqueous solution, the HPOM can be anchored onto the clay surface mainly by electrostatic interactions [40]. The Anderson unit contains three negative charges, which are neutralized in the moderate acid solution by protons in the form of acidic hydroxyl –OH2+ or NH4+ groups at the exterior of the structure. In the normal pH range, proton adsorption on a ≡Si–OH surface is very low. However, the presence of moderately acidic heteropolyanions (pH = 5) could mean an increase in the proton adsorption reaction of ≡Si–OH, forming a positively charged ≡–Si–OH2+ unit (on PILC or PCH surfaces) and –NH3+ (on PILC-F or PCH-F surfaces) [41]. Thus, HPOM clusters would assemble on the positively charged surface forming anchored RhMo6 clusters [42,43]. Fig. 4 shows an ideal representation of the electrostatic interaction between heteropolyanion RhMo6 and donor amines from organosilane groups. However as it has been well studied, during the process of impregnation using solutions containing polymolybdates, Mo speciation may occur leading to different oxyanions whose charge and size depend on the pH. The kinetics of such reactions depend on the Mo concentration of the starting solution and interface pH. It should be noted that the support of alumina/silicate may act as a pH buffer via the ionization of surface OH groups (protonation or deprotonation) for a high ratio of oxide/water used during impregnation [44]. In our case, where Mo has a concentration of 10 mg/ml, one can easily predict that protonation of basic surface OH groups will lead to an increase of pH of the impregnation solution. This pH increase in turn will lead to the partial depolymerization of [RhMo6O24H6]3− with subsequent formation of nMoO42−.
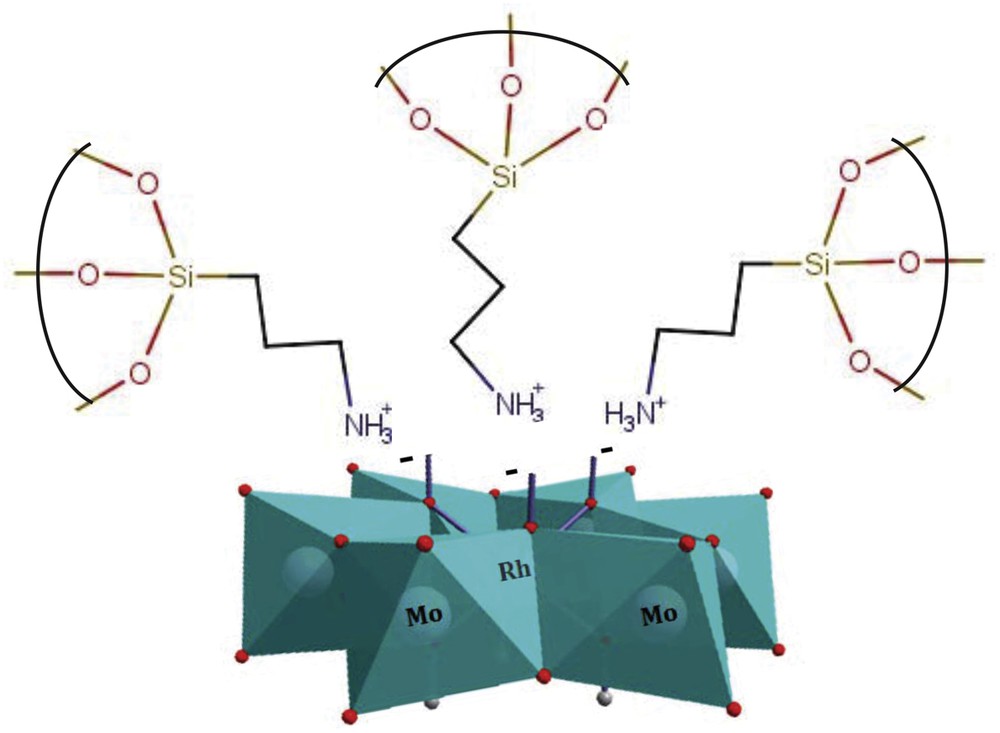
Ideal representation of the electrostatic interaction between heteropolyanion RhMo6 and donor amines from organosilane groups from clay surfaces.
The identification of molecular structures, such as those of iso- and hetero-polyoxomolybdates characterized by Mo–O–X and Mo–O–Mo bridges and terminal Mo–O bonds, can easily be accomplished by Raman spectroscopy [18].
Fig. 5 shows the comparative Raman Microprobe spectra of the original RhMo6 ammonium salt and RhMo6/PCH-F composite, which was the system with a higher content of adsorbed RhMo6. The other systems based on modified clays could not be characterized by this technique because of the lower concentration of RhMo6, low intensity lines and fluorescence under laser radiation, the typical effect observed for aluminosilicates.
