

1 Introduction
The amido complexes of late transition metals have attracted considerable interest because of their potential involvement as intermediates in metal-catalyzed carbon–nitrogen bond formation [1,2]. Recent studies on the chemistry of these complexes have focused mostly on arylamido complexes, in which the metal-amido bond may be stabilized by partial delocalization of the electron density on the amido nitrogen to an aryl substituent [2]. On the other hand, complexes with a parent-amido ligand (NH2) have attracted less attention [1,3–5]. Despite this, such species are important as feasible intermediates in metal-catalyzed amination reactions with ammonia [6]. Furthermore, considering the importance of palladium as a primary metal used in homogeneous catalysis, it is essential to develop the chemistry with this metal [6a,7].
Monomeric amido complexes have strong affinity to undergo substitutional dimerization or oligomerization, yielding amido-bridged species, which are particularly crucial for coordinatively unsaturated complexes. Therefore, a common synthetic strategy for preparing monomeric metal amides is to use a sterically hindered ancillary ligand, such as bulky tertiary phosphines and chelates including pincer-type ligands, to avoid substitutional dimerization [5–7]. An earlier report on the synthesis and thermal stability of a dimethylamido palladium(II), Pd(2,6-(Ph2PCH2)2C6H3)(NMe2), showed that the monomeric amido complex with a terdentate PCP pincer is resistant to β-hydrogen elimination and thermal decomposition at low temperatures of less than −10 °C [8a]. A previous study on the regiospecific reactivity of an arylamido platinum(II) containing the same pincer Pt(2,6-(Ph2PCH2)2C6H3)(NH(Tol-p)) toward the CC bond of acrylonitrile to yield the addition product of aminoalkyl complex, Pt(2,6-(Ph2PCH2)2C6H3)(CH(CN)CH2NH(Tol-p)) provided a mechanistic information on one of the key steps in the catalytic hydroamination of acrylonitrile with NH2(Tol-p) [8b].
This article reports on a series of dimeric and monomeric parent-amido (NH2) complexes of palladium(II) with an ancillary ligand framework of a different coordination mode as part of an ongoing study of the stability and reactivity of the parent-amido complexes. In the present study for the synthesis of a monomeric parent-amido palladium(II) complex, a sterically hindered cyclohexyl derivative of PCP pincer was used to preclude substitutional dimerization. The title complex exhibited unique reactivity toward activated olefins and acetylenes to produce regio- and diastereospecific aminated derivatives of palladium(II), respectively, via nucleophilic attack of the coordinated NH2. In this study, the catalytic hydroamination of olefins with ammonia in the presence of the relevant title complex was also examined. The mechanistic feature of the catalytic hydroamination is discussed in regard to the probed reaction profiles in palladium(II) aminated derivatives.
2 Results and discussion
2.1 NH2-bridged dimeric complexes
2.1.1 anti-[Pd(PEt3)(Ph)(μ-NH2)]2 (1) and [Pd(dppe)(NH2)]2(OTf)2 (2)
The reaction of a tetrahydrofuran (THF) solution of trans-[Pd(PEt3)2(Ph)(NH3)]OTf with NaNH2 at ambient temperature produced a gray suspension containing a single compound of palladium(II) along with dissociated triethylphosphine, as evidenced by the 31P{1H} NMR spectroscopy (Scheme 1). A pure off-white palladium(II) dimer anti-[Pd(PEt3)(Ph)(μ-NH2)]2 (1) was obtained from an n-pentane extract of the residues resulting from the reaction mixture. The 31P{1H} NMR spectrum of complex 1 in d6-benzene showed a single peak at δ 20.2, which is indicative of a single compound in solution. In the 13C{1H} NMR spectrum, the methylene carbon attached directly to the phosphorus resonates at δ 15.61 (1J(CP) = 26.7 Hz) as a doublet. In the 1H NMR spectrum, the bridged-amide protons (μ-NH2) were observed at δ −1.35 as a broad signal.
Synthesis of anti-[Pd(PEt3)(Ph)(μ-NH2)]2 (1).
Attempts to prepare a parent-amido complex with a chelate ligand dppe under similar reaction conditions as for complex 1 were unsuccessful. The reaction of [Pd(dppe)(NH3)2](OTf)2 with NaNH2 in THF at ambient temperature resulted in a deep red solution containing a palladium(0) complex Pd(dppe), which was trapped in situ by treatment with dimethyl acetylenedicarboxylate (dmad) to yield Pd(dppe)(dmad), as verified by the 31P{1H} NMR spectrum, showing a single resonance at δ 48.0 in d6-benzene [9]. The formation of Pd(dppe) from the reaction is likely a consequence of the feasible generation of a monomeric bis(amido) species Pd(dppe)(NH2)2, which undergoes facile reductive elimination by releasing hydrazine (N2H4), although electron transfer from a base (NH2−) cannot be excluded. Previous studies showed that hydrazine formation from the oxidation of coordinated ammonia to a diruthenium complex [10] or from adsorbed NH2 species on transition metal surfaces [11] is feasible. On the basis of the above result, the reaction was performed at low temperatures. The reaction of [Pd(dppe)(NH3)2](OTf)2 with NaNH2 in THF at −78 °C (dry ice/acetone) afforded a dicationic amido-bridged dimeric complex [Pd(dppe)(μ-NH2)]2(OTf)2 (2) in 67% isolated yield (Scheme 2). The 1H and 31P{1H} NMR spectra of 2 in CDCl3 exhibit an upfield broad signal at δ −0.19 corresponding to bridged-amide protons (μ-NH2) and a single 31P resonance at δ 53.2, respectively. The counter anion CF3SO3− can be established by the ν(SO3) at 1154 and 1266 cm−1 in the IR and the 19F{1H} NMR resonance at δ −78.9. The molar conductivity measurement for 2 in nitromethane reveals that the complex is a 1:2 electrolyte (ΛM = 184 Ω−1 cm2 mol−1, [2] = 0.50 × 10−3 M). The microanalytical data for 1 and 2 are consistent with the dimeric formulation (see Section 4).
Synthesis of [Pd(dppe)(μ-NH2)]2(OTf)2 (2).
