

1 Introduction
In recent years, trinuclear metal complexes, because of their unique and interesting properties have been attracting great attention, both experimentally and theoretically. Attention has been given to the syntheses and properties of bimetallic complexes because of their unique catalytic [1], optical, and electronic properties. Small gold-silver complexes have been found to be highly effective in catalysis and medicine [2]. Silver–gold complexes proved to be more effective catalysts than the pure metals because of their increased activity, resistance to poisoning, and selectivity. These materials are considered to be good candidates for electronic nanodevices and biosensors. In addition, there are many fields where these kinds of sensors can be used, such as environmental applications, electronic noses or in the chemical industry [3].
Many bimetallic gold-silver complexes have been synthesized during the past decade [4–7]. The heterobimetallic complex, AgAu(MTP)2, was synthesized and crystallographically characterized (MTP = diphenylmethylenethiophosphinate) by Fackleret al. [8]. Catalanoet al. [9] reported an Au–Ag complex that is three-coordinate at each metal center, [AuAg(DPIM)3]2+(DPIM = 2-(diphenylphosphino)-1-methylimidazole).
The trinuclear organometallic complex [Au3(HNCOH)3], which is presented in this paper, was first synthesized and characterized in 1974 [10,11].In the solid state the minimal unit shows an individual molecule of the planar trinuclear complex (Fig. 1). The intramolecular Au–Au distance is 3.308 Å. The individual molecules form a columnar structure whose intermolecular Au–Au distance is 3.346 Å [12].
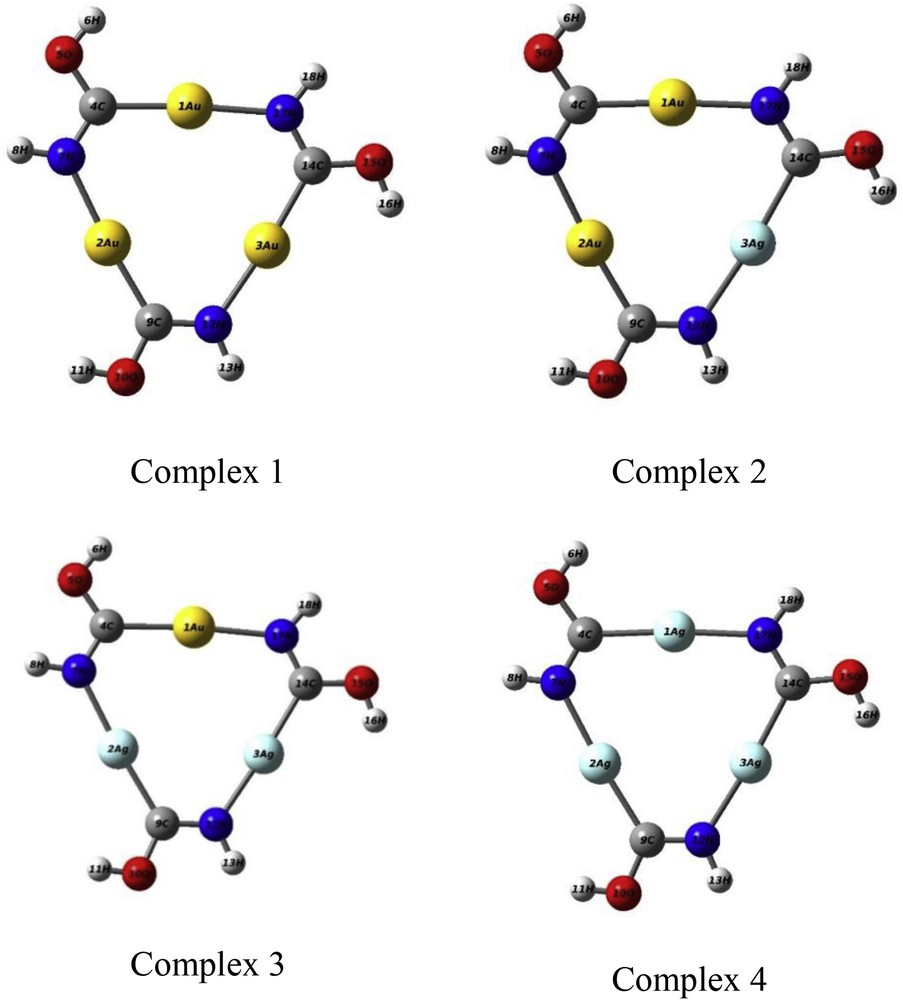
Optimized structure of pure and bimetal complexes of [AunAgm(HNCOH)3] (m + n = 3).