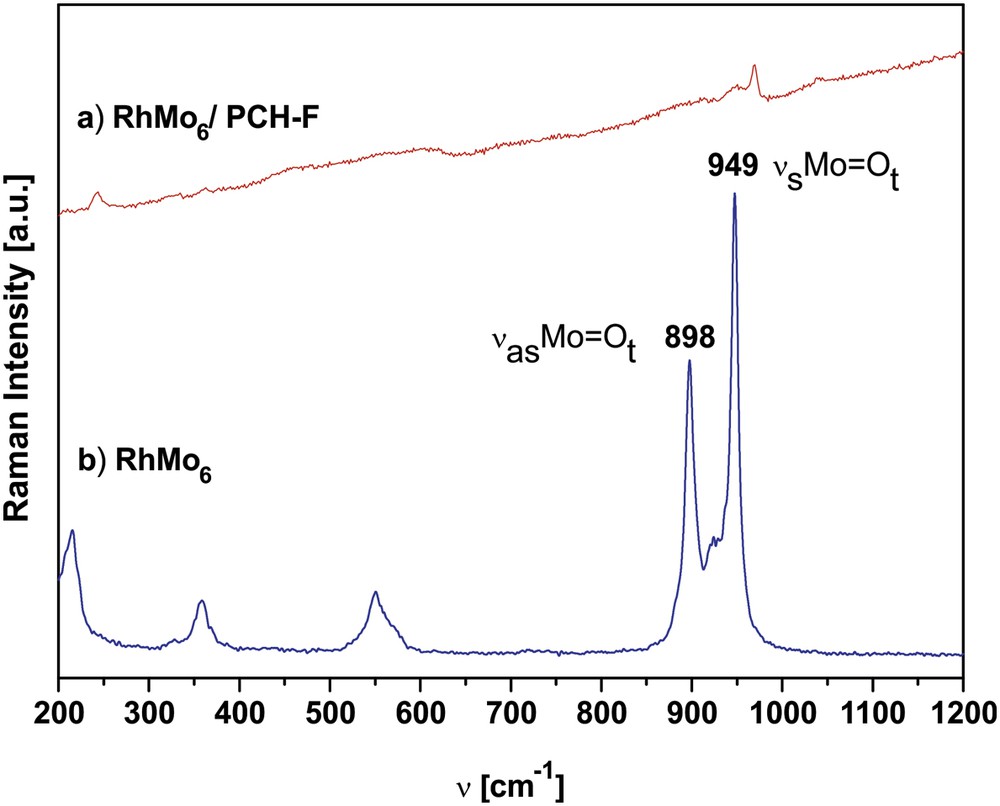
Comparative Raman microprobe spectra of original (NH4)3[RhMo6O24H6].7H2O (RhMo6) and the RhMo6/PCH-F composite calcined at 200 °C (spectral range 200–1200 cm−1).
By comparing the Raman spectrum of the RhMo6 with that of the RhMo6/PCH-F composite (previously heated at 200 °C in air), it is evident that the interaction of the Anderson phase with the support yielded band broadening and shift to higher frequencies. In fact, the lines typical of the Anderson structure are associated with the symmetric stretching mode of terminal Mo–O2t bonds at 949 cm−1 (νs) and the antisymmetric stretching at 889 and 852 cm−1 (νas). For the composite these lines merged at 970 cm−1 for νs and 949 cm−1 for νas, respectively, indicating changes in the structural symmetry and partial HPOM decomposition with the subsequent formation of more compact polyoxoanions with a stronger MoO bond. The presence of bands in the 900–960 cm−1 region indicates the presence of polymolybdic species on the surface of the support. However, due to the large width of Raman bands observed for supported Mo compounds, it is usually extremely difficult to distinguish closely similar polyoxomolybdates. Indeed, most of these species consist of edge-sharing octahedra with a cis-dioxo structure (two short adjacent terminal MoO bonds) that give rise to Mo–O vibrations (stretching or bending modes) in the same region [45].
Regarding the adsorption of the planar heteropolyanion on γ- Al2O3 (Al+3 rich surface), our studies of adsorption isotherms indicated that in the first impregnation step, a series of homogeneous and heterogeneous processes in equilibrium occurred involving phase deposition and some secondary reactions. These reactions, including the Al of the support dissolution, counter diffusion and exchange of cations [especially Rh(III) by Al(III)], have already been characterized by spectroscopic and thermal methods [17,18]. Then, the interaction between RhMo6 and Al(III) ions on the surface of the aluminosilicate should not be excluded, although the proportion of Al(III) at the clay surface is always lower compared to that of Si.
In the present work, all RhMo6/clay composites obtained were amorphous when observed with X-rays. No peaks corresponding to RhMo6 were observed, which means that the RhMo species must be well dispersed on the surface.
Fig. 6 shows the SEM image of the sample, the EDS spectrum, and the structural polyhedral representation of the polyanion RhMo6 corresponding to the (NH4)3RhMo6H6O24.7H2O salt.
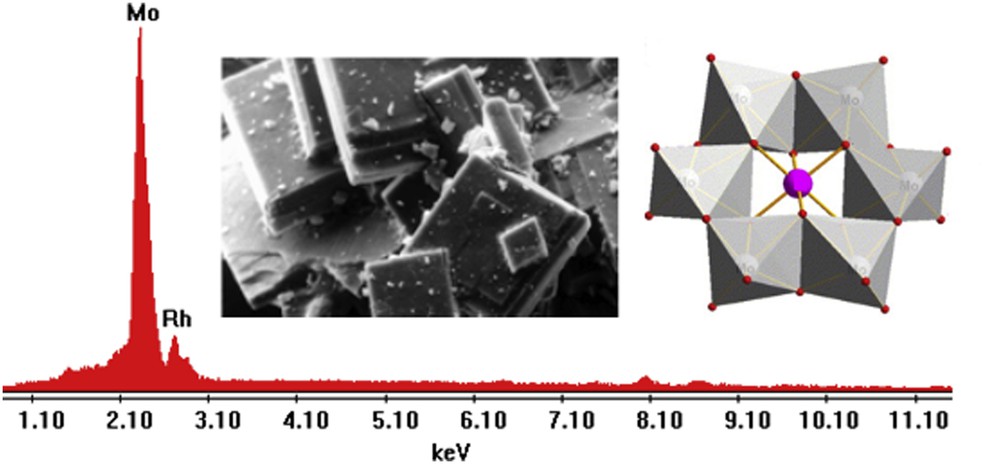
EDS signals and SEM microphotograph of the RhMo6 sample (magnification ×500, scale bar = 50 μm). Semiquantitative analysis data in element wt%: Mo: 84.09; Rh: 15.91. Theoretical values: wt%: Mo: 84.83; Rh:15.16. Polyhedral representation of the heteropolyanion.