2.1.2 Molecular structures of anti-[Pd(PEt3)(Ph)(μ-NH2)]2 (1) and [Pd(dppe)(μ-NH2)]2(OTf)2 (2)
The molecular structures of 1 and 2 were determined by single-crystal X-ray crystallography (Figs. 1 and 2). Single crystals suitable for X-ray crystallography were grown either by the slow evaporation of an n-hexane solution of 1 or by the slow diffusion of a mixture of diethyl ether and n-pentane (1:1) into a dichloromethane solution of 2. Both complexes 1 and 2 crystallize in the monoclinic space group P21/n. For 1, there are two crystallographically independent but chemically identical molecules. The two independent molecular structures of 1 contain the respective Pd2N2 rings puckered with the dihedral angles of 53.1(2)° and 55.2(2)°. The Pd–Pd distances are 2.9594(10) and 2.9401(9) Å, respectively. The cation of complex 2 contains a puckered Pd2N2 ring with a dihedral angle of 28.8(0.15)°, which is smaller than that of 1. Therefore, the Pd–Pd distance 3.0669(8) Å in 2 is slightly larger. The observed Pd–Pd distances for 1 and 2 indicate that there is no bonding between Pd and Pd [12], which are comparable to the Pt–Pt distances in the range of 3.087–3.134 Å, observed in the NH2-bridged platinum(II) dimers, anti-[Pt(POPh2)(PMePh2)(μ-NH2)]2 [4b], [Pt(PMe2Ph)2(μ-NH2)]22+ [4c], and anti-[PtMe(PPh3)(μ-NH2)]2 [5a]. The Pd–N bond lengths for 1 and 2 are in the range of 2.067(7)–2.127(7) Å. For the angles within the bridge about palladium and nitrogen, the respective Pd–N–Pd and N–Pd–N bond angles are 88.8(3)–90.4(3)° and 77.7(3)–78.3(3)° for 1 and 95.0(2)–95.6(2)° and 79.2(2)–79.6(2)° for 2. The selected bond lengths (in angstroms) and angles (in degrees) for complexes 1 and 2 are given in Tables 1 and 2, respectively. For reference, all the X-ray crystallographic data of 1 and 2 are provided in Supplementary Data, including the crystal data, atomic coordinates and equivalent isotropic displacement parameters, bond lengths and angles, anisotropic displacement parameters, and hydrogen coordinates and isotropic displacement parameters.
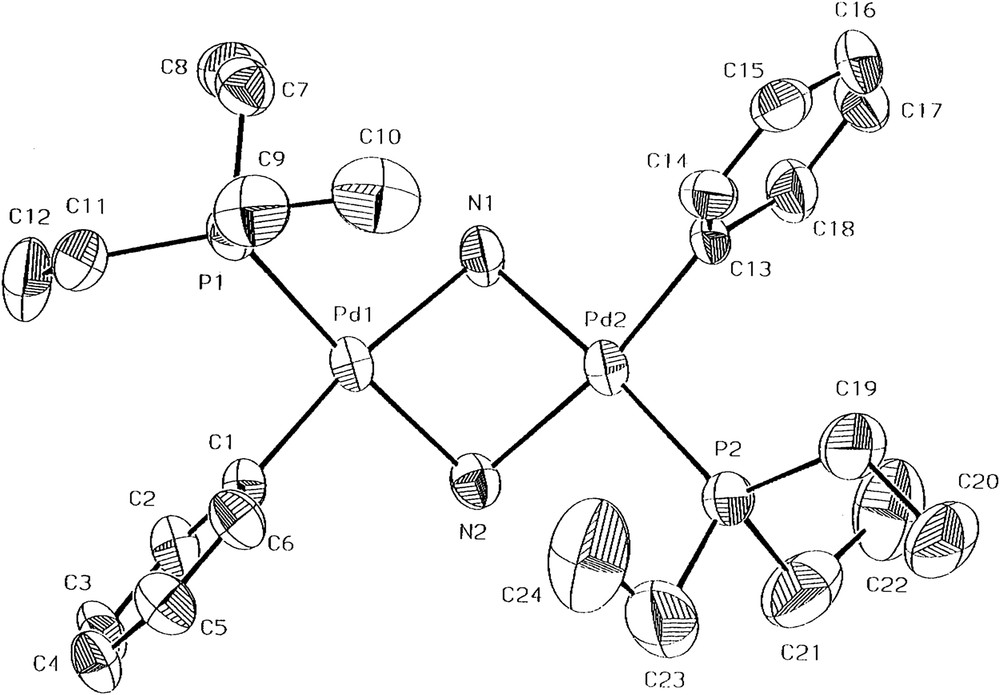
The molecular structure for one of the two crystallographically independent molecules of 1 shown with 40% thermal ellipsoids. For clarity hydrogen atoms have been omitted.
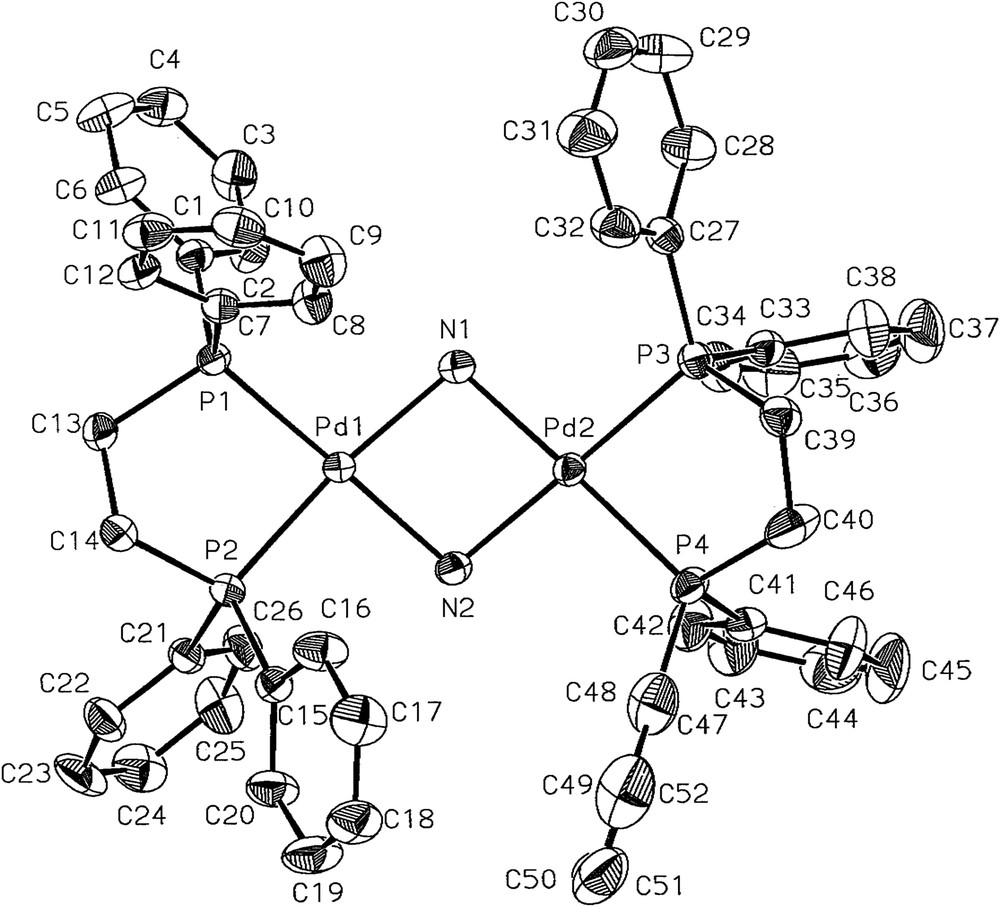
The structure of the cation of 2 shown with 40% thermal ellipsoids. The counter anions 2[CF3SO3]− and hydrogen atoms were omitted for clarity.