The structures and chemical properties of these compounds are very attractive. DFT is an affordable method for studying a variety of molecular properties and electronic structures, due to its favorable accuracy to computational cost ratio compared to accurate correlated wave function theory (WFT).There have been several reports of excellent agreement with experimental absorption and emission spectra for different gold complexes [13–16]. There are many complete theoretical studies centered on the triangular gold complex and on the electronic structures and luminescence properties at the MP2, CIS and B3LYP levels [16,17].
However, considering the few theoretical studies on these complexes, our focus in this paper is limited to the influence of silver substitution on the ionization potentials, electron affinities and other molecular properties and electronic structures of the [Au3(HNCOH)3] complex. The obtained results are compared with the corresponding properties of the [Au3(HNCOH)3] complex.
2 Computational method
The calculations of the geometry and electronic structure were performed using Gaussian 09 at the spin unrestricted DFT wave function (B3LYP) level, i.e., the Becke three-parameter exchange function in combination with the LYP correlation function of Lee, Yang and Parr with a 6-311G* basis set for C, H, N and O atoms, and effective core potential basis set LanL2DZ for Au and Ag atoms. The optimized calculation was confirmed to be local minima by performing harmonic vibration frequency analyses (no imaginary frequency found). No symmetry constrains were applied, and only the default convergence criteria were undertaken during optimization.
Natural bond orbitals (NBO) have been applied to gain insight into the electronic structure of the molecules and the nature of metal- metal and metal-ligand bonds. The popularity of the NBO method comes from its ability to transform the delocalized molecular orbitals into equivalent localized molecular orbitals that recover the expected Lewis bond patterns.
3 Results and discussion
3.1 Geometrical properties
This part of the study is started with the substitution of one silver atom for gold, to establish a series of gold-silver complexes with varying compositions. This replacement continues until the pure silver complex is reached. As the first step, the equilibrium geometries of all [AunAgm(HNCOH)3] (m + n = 3) complexes are investigated. The results of geometry optimization are reported in Fig. 1.
The results of the optimization process (Fig. 1) show that the most stable structures of pure Au and Ag complexes are almost similar. All the structures are planar. Just the H (hydrogen) atoms are not in the plane. The symmetry point group of these structures is C3h. Table 1 lists the distances and angles along with the experimental values. Accordingly, comparison with the experimental data reveals a good agreement. The average error in the bond distance was determined to be 0.300 A°. The larger difference, 0.064 A°, is in the Au–N distance. In all of the structures, the C–N bond length is reasonable for a double bond. There is a slight dispersion, considering the angles, compared with the experimental results although the agreement is good. The angles centered at the carbon atom are distorted with respect to the sp2 hybridization. On the other hand, the angles centered at N are close to the true value. Therefore, the presence of the metal atom distorts the hybridization of the close adjacent C atom. For these complexes, the intramolecular metal–metal distances with bridging-ligand bonding are in the order of Au–Au < Au–Ag < Ag–Ag. The results were in agreement with the experimentally determined equilibrium geometry. Mohamed et al. [18] have reported the metal–metal distances in the tri-icosahedral structure of the cluster [(PPh3)12Au12Ag13Cl6]m+ following the trend of Au–Au < Au–Ag < Ag–Ag.
Selected bond distances (Å) and angles (deg) with the experimental results for all complexes.