The analysis by AAS revealed contents of approximately 1 wt% of Mo in the systems based on unfunctionalized PILC and PCH. However, when the phase was supported on either functionalized material, this percentage reached 7 wt% and 3 wt% for RhMo6/PCH-F and RhMo6/PILC-F, respectively.
Table 2 shows data from the Mo chemical analysis by AAS and EDS data of Mo, Rh, Si, Al and Fe for RhMo6/PILC/PILC-F and RhMo6/PCH/PCH-F. Values for Si and Al agree with the expected amounts for the final materials considering the additional incorporation of Al (for PILC) and Si (for PCH or F-systems) during the respective chemical modifications undergone by the original clay.
Chemical data of Mo% by AAS and major elements % by SEM-EDS for RhMo6/PILC/PCH and RhMo6PILC-F/PCH-F systems.
| Catalyst | Mo% (AAS) | Mo% (EDS) | Rh% (EDS) | Si% (EDS) | Al% (EDS) | Fe% (EDS) |
| RhMo6/PCH-F | 7.00 | 18.07 | 6.18 | 65.65 | 4.49 | 5.61 |
| RhMo6/PCH | 1.30 | 2.30 | 0.86 | 81.13 | 10.30 | 5.41 |
| RhMo6/PILC-F | 2.00 | 1.10 | 0.25 | 69.92 | 23.46 | 5.27 |
| RhMo6/PILC | 1.00 | 2.20 | 0.80 | 67.25 | 24.55 | 5.20 |
Fig. 7 (a and b) show the EDS spectra of the functionalized system (PCH-F) and the supported RhMo6 phase (RhMo6/PCH-F), respectively. The lines of the major elements, Al and Si, are observed. Variation in the Si/Al ratio was observed for the different samples because of the treatment of the original material with silane-based surfactants and additional RhMo6 impregnation. Also, the spectral lines corresponding to some typical original metallic mineral ions (Ti, Mg, Fe) were observed. After equilibrium impregnation with RhMo6 ammonium salt aqueous solution, the material showed additional signals corresponding to Mo and Rh of the heteropolyanion.
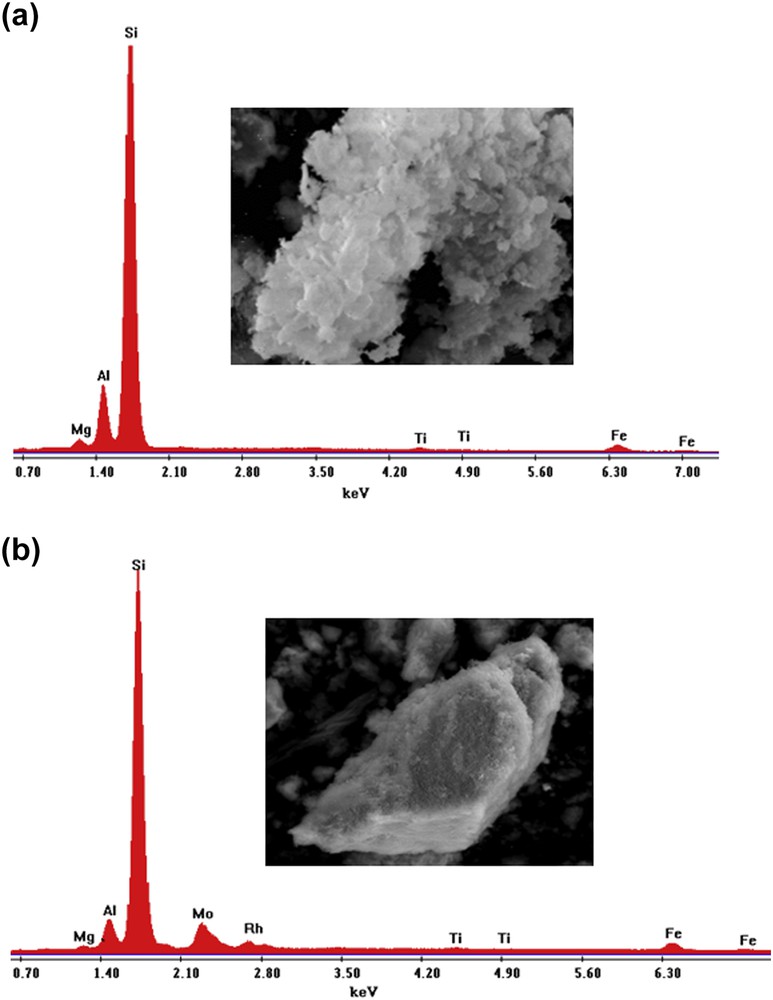
(a). EDS signals and microphotograph of the PCH-F sample (magnification ×2500, scale bar = 20 μm). Semiquantitative EDS analysis data in element wt %: Mg: 1.50; Al: 9.64; Si: 83.73; Ti: 0.78; Fe: 4.35. Total: 100. (b). EDS signals and microphotograph of the RhMo6/PCH-F sample (magnification ×2500, scale bar = 20 μm).