Selected bond lengths (Å) and angles (°) for one of the two crystallographically independent molecules of 1.
| Pd(1)–N(1) | 2.104(7) | Pd(1)–C(1) | 2.000(9) |
| Pd(1)–N(2) | 2.085(7) | Pd(2)–C(13) | 1.998(9) |
| Pd(2)–N(1) | 2.067(7) | Pd(1)–P(1) | 2.233(3) |
| Pd(2)–N(2) | 2.127(7) | Pd(2)–P(2) | 2.235(3) |
| Pd(2)–N(1)–Pd(1) | 90.4(3) | N(2)–Pd(1)–P(1) | 174.3(2) |
| Pd(1)–N(2)–Pd(2) | 89.3(3) | N(1)–Pd(1)–P(1) | 99.5(2) |
| N(2)–Pd(1)–N(1) | 77.8(3) | C(13)–Pd(2)–N(1) | 92.8(3) |
| N(1)–Pd(2)–N(2) | 77.7(3) | C(13)–Pd(2)–N(2) | 170.4(3) |
| C(1)–Pd(1)–N(2) | 93.5(3) | C(13)–Pd(2)–P(2) | 91.7(3) |
| C(1)–Pd(1)–N(1) | 171.3(3) | N(1)–Pd(2)–P(2) | 175.1(2) |
| C(1)–Pd(1)–P(1) | 89.0(3) | N(2)–Pd(2)–P(2) | 97.7(2) |
Selected bond lengths (Å) and angles (°) for 2.
| Pd(1)–N(1) | 2.089(6) | Pd(1)–P(1) | 2.255(2) |
| Pd(1)–N(2) | 2.067(6) | Pd(1)–P(2) | 2.240(2) |
| Pd(2)–N(1) | 2.069(6) | Pd(2)–P(3) | 2.240(2) |
| Pd(2)–N(2) | 2.075(6) | Pd(2)–P(4) | 2.244(2) |
| Pd(2)–N(1)–Pd(1) | 95.0(2) | N(1)–Pd(1)–P(1) | 101.3(2) |
| Pd(1)–N(2)–Pd(2) | 95.6(2) | P(2)–Pd(1)–P(1) | 85.69(7) |
| N(2)–Pd(1)–N(1) | 79.6(2) | N(1)–Pd(2)–P(3) | 98.8(2) |
| N(1)–Pd(2)–N(2) | 79.9(2) | N(2)–Pd(2)–P(3) | 174.6(2) |
| N(2)–Pd(1)–P(2) | 93.4(2) | N(1)–Pd(2)–P(4) | 176.9(2) |
| N(1)–Pd(1)–P(2) | 173.0(2) | N(2)–Pd(2)–P(4) | 98.1(2) |
| N(2)–Pd(1)–P(1) | 179.1(2) | P(3)–Pd(2)–P(4) | 83.03(8) |
The amido-bridged dimeric complexes 1 and 2 in the solid state and in solution are stable in air for a period of days. No reactions with unsaturated molecules, such as CO2, CH2CHCN, dimethyl acetylenedicarboxylate, diethyl maleate, and cyclohexene were observed, indicating a lack of nucleophilicity of the bridging NH2. Considering the observed stability, it is unanticipated that although the few analogous platinum(II) dimers have been reported [4,5a], the title complexes are, to the best of our knowledge, the first structurally determined NH2-bridged palladium(II) dimers [13].
2.2 Monomeric amido complex Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (3) and its reactivity
Because the formation of amido-bridged dimeric species resulted from substitutional dimerization, a palladium(II) ammine complex containing a trans-spanning terdentate ligand was as a synthetic precursor for the preparation of a monomeric parent-amido palladium(II) complex. In a similar synthetic manner, as for 1, treatment of the cationic ammine complex [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)](OTf) with NaNH2 afforded the monomeric palladium(II) amide Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (3). When NaH was used as a deprotonating agent in place of NaNH2, the reaction yielded a mixture of two products, including mainly complex 3 along with a small amount of palladium(II) hydride (2,6-(Cy2PCH2)2C6H3)PdH. The formation of the palladium(II) hydride was verified by the observation of upfield hydride resonance at δ −3.73 (t, 2J(PH) = 16 Hz) in the 1H NMR spectrum [8a,14]. On the other hand, the metathetical replacement of the triflate ligand in Pd(2,6-(Cy2PCH2)2C6H3)(OTf) with NaNH2 yielded the monomeric palladium(II) amide, exclusively. Scheme 3 represents the used synthetic routes. The formation of Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (3) was established by the upfield shift of NH2 resonance at δ −0.20 and the relative intensity of the methylene protons in the pincer ligand and NH2 resonances in the 1H NMR spectrum. On diluting a d6-benzene solution of 3, the NH2 resonance shifts upfield, implicating intermolecular hydrogen bonding in the complexes.
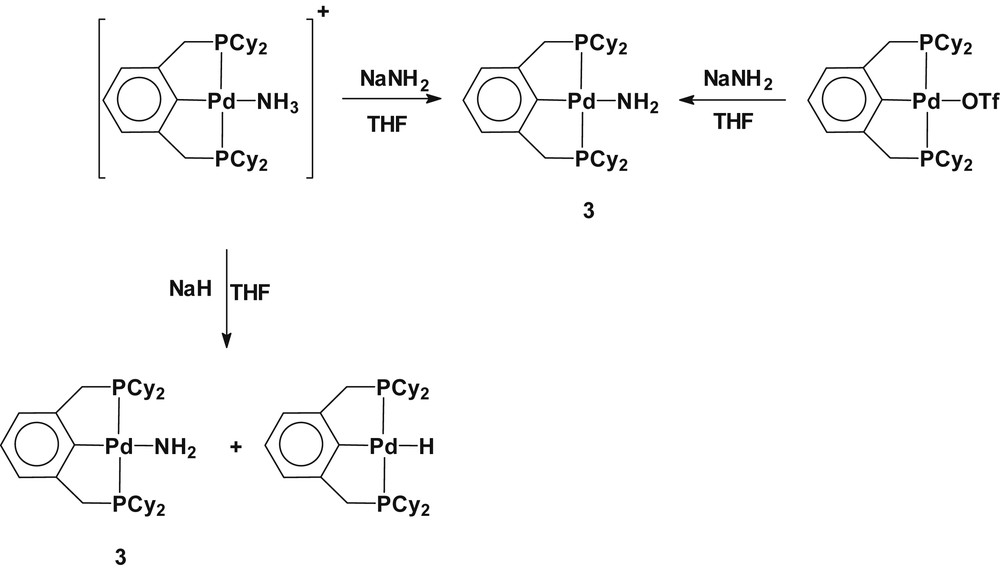
Synthesis of Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (3).