| Complex 1 | |||||
| Bond distances (Å)/[exp] | |||||
| Au(1)⋯Au(2) | 3.494 | Au(3)–C(14) | 2.013/[2.00] | Au(1)–C(4) | 2.013/[2.00] |
| Au(1)⋯Au(3) | 3.494 | Au(1)–N(17) | 2.093/[2.03] | Au(2)–C(9) | 2.013/[2.00] |
| Au(2)⋯Au(3) | 3.495 | Au(2)–N(7) | 2.094/[2.03] | Au(3)–N(12) | 2.094/[2.03] |
| Bond angles (deg)/[exp] | |||||
| N(17)–Au(1)–C(4) | 178.4/[180.0] | Au(1)–Au(2)–Au(3) | 60.00/[60.0] | Au(1)–C(4)–O(5) | 123.7/[121.4] |
| Au(1)–C(4)–N(7) | 122.3/[119.4] | N(17)–Au(1)–C(4)–N(7) | 0.22/[0.0] | ||
| C(4)–N(7)–Au(2) | 119.5/[120.6] | Au(1)–C(4)–N(7)–Au(2) | 0.01/[0.0] | ||
| Complex 2 | |||||
| Bond distances (Å) | |||||
| Au(1)⋯Au(2) | 3.516 | Ag(3)–C(14) | 2.113 | Au(1)–C(4) | 2.015 |
| Au(1)⋯Ag(3) | 3.503 | Au(1)–N(17) | 2.088 | Au(2)–C(9) | 2.013 |
| Au(2)⋯Ag(3) | 3.490 | Au(2)–N(7) | 2.097 | Ag(3)–N(12) | 2.136 |
| Bond angles (deg) | |||||
| N(17)–Au(1)–C(4) | 176.39 | Au(1)–Au(2)–Ag(3) | 60.00 | Au(1)–C(4)–O(5) | 122.42 |
| Au(1)–C(4)–N(7) | 122.80 | N(17)–Au(1)–C(4)–N(7) | 1.22 | ||
| C(4)–N(7)–Au(2) | 122.42 | Au(1)–C(4)–N(7)–Au(2) | 0.00 | ||
| Complex 3 | |||||
| Bond distances (Å) | |||||
| Ag(1)⋯Ag(2) | 3.521 | Au(3)–C(14) | 2.015 | Ag(1)–C(4) | 2.108 |
| Ag(1)⋯Au(3) | 3.524 | Ag(1)–N(17) | 2.136 | Ag(2)–C(9) | 2.111 |
| Ag(2)⋯Au(3) | 3.526 | Ag(2)–N(7) | 2.127 | Au(3)–N(12) | 2.090 |
| Bond angles (deg) | |||||
| N(17)–Ag(1)–C(4) | 175.05 | Ag(1)–Ag(2)–Au(3) | 60.00 | Ag(1)–C(4)–O(5) | 124.54 |
| Ag(1)–C(4)–N(7) | 121.08 | N(17)–Ag(1)–C(4)–N(7) | 0.89 | ||
| C(4)–N(7)–Ag(2) | 122.24 | Ag(1)–C(4)–N(7)–Ag(2) | −0.36 | ||
| Complex 4 | |||||
| Bond distances (Å) | |||||
| Ag(1)⋯Ag(2) | 3.549 | Ag(3)–C(14) | 2.107 | Ag(1)–C(4) | 2.108 |
| Ag(1)⋯Ag(3) | 3.548 | Ag(1)–N(17) | 2.129 | Ag(2)–C(9) | 2.109 |
| Ag(2)⋯Ag(3) | 3.547 | Ag(2)–N(7) | 2.128 | Ag(3)–N(12) | 2.130 |
| Bond angles (deg) | |||||
| N(17)–Ag(1)–C(4) | 175.80 | Ag(1)–Ag(2)–Ag(3) | 60.00 | Ag(1)–C(4)–O(5) | 124.00 |
| Ag(1)–C(4)–N(7) | 121.64 | N(17)–Ag(1)–C(4)–N(7) | 4.33 | ||
| C(4)–N(7)–Ag(2) | 122.54 | Ag(1)–C(4)–N(7)–Ag(2) | 0.15 |
3.2 NBO charge
The obtained total atomic charge values by natural bond orbital analyses (NBO) are reported in Table 2.As can be seen in the results, all the metal atoms have a net positive charge while nitrogen atoms are negative. The charge of the oxygen atoms is smaller than the charge of the N for all compounds. According to the NBO method, the obtained atomic charge for all complexes shows that the metal atoms have bigger positive atomic charges than the other atoms.
NBO charges of the complexes in the ground state.