With regard to the values of Mo and Rh obtained by EDS, the Rh/Mo ratio of about 0.3 wt% obtained duplicates the ideal value of 0.17 wt%, suggesting the decomposition of the structure, resulting in different oxo-anionic species of Rh and Mo anchored to the surface. In order to investigate this effect the XPS technique was used. This analysis provided some insight into the chemical and dispersion degree of the surface species both for the unsupported RhMo6 compound and for the composite with a higher content of RhMo6, RhMo6/PCH-F, which was previously treated at 150 °C in an inert atmosphere. Another measurement was performed after “in situ” treating this sample at 350 °C in a reducing atmosphere, simulating the pretreatment to which the sample is submitted before being used in the catalytic reaction.
Data for the pure RhMo6 phase revealed a Rh3d5/2 signal at 310.8 ± 0.2 eV and a Mo3d5/2 signal at 233.0 ± 0.2 eV, corresponding to Rh(III) and Mo(VI), respectively. Because of the overlapping of N1s with the Mo3p3/2 component, the state and amount of N could not be determined. The surface atomic composition nRh/nMo = 0.17, derived from the Rh/Mo intensity ratio [30], is in excellent agreement with the bulk.
Data for the RhMo6/PCH-F composite (pre-treated at 150 °C in an inert atmosphere) showed Rh3d5/2 (309.6 ± 0.2 eV) and Mo3d5/2 (232.1 ± 0.2 eV) binding energies that are characteristic of Rh(III) and Mo(VI), respectively. However, in this case the Rh/Mo ratio derived from the intensity ratio was much higher than the ideal relationship, and also greater than the data obtained by EDS (Table 3). This discrepancy indicates partial decomposition of the Anderson phase structure, as it was observed by Raman Microprobe studies.
Binding energies (eV) and surface composition of pure RhMo6 and RhMo6/PCH-F (heated at 150 °C and a reduced sample at 350 °C).
| Sample | Rh3d5/2 (eV) | Mo3d5/2 (eV) | Sample composition* | |||
| %MoS | %RhS | (nRh/nMo)S | (nRh/nMo)A | |||
| RhMo6 | 310.8 | 233.0 | 0.17 | 0.17 | ||
| RhMo6/PCH-F | 309.6 | 232.1 | 4.6 | 2.6 | 0.56 | 0.34 |
| RhMo6/PCH-F(r) | 307.4 | 232.2 (MoVI) | 1.9 | 2.1 | 0.53 | 0.34 |
| 229.1 (Mo°) | 2.1 |
XPS data for the RhMo6/PCH-F composite (reduced at 350 °C) revealed a Rh3d5/2 signal at 307.4 ± 0.2 eV corresponding to Rh° and two Mo3d5/2 signals at 232.0 ± 0.2 eV and 229.1 ± 0.2 eV corresponding to Mo(VI) and Mo° respectively. These results confirmed the presence of Mo from different polyanionic species, where more than 50% of Mo is reduced. Taking into account our studies about the reducibility of Mo in Anderson Phases [17], this result indicated that the Mo content reduced at temperatures as low as 350 °C could belong to an Anderson polymolybdate but the oxidized Mo content could belong to a species molybdate with a lower degree of polymerization.
The behavior of RhMo6 in a reducing atmosphere (TPR) as well as other isomorphic phases containing other heteroatoms (Co, Ni, Cu, Cr, etc.) has been widely investigated in previous work at our laboratory [15]. Fig. 8 shows comparative TPR diagrams corresponding to the original RhMo6 phase and the same phase supported on both modified materials. The profile of the pure phase (curve b) displays an intense peak at around 285 °C, and two signals of lower intensity at approximately 370 and 707 °C. The first high signal includes the reduction of Rh (III) to Rh° and some Mo reduction steps, whose normal reduction stages are as follows: Mo(VI)→Mo(IV)→Mo° [17]. By examining the behavior of other Anderson phases containing different heteroatoms, it was found that the presence of metallic Rh induces the complete reduction of Mo at lower temperatures (∼250 °C). This effect can be observed both in the pure phase and in the supported ones. The high reducibility of RhMo6 can be associated with the oxidant character of the noble metal.
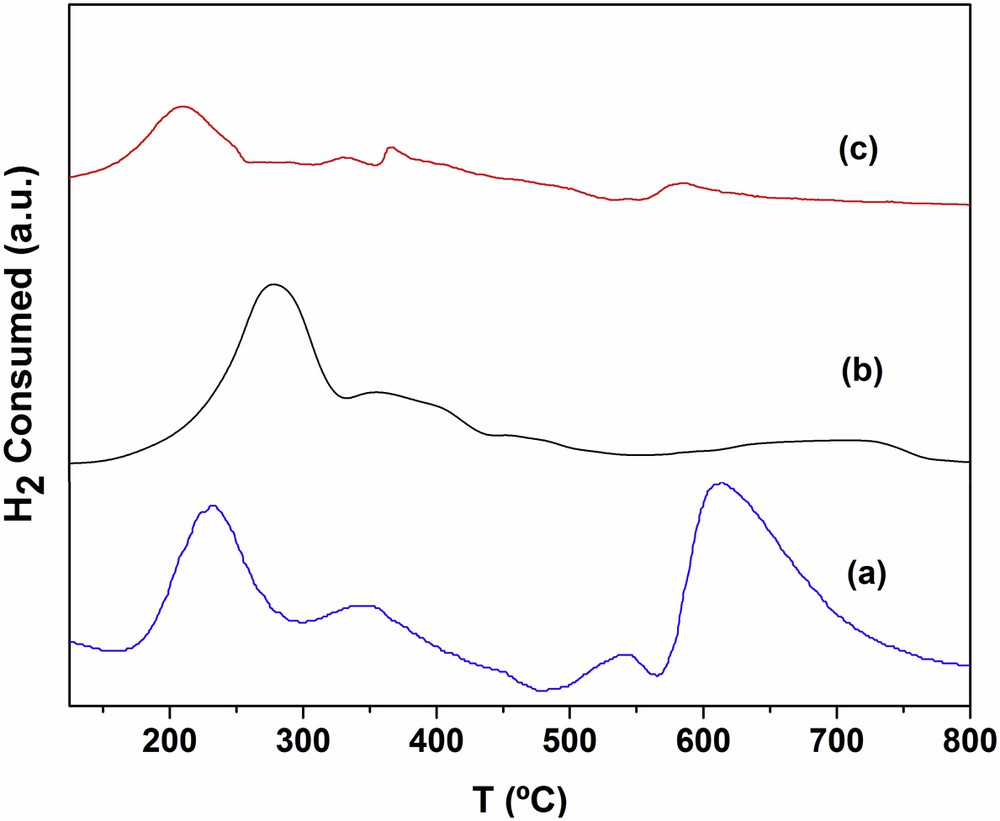
Diagrams of TPR corresponding to (a) RhMo6/PCH-F, (b) RhMo6 original phase and (c) RhMo6/PILC-F.