Complex 3 is highly sensitive to air and moisture. The monomeric amido complex slowly (over the course of days) reacts with trace amounts of water to yield a hydroxo complex Pd(2,6-(Cy2PCH2)2C6H3)(OH) (4) (Scheme 4). The hydroxo complex can be verified by the observation of upfield triplet resonance for the coordinated hydroxide (Pd–OH) at δ −1.22 with the small value of 3J(PH) = 3.3 Hz in the 1H NMR spectrum along with a single 31P NMR resonance at δ 48.7 [15,16]. For further characterization, the hydroxo complex Pd(2,6-(Cy2PCH2)2C6H3)(OH) was prepared independently by metathesis from Pd(2,6-(Cy2PCH2)2C6H3)(OTf) and KOH in THF (see Section 4). The observed σ-ligand exchange reaction of 3 with H2O compares well with a previous report on the reactivity of a parent-amido Ni(II) complex toward H2O [17]. Exposing the complex 3 to an excess of water resulted in the immediate conversion of the complex into two species, which exhibit 31P NMR resonance at δ 52.7 and δ 54.7 (predominant), respectively. The former is assigned to the cationic ammine species [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)]+, which was established by the observation of an identical 31P{1H} NMR resonance with the complex [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)](OTf). The latter can be attributed to the cationic aqua complex [Pd(2,6-(Cy2PCH2)2C6H3)(OH2)]+, as evidenced by the identical 31P{1H} NMR resonance at δ 54.7 with a complex prepared from Pd(2,6-(Cy2PCH2)2C6H3)(BF4) and H2O in d6-benzene. No formation of the hydroxo complex was observed from the reaction of 3 with an excess of water. Therefore, the observed results can be explained by a sequence of reactions involving the σ-ligand exchange of the amido complex with H2O to give the hydroxo complex, which then protonates from excess water molecules to generate the observed cationic aqua complex (Scheme 4). The formation of a small amount of cationic ammine complex can be attributed to the ligand substitution of the cationic aqua complex with the liberated NH3.
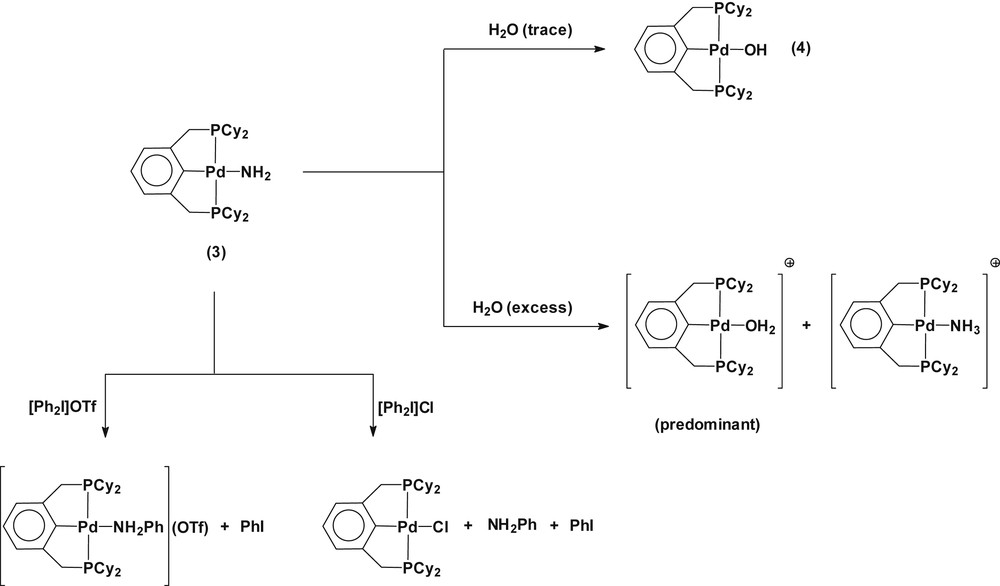
Reactions of 3 with H2O and [Ph2I]X (X = OTf, Cl).
A d6-benzene solution of 3 reacted with diphenyliodonium triflate ([Ph2I]OTf) to give an N-phenylated amine complex [Pd(2,6-(Cy2PCH2)2C6H3)(NH2Ph)]OTf (Scheme 4). The formation of the cationic aniline complex was confirmed by its independent preparation from a reaction of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) with NH2Ph in d6-benzene (see Section 4). The reaction of a d6-benzene solution of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) with NH2Ph produced [Pd(2,6-(Cy2PCH2)2C6H3)(NH2Ph)]OTf, displaying a rather broad single 31P{1H} NMR resonance at δ 52.1 in accordance with that of the species generated by the reaction of 3 with [Ph2I]OTf. An attempt to isolate the cationic aniline species was unsuccessful because of dissociation of the coordinated aniline, converting to Pd(2,6-(Cy2PCH2)2C6H3)(OTf). In the meanwhile, reaction of 3 with the chloride salt of [Ph2I]Cl generated Pd(2,6-(Cy2PCH2)2C6H3)Cl along with the released NH2Ph, which was identified by GC–MS (NH2Ph: m/z = 93, 66, 39).
The amido complex 3 reacted readily with phenylacetylene (HCCPh) to quantitatively yield a palladium(II) acetylenide Pd(2,6-(Cy2PCH2)2C6H3)(CCPh) (5) along with the liberation of ammonia, which was detected in the 1H NMR spectrum (broad 1:1:1 triplet, at δ −0.15, 1J(NH) = 41 Hz) (Scheme 5) [17]. Complex 5 was prepared from preparative scale experiment and fully characterized (see Section 4). Complex 5 shows its characteristic absorption peak for the ν(CC) at 2096 cm−1 in the IR spectrum, and the 13C{1H} NMR resonances for the coordinated acetylenide at δ 113.1 (t, Pd–CC, 2J(CP) = 12 Hz) and at δ 118.3 (Pd–CC). These spectroscopic data are in good agreement with those reported for trans-Pd(PEt3)2(C6H4–Me-p)(CCPh) at 2092 cm−1 in the IR, and at δ 119.8 (t, Pd–CC, 2J(CP) = 20 Hz) and at δ 111.3 (Pd–CC) in the 13C{1H} NMR spectrum [18]. The observed reactivity for 3 toward phenylacetylene reveals that the coordinated NH2 is highly basic for the complex to undergo σ-ligand exchange via protonation rather than the migratory insertion of the CC triple bond into the Pd–NH2 via nucleophilic attack on the sp-carbon of HCCPh. In view of the thermodynamic aspect, the driving force for the formation of the palladium(II) acetylenide parallels the relative ground state stability in the order of M-C(sp) > M-NH2(sp3) or M-C(sp2, vinylic) [19,20].
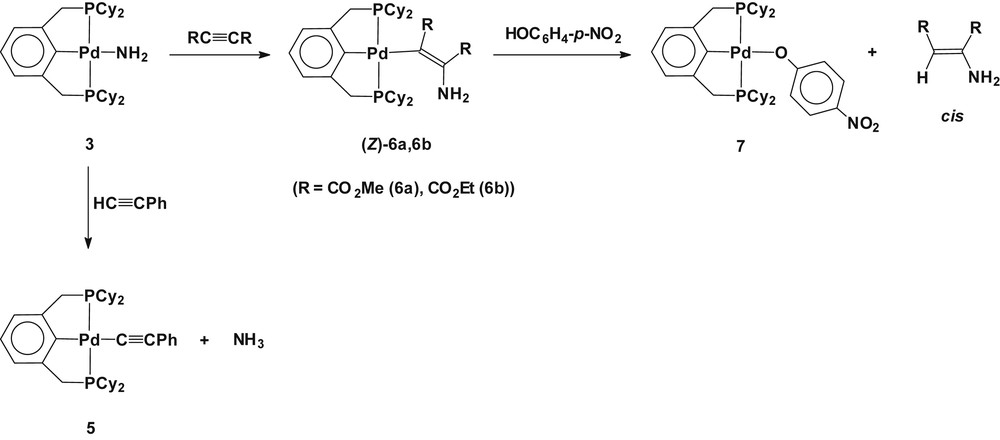
Reactions of 3 with RCCR (R = CO2Me, CO2Et) and HCCPh.