| Complex 1 | Complex 2 | Complex 3 | Complex 4 | ||||
| Atom | Charge | Atom | Charge | Atom | Charge | Atom | Charge |
| Au1 | 0.365 | Au1 | 0.359 | Ag1 | 0.535 | Ag1 | 0.484 |
| Au2 | 0.365 | Au2 | 0.351 | Ag2 | 0.540 | Ag2 | 0.484 |
| Au3 | 0.365 | Ag3 | 0.508 | Au3 | 0.380 | Ag3 | 0.484 |
| C4 | 0.299 | C4 | 0.298 | C4 | 0.323 | C4 | 0.236 |
| O5 | −0.696 | O5 | −0.698 | O5 | −0.354 | O5 | −0.708 |
| N7 | −0.875 | N7 | −0.874 | N6 | −0.883 | N7 | −0.889 |
| C9 | 0.299 | C9 | 0.294 | C8 | 0.335 | C9 | 0.235 |
| O10 | −0.696 | O10 | −0.702 | O9 | −0.348 | O10 | −0.708 |
| N12 | −0.875 | N12 | −0.886 | N10 | −0.876 | N12 | −0.889 |
| C14 | 0.299 | C14 | 0.242 | N12 | −0.871 | C14 | 0.235 |
| O15 | −0.696 | O15 | −0.702 | C14 | 0.377 | O15 | −0.708 |
| N17 | −0.875 | N17 | −0.881 | O15 | −0.345 | N17 | −0.889 |
Molecular electrostatic potential (MEP) at a point in space around a molecule gives information about the net electrostatic effect produced at that point by the total charge distribution of the molecule[18]. The interaction between the positive charge and some point in the molecule will be attractive if the point is negatively charged; repulsive if it is positively charged, and the strength of interaction will depend on the magnitude of the charge. It is convenient to display this map using the colors of the rainbow from red to blue. Red is the electron-rich end and blue is the electron-poor end.
The MEP surface provides necessary information about the reactive sites. Fig. 2 shows the electrostatic potential counter map of complexes.
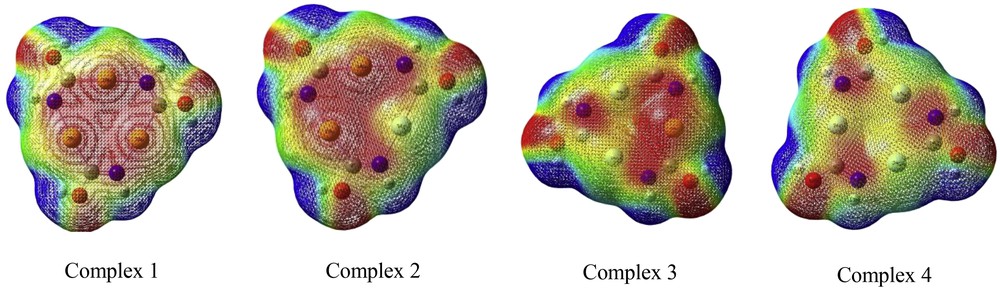
Calculated 3D molecular electrostatic potential contour map of complexes.
3.3 HOMO–LUMO gap
The highest occupied molecular orbitals (HOMOs) and the lowest-lying unoccupied molecular orbitals (LUMOs) are named as frontier molecular orbitals (FMOs). HOMO and LUMO are very important parameters for quantum chemistry. The HOMO and LUMO orbitals are plotted in Fig. 3. In complexes 1,2 and 3,the HOMO belongs to the d atomic orbital of the one gold atom. In complex 4, it belongs to the atomic orbital of the silver atom. Also for all of the complexes, the irreducible representation of the LUMO is shown with e″. As well as the LUMO is located at the carbon and nitrogen atoms, largely.
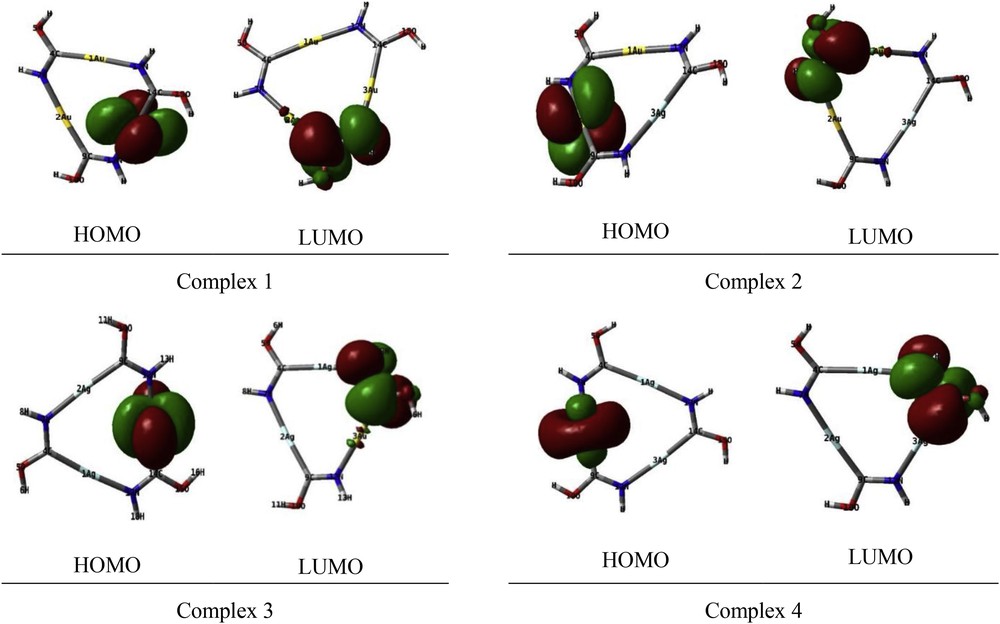
HOMO and LUMO orbitals of complexes.