The TPR of the PCH-F-supported system (Fig. 8, curve a) shows a strong signal centered at 235 °C, corresponding to the reduction of Rh, while the signals at 350 °C and at 620 °C are assigned to the reduction of Mo from different oxide species which interact differently with the support. A signal of low intensity at 540 °C may be assigned to the reduction of Fe belonging to the original clay. The temperature values are slightly lower than those of the pure phase (Fig. 8, curve b), though the signals are more intense; this effect is caused by the interaction with the support [18]. Regarding RhMo6/PILC-F (Fig. 8, curve c), due to the low concentration of the supported phase, only one peak was observed at approximately 200 °C and corresponded to species that interact weakly with the support. This signal may be assigned to both Rh and primary Mo reduction steps. The 2nd weak signal between 600 and 700 °C corresponds to the Mo final reduction step. Such behavior means that the Rh/Mo oxidic phases present different degrees of interaction according to their structural properties and the support characteristics and suggests that for the PCH-F support the HPOM interaction appears to be higher due to a higher reduction temperature than for the RhMo6/PILC-F system, which presented lower adsorbed Rh and Mo concentration.
The acid properties of the prepared supports and catalysts were investigated by the method of isopropanol (IPA) decomposition followed by TPSR. It is well known that the dehydration of IPA on solid acids leads to the formation of propene and 2-isopropyl ether, whereas its dehydrogenation leads to the formation of acetone [46]. The dehydration/dehydrogenation selectivity depends, among other factors, on the surface structure of the solid. The dehydration reaction is the main process when strong Brönsted (H+) and Lewis acid sites are present. The majority of primary alcohols react through an E2 concerted mechanism, whereas an E1 mechanism can be found for secondary alcohols, as is the case of IPA [47,48]. According to Scheme 3, the E1 mechanism of olefin formation involves one acid site, on which a carbonium ion intermediate is formed; this ionic species is then transformed into an olefin molecule. For the E2 mechanism to be developed, both acid and basic adjacent sites have to be present on the solid, leading to the dehydration to either ether or olefin.
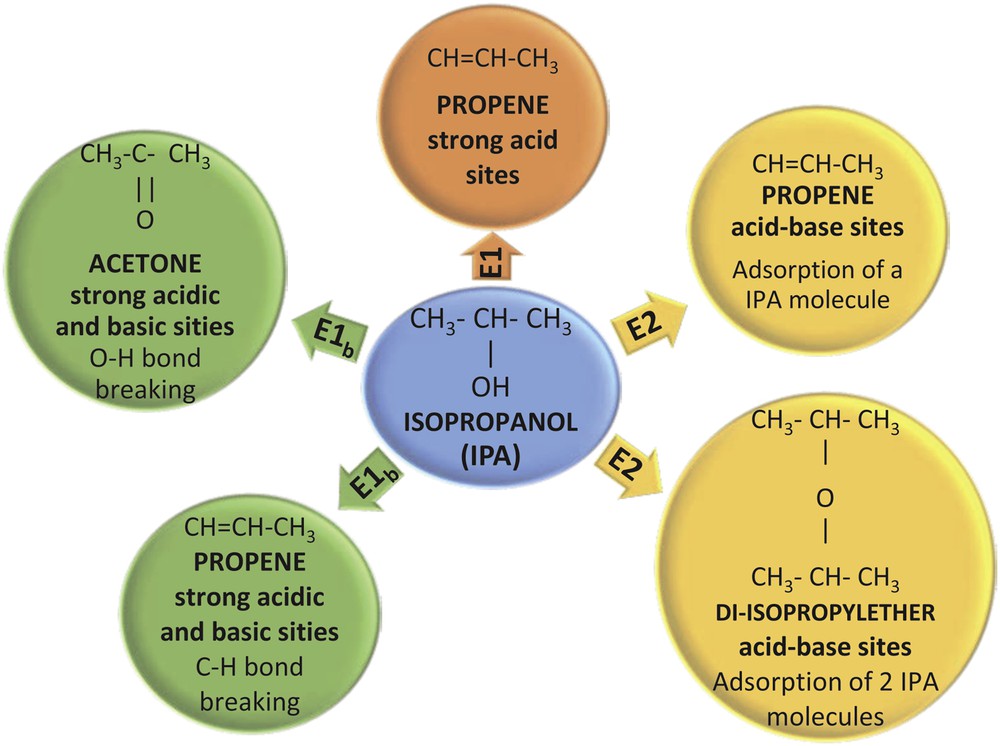
Isopropanol decomposition followed by TPSR.