Complex 3 undergoes clean reactions with activated acetylenes, such as dialkyl acetylenedicarboxylate (RCCR; R = CO2Me, CO2Et), to produce the diastereospecific insertion derivatives of Pd(2,6-(Cy2PCH2)2C6H3)(CRCR(NH2)) (R = CO2Me (6a), CO2Et (6b)) (Scheme 5). Complexes 6a and 6b have been fully characterized by multinuclear NMR (1H, 13C, and 31P) and fast atom bombardment mass spectrometry (FABMS). The formation of aminovinylic complexes was verified readily by the observation of resonances for the corresponding two methyl and two ethyl groups for the moiety of Pd(CRCR(NH2)) (R = CO2Me, CO2Et) in the 1H and 13C{1H} NMR spectra. The amino protons (NH2) resonate around δ 3.9 as a broad peak, which was confirmed by the D2O exchange experiment. In the 1H NMR spectra, the virtual triplet methylene protons (PCH2) in the PCP pincer ligand were observed to be diastereotopic at δ 3.1 and 3.3 as a doublet of triplet (2J(HaHb) = 17.6 Hz,∣2J(PH) + 4J(PH)∣ ≅ 8 Hz), respectively. In solution, no coordination of the amino group in the complex was observed, as evidenced by 1H and 31P{1H} NMR spectroscopy; all signals were intact on the addition of coordinating molecules, such as pyridine and PPh3, to the d6-benzene solution of 6a and 6b. Endeavors to isolate complexes 6a and 6b from solution were futile because of the high solubility in most organic solvents including n-pentane. Therefore, the removal of all volatiles from solution under high vacuum resulted in yellow solids, which afforded satisfactory FABMS data, displaying a parent molecular ion peak in good accordance with the calculated molecular weight in addition to the expected peaks because of molecular fragmentation. Although the absolute diastereomeric configuration of 6a and 6b could not be determined because of the failure to obtain suitable single crystals for an X-ray structural study, the stereochemistry of complexes 6a and 6b (Z)-isomer was assigned by performing subsequent reactions. A further reaction of 6a and 6b with an acidic phenol HOC6H4-p-NO2 produced only a single isomeric product cis-CHRCR(NH2) (R = CO2Me, CO2Et) with the retention of the configuration, along with the palladium(II) p-nitrophenoxide Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4-p-NO2) (7) (Scheme 5). The formation of Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4-p-NO2) (7) was verified by its independent synthesis from the reaction of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) and Na(OC6H4-p-NO2) in THF (see Section 4). As a control, reaction of a d6-benzene solution of dialkyl acetylenedicarboxylate with gaseous ammonia produces an isomeric mixture of cis- and trans-(CHRCR(NH2)). The diastereoselective formation of (Z)-Pd(2,6-(Cy2PCH2)2C6H3)(CRCR(NH2)) (6a, 6b) resulting from the reaction of Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (3) and RCCR implies that the insertion reaction presumably involves a concerted pathway, apparently excluding dissociative nucleophilic addition. The observed reactivity for the palladium(II) aminovinylic complexes toward HOC6H4-p-NO2 in the ligand exchange reaction reveals that the σ-vinylic ligand is more basic than the σ-phenoxide, liberating stereoselective olefinic derivatives with retention of configuration. In view of the importance of using ammonia as a building block for the production of nitrogen-containing compounds [6], this study on the reaction profile of the syn-insertion of activated acetylene into the Pd–NH2 bond in the title complex is noteworthy. Prior examples of the syn-insertion of alkynes and alkenes into the Pd–N bonds have rarely been found in arylamido complexes [21].
The insertion reaction of the CC bond of dialkyl maleate (cis-(CO2R)CHCH(CO2R)) (R = CH3, CH2CH3) into the Pd–NH2 bond of complex 3 was attempted, resulting in no formation of the insertion derivatives. Instead, dialkyl maleate isomerizes into dialkyl fumarate (trans-(CO2R)CHCH(CO2R)) in the presence of a catalytic amount of 3, as evidenced by 1H NMR spectroscopy. The reaction proceeds rather slowly at ambient temperature, resulting in an isomeric trans/cis ratio of 0.8 (for 4 h) and 8.9 (for 24 h) for the dimethyl derivative (CO2Me)CHCH(CO2Me). The conversion of dialkyl maleate (cis-isomer) into dialkyl fumarate (trans-isomer) was completed over a period of 5 days at ambient temperature. Similarly, a reaction of the parent-amido complex 3 with cis-stilbene (cis-CHPhCHPh) was attempted, resulting in no reaction at ambient temperature. On the other hand, at an increased temperature of 50 °C, the complex 3 catalyzed the isomerization of cis-stilbene to trans-stilbene, resulting in a trans/cis ratio of 2.0 for 2 h. No insertion product was observed in the course of the catalytic isomerization. Isomerization did not occur in the absence of the parent-amido complex. As a control, no isomerization was observed in the reaction via a base, such as NH3 or p-toluidine. The migratory insertion of the cis-isomers into the Pd–NH2 bond followed by deinsertion cannot lead to the formation of trans-isomers because rotation of the C–C single bond in the insertion species erythro-Pd(CHXCHXNH2) cannot invert the configuration of the β-carbon atom. Therefore, the observed diastereomeric isomerization catalyzed by complex 3 can most likely be explained by a sequence of reactions as follows. Migratory insertion of the CC bond of the cis-isomers into the Pd–NH2 bond leads to a transient aminoalkyl species erythro-Pd(CHXCHXNH2), which undergoes β-H elimination to generate a Pd(II)-hydride and cis-CHXCXNH2. The Pd(II)-hydride reacts with cis-CHXCXNH2 with the opposite regiochemistry to generate Pd(C(NH2)XCH2X), in which the rotation of the C–C bond followed by elimination of the other β-H leads to the generation of a Pd(II)-hydride again along with trans-CHXCXNH2. The Pd(II)-hydride reacts with trans-CHXCXNH2 to afford threo-Pd(CHXCHXNH2), from which the isomerized trans-olefin can be released via deinsertion to regenerate complex 3. A previous theoretical study demonstrated that β-H elimination from the ammonioalkyl complexes of group 10 metals is kinetically competitive in the catalytic hydroamination of ethylene with ammonia [22].