The computed HOMO–LUMO energy gaps of 1, 2, 3 and 4 complexes were found to be 0.28, 0.30, 0.31 and 0.34 eV, respectively, at the B3LYP/GEN level. Thus the energy gap was found to be significantly increased with increase in the number of silver atoms in the [AunAgm(HNCOH)3] (m + n = 3) complexes compared to that of complex 1.
3.4 Ionization potential and electron affinity
The ionization potential (IP) is an important parameter to understand the stability towards ejecting out one electron from its HOMO energy level to the continuum. The vertical ionization potentials were computed as the energy required to release an electron from the system at the ground state reference geometry.
The computed first vertical ionization potential of 1, 2, 3, and 4 complexes was found to be 8.50, 8.28, 8.12 and 8.02 eV, respectively. Thus, the first ionization potential is decreased on going from the [Au3(HNCOH)3] complex to other [AunAgm(HNCOH)3] (m + n = 3) complexes; the largest decrease is revealed for the [Ag3(HNCOH)3] complex. The vertical electron affinity (EA) of studied systems was also computed. The EA is defined as the amount of energy released when an electron is attached to the system. The computed electron affinities were found to be −0.55, −0.43, −0.39 and −0.32 eV for the 1, 2, 3 and 4 complexes, respectively. Thus, the electron affinity is significantly increased with the increase in the number of Ag atoms.
3.5 Chemical hardness and chemical potential
The computed hardness and chemical potentials of the complexes are listed in Table 3. Chemical hardness[18] which demonstrates the resistance to alteration in electron distribution is given by:
| (1) |
The obtained ionization potentials, electron affinities, HOMO–LUMO gaps, hardness and chemical potentials of complexes.
| Sample | IP (eV) | EA (eV) | HLG (eV) | Hardness (eV) | Chemical potential (eV) |
| Complex 1 | 8.50 | −0.55 | 0.28 | 4.53 | −3.98 |
| Complex 2 | 8.28 | −0.43 | 0.30 | 4.36 | −3.93 |
| Complex 3 | 8.12 | −0.39 | 0.31 | 4.25 | −3.86 |
| Complex 4 | 8.02 | −0.32 | 0.34 | 4.22 | −3.80 |
Following Parr and Pearson[18], the electronic chemical potential, describing the escaping tendency of an electron from a stable system can be calculated as
| (2) |
We reported here the results of DFT calculations for the global reactivity descriptors (Table 3).The listed values showed that the softest complex was [Ag3(HNCOH)3] with the hardness of 4.22 eV, while the maximum hardness of 4.53 eV was found for the [Au3(HNCOH)3] complex. The less polarizable species have higher hardness values. The zero hardness corresponds to the most polarizable structure. Considering the chemical hardness, if one molecule has a large HOMO–LUMO gap, it is a hard molecule or if one has a small HOMO–LUMO gap it is a soft molecule. One can also relate the stability of a molecule to hardness, which means that the molecule with the least HOMO–LUMO gap means it is more reactive.
With the Mulliken definition for the chemical potential, the negative μ values correlate with a more stable or less-reactive compound. The μ values (Table 3), indicated that the [Au3(HNCOH)3] complex, with μ = −3.98 eV, was the least reactive in this series of compounds. On the other hand, the value of the chemical potential characterizing the most reactive complex, [Ag3(HNCOH)3], in this series was −3.80 eV.
The chemical reactivity of [AunAgm(HNCOH)3](n + m = 3) followed the order of [Au3(HNCOH)3] < [Au2Ag1(HNCOH)3] < [Au1Ag2(HNCOH)3] < [Ag3(HNCOH)3], which is in agreement with the experimental results. For instance, Hettiarachchi et al. [19] reported the synthesis and characterization of d10–d10 heterometallic compounds containing gold and silver, K[AuxAg1−x(CN)2], and compared the results with the corresponding pure systems. They observed that the emission bands in the mixed-metal complexes lay between the emission bands of the pure K[Au-(CN)2] and K[Ag(CN)2].