Fig. 9 reports the results of the TPSR analysis for the pure PILC-F support and for the fresh RhMo6/PILC-F catalyst. No desorption of propene was observed for the PILC-F support, indicating an inactive surface toward IPA decomposition. The deposit of the RhMo6 Anderson phase onto this support incorporates Lewis acid sites to the surface. This fact explains desorption of propene observed. Regarding the PCH-F support (Fig. 10), the formation of propene with a maximum at 110 °C was detected. This result suggested the existence of acid sites on the surface according to the E1 mechanism. This fact has usually been reported for pure acidic solids, such as silica-alumina and zeolites, [48] and could be associated with higher values of specific area (Table 1). The subsequent impregnation of these supports with RhMo6 heteropolyanion resulted in the addition of slightly stronger new acid sites where propene was desorbed at higher temperature, 160 °C (Figs. 10 and 11). The presence of RhMo6 on both supports led to the desorption of propene at a similar temperature, indicating sites of similar acid strength. On the other hand, a small desorption band of CO2 at 100 °C was observed on RhMo6/PCH-F, implying the presence of some basic sites in the system (Fig. 11). No desorption of acetone was observed, indicating the absence of dehydrogenation reactions in the studied systems.
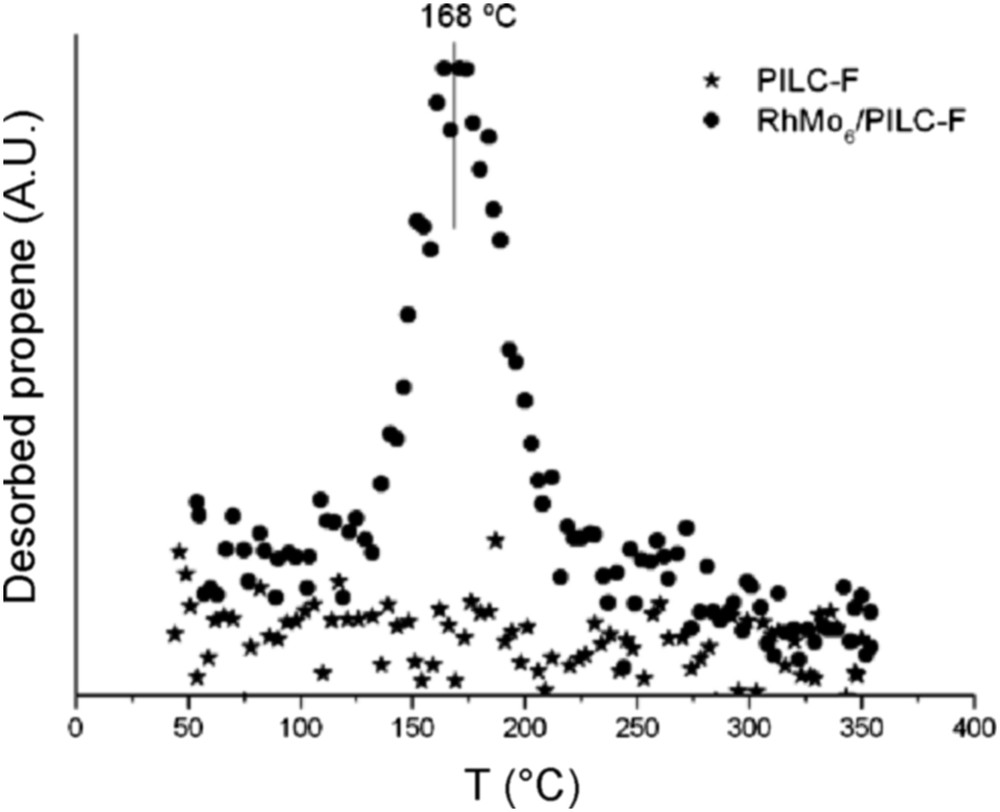
Desorption of propene as a function of temperature (TPSR), of the PILC-F support and RhMo6/PILC-F catalyst.
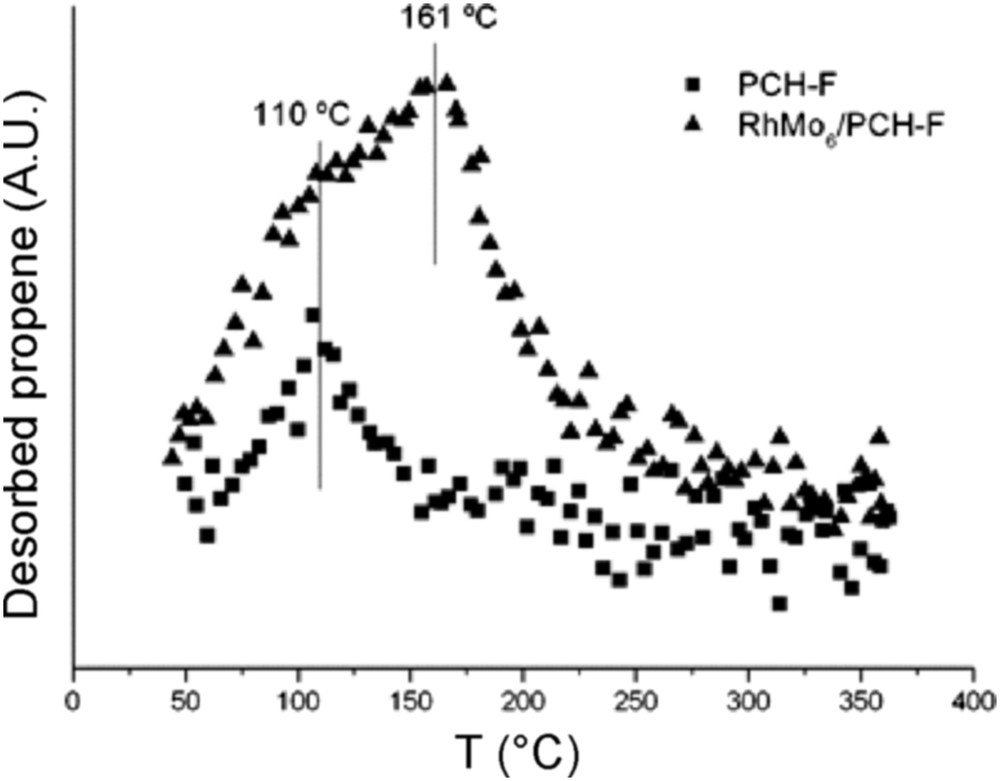
Desorption of propene as a function of temperature (TPSR), of the PCH-F support and RhMo6/PCH-F catalyst.