A stoichiometric reaction of 3 with acrylonitrile (CH2CHCN) generated a metastable insertion product, Pd(2,6-(Cy2PCH2)2C6H3)(CH(CN)CH2NH2), which was detected by 1H and 31P NMR spectroscopy (Scheme 6). A d6-benzene solution of the reaction mixture showed anticipating proton resonances for the methyne (CH) and methylene (CH2) protons of the Pd–CH(CN)CH2NH2 moiety around at δ 2.2, 3.1, and 3.5 as multiplets in the 1H NMR spectrum and a new single resonance at δ 49.0 in the 31P{1H} NMR spectrum. The observed proton resonances of the Pd(CH(CN)CH2NH2) moiety are in good agreement with those of the analogous aminoalkyl platinum(II) complex, Pt(2,6-(Ph2PCH2)2C6H3)(CH(CN)CH2NHTol-p) at δ 2.75 (m, 1H, CH), 3.08 (m, 1H, CHa), and 3.45 (m, 1H, CHb), respectively [8b]. Attempts to isolate the insertion product from the solution were unsuccessful because of decomposition to a couple of unidentified species. The reaction of 3 with an excess of acrylonitrile slowly produced polymeric species of acrylonitrile at ambient temperature, which precipitated in solution. The 31P NMR resonance at δ 49.0 because of an aminoalkyl palladium(II) derivative observed at an early stage of the reaction mostly disappeared to generate a couple of new 31P NMR resonances, one at δ 59.2 corresponding to a cationic acrylonitrile complex [Pd(2,6-(Cy2PCH2)2C6H3)(CH2CHCN)]+ (vide infra, catalytic hydroamination of olefins with ammonia). The isolated polymer from the reaction mixture shows the characteristic absorption peaks for the ν(CN) at 2245 and 2204 cm−1 and for the ν(NH2) at 3300 and 3360 cm−1 in the IR spectrum. As a control, the reaction of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) with an excess acrylonitrile generated [Pd(2,6-(Cy2PCH2)2C6H3)(CH2CHCN)](OTf) without producing any polymeric species. Therefore, the observed results can be explained by a sequence of reactions involving migratory insertion of the CC bond of acrylonitrile into the Pd–NH2 bond at an early stages of the reaction to generate a metastable aminoalkyl palladium(II) complex, which undergoes the consecutive insertion of acrylonitrile into the Pd–C bond followed by the liberation of polymeric species (Scheme 6). The present result is comparable to a previous study on an aminoalkyl platinum(II) complex, which exhibited no further reactivity toward acrylonitrile, and is in line with the migratory insertion barriers for the σ-alkyl complexes in the order Pd(II) < Pt(II) [23]. Attempted reactions of 3 with unactivated olefins, such as 1-hexene, cyclohexene, and styrene, were futile presumably because of the insufficient nucleophilicity of the coordinated amide along with the rigidity of the sterically hindered pincer ligand in the title complex.
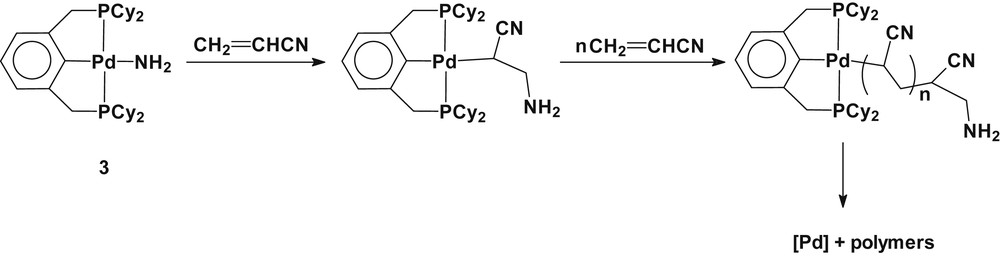
Reaction of 3 with acrylonitrile.
2.3 Catalytic hydroamination of olefins with ammonia
The catalytic hydroamination of various olefins with NH3 was examined in the presence of Pd(2,6-(Cy2PCH2)2C6H3)(OTf). Unless otherwise noted, the catalytic reactions were typically performed at a fixed concentration of olefin substrates (100 equiv) with various concentrations of NH3 in the presence of a catalyst (1.0 equiv) for 12 h at 60 °C. The results are summarized in Table 3. For the catalytic hydroamination of acrylonitrile (CH(CN)CH2) with NH3 (entries 1–5), a range of hydroaminated products of primary (CH2(CN)CH2NH2), secondary ((CH2(CN)CH2)2NH), and tertiary ((CH2(CN)CH2)3N) amines with different molar ratios were produced in an overall yield of more than 99%, indicating that the produced amine derivatives compete with ammonia as nucleophilic substrates. The catalytic overall yield increased considerably with increasing concentration of NH3 (entries 2–5). The mole percent ratios of the produced amine derivatives (primary/secondary) generally increased with increasing concentration of ammonia (entries 3–5). On the other hand, at a relatively low concentration of ammonia compared to acrylonitrile (NH3/acrylonitrile = 4/5), the tertiary amine derivative ((CH2(CN)CH2)3N), 11 mol %) was produced along with the predominant formation of secondary amine ((CH2(CN)CH2)2NH, 57 mol %) (entry 2). Hydroaminated products were not produced at a significantly low ratio of ammonia-to-acrylonitrile (NH3/acrylonitrile = 1/5, entry 1). For the hydroamination of methyl acrylate (CH2CHCO2Me) with ammonia, the reaction rate considerably decreased in comparison with those from acrylonitrile with ammonia, resulting in an overall yield of 47% at 80 °C for 18 days (entry 6). The resulting products were only a mixture of the secondary ((CH2(CO2CH3)CH2)2NH, 83 mol %) and the tertiary ((CH2(CO2CH3)CH2)3N, 17 mol %) amines without the primary amine (CH2(CO2CH3)CH2NH2), apparently revealing that the generated primary amine (more basic than NH3) subsequently attacks methyl acrylate to produce the secondary amine predominantly. For the hydroamination of crotononitrile (MeCHCHCN, a mixture of cis- and trans-isomer with a ratio of ca. 1.0) with ammonia (entries 7–9), the reaction rate was much slower than that from acrylonitrile (or methyl acrylate) with ammonia. On the other hand, the resulting product was exclusively the primary amine (CH2(CN)CH(CH3)NH2). No formation of the secondary and tertiary amines from the catalytic reaction can be attributable mostly to the steric effect of the produced primary amine. The catalytic hydroamination of other unsaturated hydrocarbons, such as cyclohexene, styrene, 1-hexene, 1-hexyne, and diphenylacetylene, with ammonia was attempted without success.