3.5.1 Second order perturbation analysis
The natural bond orbital (NBO) analysis shows the intermolecular orbital interaction. In this part of study, we examine all possible interactions between donor Lewis-type and acceptor non-Lewis-type NBOs. These interactions lead to donation of occupancy from the localized NBOs of the idealized Lewis structure into the empty non-Lewis orbitals. For this reason, they are denoted as electron delocalization corrections to the zeroth-order natural Lewis structure[20]. The stabilization energy E(2) value for each Lewis-type donor (i) and non-Lewis-type acceptor (j), associated with the electron delocalization between i and j (i→j), is estimated as
Second order perturbation theory analysis of the fock matrix in the NBO basis of complexes; E(2) is the mean energy of hyper conjugative interactions, E(j)–E(i) is the energy difference between the donor and acceptor i and j NBO orbitals and F(i, j) is the Fock matrix elements between i and j NBO orbitals
| Sample | Donor NBO (i) | Acceptor NBO (j) | E(2) (kcal/mol) | E(j)–E(i) (a.u.) | F(i,j) (a.u.) |
| Complex 1 | LPO5 | BD*(C4-N7) | 22.73 | 0.33 | 0.111 |
| BD*(Au1-C4) | LP* Au1 | 23.56 | 0.03 | 0.101 | |
| LP N7 | BD*(Au2-C9) | 38.34 | 0.54 | 0.184 | |
| LP O10 | BD*(C9-N12) | 22.72 | 0.33 | 0.11 | |
| BD*(Au2-C9) | LP*Au2 | 23.39 | 0.03 | 0.101 | |
| LP N12 | BD*(Au3-C14) | 38.35 | 0.54 | 0.184 | |
| LP N17 | BD*(Au1-C4) | 38.42 | 0.54 | 0.184 | |
| BD*(Au3-C14) | LP* Au3 | 23.36 | 0.03 | 0.101 | |
| Complex 2 | LP O5 | BD*(C4-N7) | 22.58 | 0.33 | 0.11 |
| BD*(Au1-C4) | LP*Au1 | 52.25 | 0.01 | 0.097 | |
| LPN7 | BD*(Au2-C9) | 38.28 | 0.54 | 0.184 | |
| LP O10 | BD*(C9-N12) | 21.99 | 0.33 | 0.109 | |
| LP N17 | BD*(Au1-C4) | 39.64 | 0.54 | 0.187 | |
| LP O15 | BD*(C14-N17) | 22.22 | 0.33 | 0.109 | |
| Complex 3 | LP O5 | BD*(C4-N7) | 21.49 | 0.34 | 0.108 |
| LP N7 | BD*(Ag2-C9) | 20.46 | 0.53 | 0.134 | |
| LP O10 | BD*(C9-N12) | 22.12 | 0.33 | 0.109 | |
| LP N12 | BD*(Au3-C14) | 39.68 | 0.54 | 0.187 | |
| LP O15 | BD*(C14-N17) | 21.88 | 0.33 | 0.109 | |
| Complex 4 | LP O5 | BD*(C4-N7) | 21.42 | 0.34 | 0.108 |
| LP O10 | BD*(C9-N12) | 21.41 | 0.34 | 0.108 | |
| LP O15 | BD*(C14-N17) | 21.41 | 0.34 | 0.108 |
4 Conclusions
In this study, we have performed detailed DFT calculations to study the effect of Ag on the electronic structure and molecular properties of the [Au3(HNCOH)3] complex. All the structures are planar. Just the H (hydrogen) atoms are not in the plane. The symmetry point group of these structures is C3h.
The ionization potential was found to decrease while the electron affinity was found to increase significantly on going from the [Au3(HNCOH)3] complex to other [AunAgm(HNCOH)3] (m + n = 3) complexes; the largest decrease is revealed for the [Ag3(HNCOH)3] complex. The [Au3(HNCOH)3] complex may protect certain cellular systems which have lower electron affinity and higher ionization potential than that of the studied structures against ionizing radiation and free electrons by ionizing itself or by accepting the electron.
We reported here the results of DFT calculations for the global reactivity descriptors. The results showed that the softest complex was [Ag3(HNCOH)3], while the maximum hardness was found for the [Au3(HNCOH)3] complex. The μ values indicated that the [Au3(HNCOH)3] complex was the least reactive in this series of compounds. Also the donor–acceptor interactions of complexes were investigated.
Acknowledgments
Financial support from the University of Mazandaran is gratefully acknowledged.