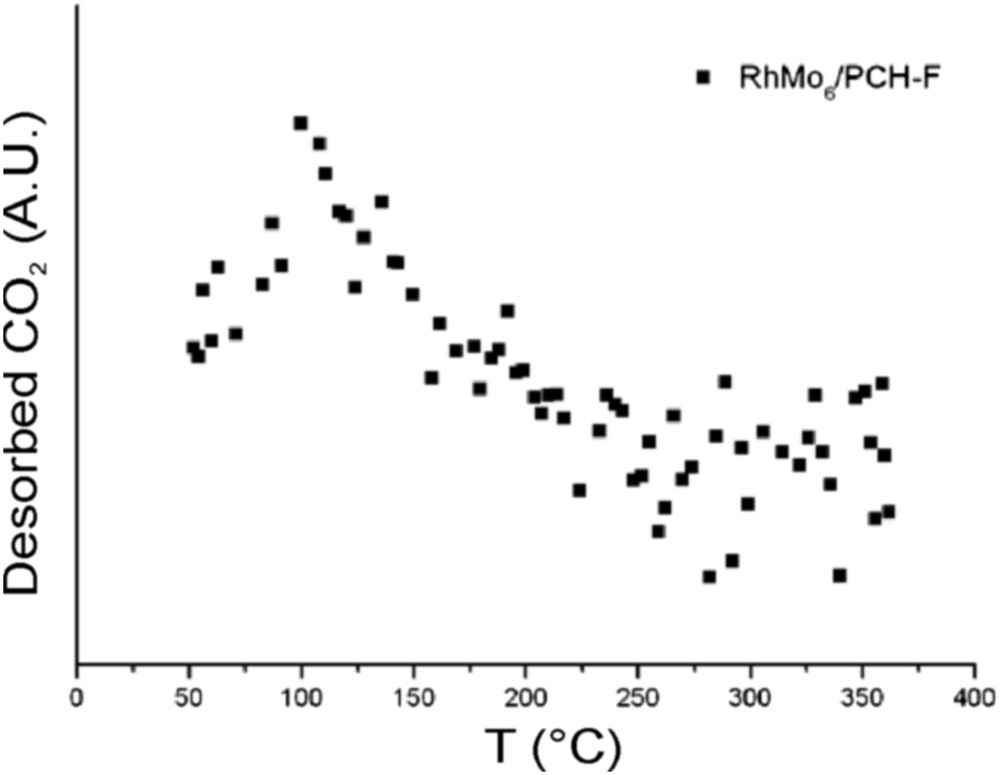
Desorption of CO2 for the RhMo6/PCH-F catalyst.
3.2 Catalytic evaluation
Scheme 4 shows the reaction for the hydrogenation of cinnamaldehyde (CAL). The most important products obtained from the hydrogenation of CC bond and/or CO group/groups are cinnamic alcohol (CA), hydrocinnamaldehyde (HCAL) and hydrocinnamic alcohol or 3- phenylpropanol (PP).
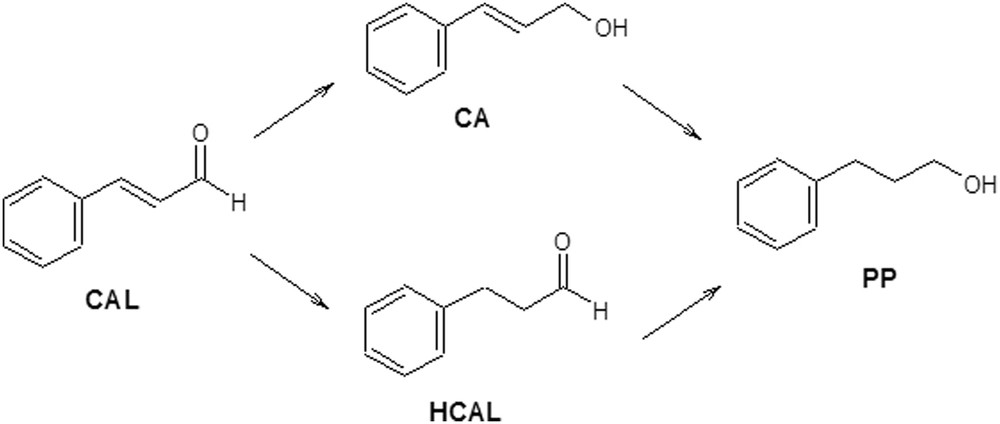
Reaction scheme of the cinnamaldehyde hydrogenation.