Hydroamination of olefins with NH3 catalyzed by Pd(2,6-(Cy2PCH2)2C6H3)(OTf).a
| Entry | Olefin | NH3 (molar equivb or atm) | Overall yield (%)c | HA products (mol %)c | ||
| n = 1 | n = 2 | n = 3 | ||||
| 1 | CH2CHCN | 20d | 0e | |||
| 2 | CH2CHCN | 80d | 12 | 32 | 57 | 11 |
| 3 | CH2CHCN | 100d | 17 | 21 | 79 | 0 |
| 4 | CH2CHCN | 120d | 20 | 29 | 71 | 0 |
| 5 | CH2CHCN | 7 atmf | >99 | 84 | 15 | 0 |
| 6 | CH2CHCO2Me | 100d | 47g | 0 | 83 | 17 |
| 7 | MeCHCHCNh | 300d | 11i | 100 | 0 | 0 |
| 8 | MeCHCHCN | 7 atmf | 9 | 100 | 0 | 0 |
| 9 | MeCHCHCN | 25 atmf | 29 | 100 | 0 | 0 |
a Reaction conditions: Pd(2,6-(Cy2PCH2)2C6H3)(OTf) (3.0 mg, 4.0 × 10−3 mmol), [olefin] = 100 × [Pd].
b Molar equivalent to [Pd].
c 1H NMR integration or GC-based yield for 12 h at 60 °C, unless otherwise noted. The overall yield is relative to the olefin.
d In a vacuum NMR tube.
e For 24 h.
f In a high pressure reactor.
g For 18 days at 80 °C.
h 150 equiv.
i For 4.5 days.
For a mechanistic feature, further experiments were conducted by NMR spectroscopy. The 1H NMR spectrum of a CDCl3 solution of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) in the presence of acrylonitrile with a slight excess displayed broad resonances at ambient temperature. The variable temperature experiments for the 1H NMR spectra of the CDCl3 solution revealed a dynamic process attributable to the rapid ligand exchange between the triflate and acrylonitrile in the complex (Fig. 3). The rate of exchange process is fast on the NMR time scale at >10 °C and decreases at low temperatures. The proton resonances for the coordinated acrylonitrile cleanly resolved at −22 °C, indicating the formation of a stable olefinic complex in solution. On the other hand, on the addition of a large excess amount of acrylonitrile (ca. 30 equiv) into the solution, the proton resonances for the free acrylonitrile displayed well-resolved sharp peaks at ambient temperature. The 31P{1H} NMR spectrum of this solution exhibited a sharp single resonance at δ 59.4, which is assignable to the cationic acrylonitrile complex [Pd(2,6-(Cy2PCH2)2C6H3)(CH2CHCN)](OTf) analogous to the reported platinum(II) complexes [8b]. Attempts to isolate the olefinic complex from the solution were unsuccessful because of conversion into the palladium triflate Pd(2,6-(Cy2PCH2)2C6H3)(OTf). The resulting cationic olefin complex is stable in solution in the presence of excess acrylonitrile for a period of several days at ambient or increased temperature (60 °C) without producing polymeric species or any other side products. The addition of NH3 (excess) to this solution, however, immediately generated the cationic ammine complex [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)](OTf) along with the acrylonitrile complex. Subsequently, the reaction mixture slowly undergoes a catalytic reaction to produce a mixture of hydroaminated products. No polymeric species was produced from the catalytic reaction.
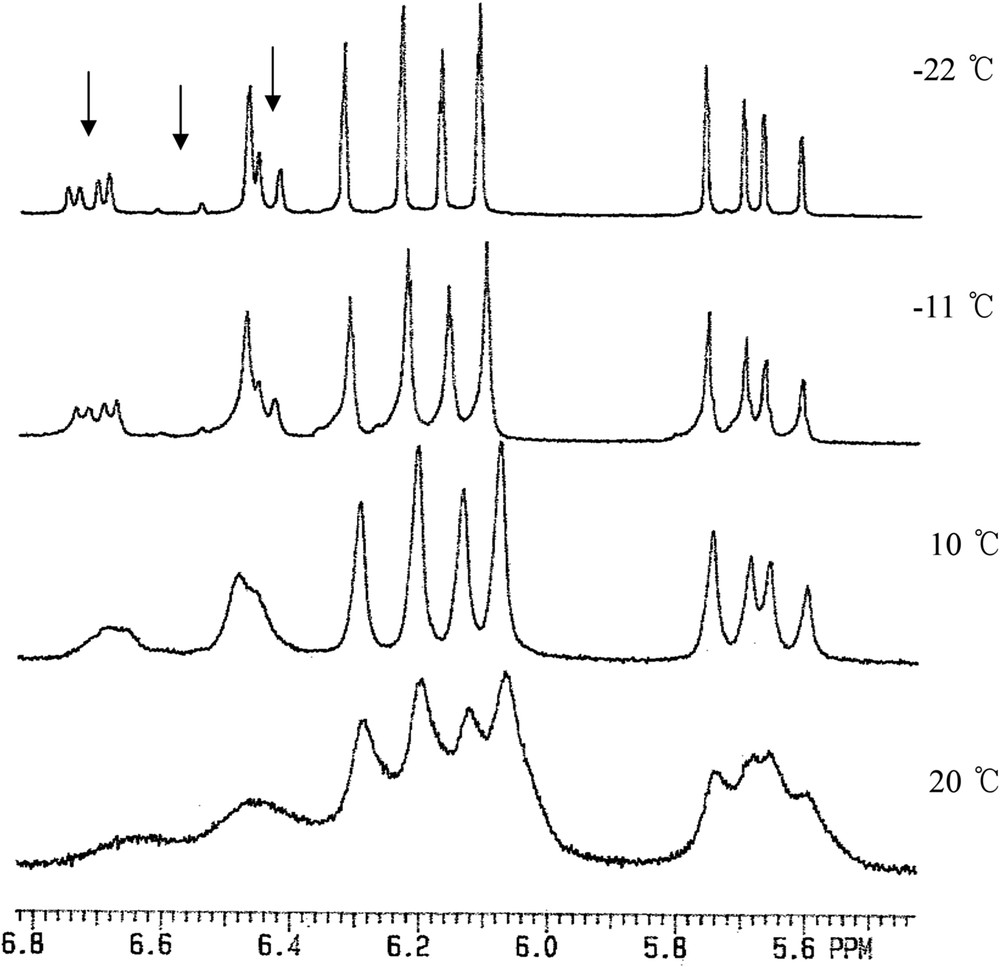
The VT 1H NMR spectra of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) in the presence of CH2CHCN in CDCl3 show that there is a fast ligand exchange between the triflate and acrylonitrile in the complex on the NMR time scale at >10 °C. The resonances of the coordinated acrylonitrile resolve at −22 °C, being marked with arrows.