Fig. 12 shows the conversion attained by each of the systems studied at 360 min of reaction. As can be seen, the systems supported on PCH and PCH-F were found to be more active than those supported on PILC and PILC-F, whose activities were not higher than 5%. This result may be attributed to the higher percentage of Rh and Mo found on both PCH supports. Also, it is possible to observe that the effect of functionalization on the PCH support led to an increase in activity with respect to the unfunctionalized system. Thus, after 360 min of reaction the conversion obtained was 48% and 56% for RhMo6/PCH and RhMo6/PCH-F, respectively. This effect may also be attributed to the higher amount of Rh and Mo in the catalyst supported on PCH-F. However, the increase in activity was lower than expected, probably due to the fact that metallic ion sites were not available enough for the reagent.
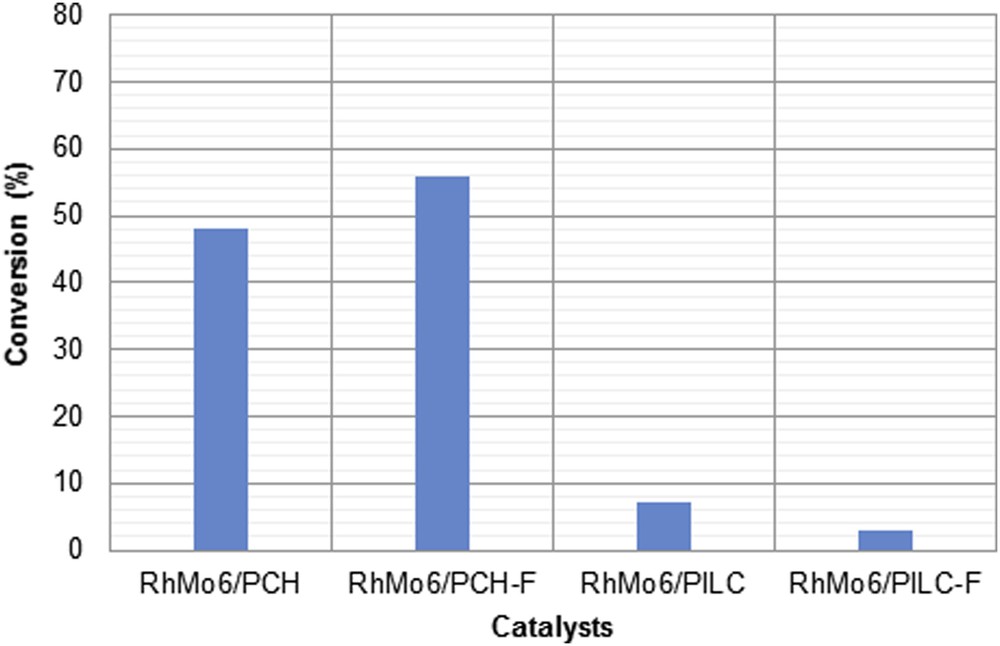
Activity of the cinnamaldehyde hydrogenation at 360 min of reaction catalyzed by RhMo6/PCH, RhMo6/PCH-F, RhMo6/PILC and RhMo6/PILC-F.
Table 4 shows the selectivities at 25% of conversion and the different reaction products obtained for the most active systems.
Selectivity of CAL hydrogenation to HCAL, PP and CA (at 25% of conversion).
| Catalyst | SHCAL (%) | SPP(%) | SCA(%) |
| RhMo6/PCH | 62 | 20 | 18 |
| RhMo6/PCH-F | 77 | 11 | 12 |
As can be seen, both PCH and PCH-F systems show a good selectivity for the product (HCAL). The use of the RhMo6/PCH-F catalyst strongly increased the selectivity to HCAL and it showed similar selectivity to minority products (PP and CA) compared with the same unfunctionalized system. This behavior can be associated with Lewis acid characteristics (shown in the study by TPSR) responsible for the hydrogenation of the CO group. The distribution of minority products, CA and PP, showed by RhMo6/PCH-F can be explained by acidic features provided by the heteropolyanion. This property increased for the functionalized system due to the natural acidity of the support surface. Thus a combined effect of Brönsted and Lewis acid sites should not be discarded because the acidity of the support has a strong influence in this catalytic reaction, as demonstrated recently in our work on RhMo6/alumina supports of different surface acidity values [18]. Further studies about these new catalysts are in progress to better define their preparation and the reaction conditions.
4 Conclusions
This study has shown that it was possible to obtain Rh-Mo catalysts from the composites of Anderson-type heteropolymolybdates [RhMo6O24H6]3-and two types of heterostructured and functionalized Argentinean clays: PILC-F and PCH-F. The functionalization process leads to a support which is able to anchor polymetalate active phases and provides an interesting approach in the field of immobilized catalysts.
These materials, unlike other conventional catalysts, combine the advantages of both heteropolycompounds and clays such as, low cost, easy control of metal loading and acidity levels.
These composites were evaluated in the liquid phase hydrogenation of cinnamaldehyde. It was found that RhMo6/PCH and RhMo6/PCH-F catalysts are more active because of a higher concentration of heteropolyanions. Prereduced catalysts led to a system where the hydrogenating capacity (Rh), the redox sites (Mo) and different acid/base sites coexist to produce a good performance to HCAL.
Acknowledgements
We greatly appreciate the Prof. Edmond Payen’s cooperation allowing the use of Raman equipment at “Unitè de Catalyse et de Chimie du Solide”, UMR, CNRS France, and for his valuable comments. We are grateful to Lic. Ma. Fernanda Mori for their contribution in XPS measurements and to Dr. Daniela Lick for TG-DSC measurements.
The authors would like to thank the following institutions for funding this work: ANPCyT for the purchase of the SPECS multitechnique analysis instrument (PME8-2003)”; (CONICET) (PIP 0185); CICPBA (Project 832/14) and Universidad Nacional de La Plata (Projects X 633, X 700 and I 172).