In the catalytic reaction, a mechanism involving the coordination of ammonia to a palladium(II) sphere followed by deprotonation to generate a parent-amido complex can be excluded because polymeric species, which could be derived from a migratory insertion derivative of palladium(II), that is, an aminoalkyl complex, was not formed from the catalytic reaction (vide supra). A further experiment to examine the ability of ammonia to deprotonate the coordinated ammonia in [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)](OTf) was conducted. The treatment of the amido complex 3 with NH4OTf in d6-benzene produced [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)](OTf) and NH3, quantitatively, as evidenced by 1H and 31P{1H} NMR spectroscopy, apparently excluding the involvement of a Pd(II) amido species in the catalytic reaction. Therefore, the present catalytic reaction may proceed via the nucleophilic attack of ammonia on the precoordinated olefin to palladium(II) to generate a transient alkylammonium complex [Pd(2,6-(Cy2PCH2)2C6H3)(CH2(CN)CH2NH3)]+ in parallel to a previous report on catalytic hydroamination by platinum(II) catalysts [8b]. A high concentration of ammonia would act as a base to transfer a proton from the suggested species to produce hydroaminated products (pathway A in Scheme 7). The relative stabilities of the σ-donor amine versus the π-olefinic acrylonitrile complexes showed that acrylonitrile practically competes with amine in the coordination spheres of palladium(II) [24] and platinum(II) [8b,25]. The nucleophilic addition of amines on the coordinated olefin to palladium(II) complexes has been well recognized [26]. An alternative pathway for the catalytic reaction likely involves a 5-coordinate Pd(IV)-hydride species, from which C–H reductive elimination leads to hydroaminated products (pathway B in Scheme 7). Previous computational studies of the hydroamination of ethylene with ammonia or aniline catalyzed by d8-metal complexes illustrated that intramolecular transfer of an alkylammonium proton to the metal center to generate a 5-coordinate (16-electron) metal-hydride intermediate is energetically feasible [22,27]. Scheme 7 presents plausible reaction pathways for the observed catalytic hydroamination of olefins with ammonia in the presence of the title complex.
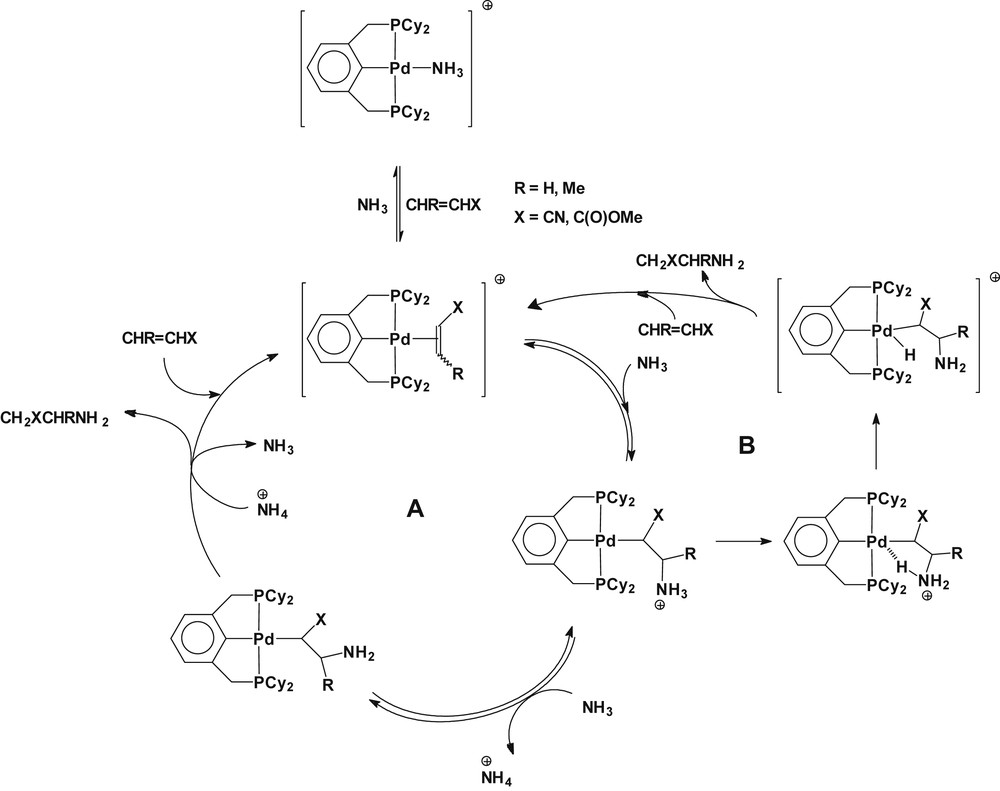
Plausible reaction pathways for the observed hydroamination of olefins with ammonia in the presence of Pd(2,6-(Cy2PCH2)2C6H3)(OTf), involving an aminoalkyl Pd(II) species (pathway A) or a Pd(IV) hydride (pathway B).
3 Summary
Novel parent-amido complexes of palladium(II) anti-[Pd(PEt3)(Ph)(μ-NH2)]2, [Pd(dppe)(μ-NH2)]2(OTf)2, and Pd(2,6-(Cy2PCH2)2C6H3)(NH2) were prepared from the respective ammine complexes by deprotonation of the coordinated ammonia. The amido-bridged dimeric complexes are inert to unsaturated molecules, such as CO2, activated olefins, and acetylenes, revealing a lack of nucleophilicity of the bridging NH2. The monomeric parent-amido complex Pd(2,6-(Cy2PCH2)2C6H3)(NH2) showed the strong nucleophilicity of the coordinated amide toward a range of electrophiles to undergo σ-ligand exchange or addition reactions with activated acetylenes and olefins to yield diastereo- and regiospecific aminated derivatives of palladium(II) complexes. The palladium(II) triflate Pd(2,6-(Cy2PCH2)2C6H3)(OTf) catalyzes the hydroamination of olefins with ammonia to produce a range of amine derivatives. A mechanistic feature for the observed catalytic reaction was discussed with respect to the probed aminoalkyl derivative of palladium(II).
4 Experimental
4.1 General methods and materials
All preparations of air sensitive compounds were performed on a standard Schlenk line or in an inert atmosphere glovebox under argon. THF and diethyl ether were freshly distilled from sodium/benzophenone ketyl under nitrogen and then stored over molecular sieves. Benzene, n-hexane, and n-pentane were distilled from sodium/benzophenone ketyl in the presence of tetraglyme. CH2Cl2 was dried by refluxing over LiAlH4 or CaH2 under N2. PdCl2 was supplied by the Pressure Chemical Company and used as received. 1,5-Cyclooctadiene, dppe, AgOTf, NH4OTf, α,α′-dibromo-m-xylene, dimethyl acetylenedicarboxylate (dmad), diethyl acetylenedicarboxylate (dead), phenylacetylene, acrylonitrile, dimethyl maleate, diethyl maleate, cis-stilbene, and NMR solvents (CDCl3, C6D6) were purchased from Aldrich Chemical Company and used as supplied. Dicyclohexylphosphine was obtained from Strem Chemicals Inc. All other reagents were acquired from various commercial companies. Pd(dppe)Cl2 was prepared by the displacement of cyclooctadiene from Pd(cod)Cl2 with dppe. Complexes of trans-Pd(PEt3)2(Ph)Cl [28], Pd(2,6-(Cy2PCH2)2C6H3)(OTf) [29], trans-[Pd(PEt3)2(Ph)(NH3)]OTf, [Pd(dppe)(NH3)2](OTf)2, and [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)]OTf were prepared using the procedures reported elsewhere [30].
4.2 Physical measurements
The IR spectra were recorded on a Bruker (Tensor 37) FT-IR spectrometer, as a pressed KBr pellets. The 1H, 13C{1H}, 19F{1H}, and 31P{1H} NMR spectra were measured on a Varian Gemini-2000 spectrometer (1H (199.975 MHz), 13C{1H} (50.288 MHz), 19F{1H} (188.140 MHz), 31P{1H} (80.950 MHz)), using the deuterium signal of the solvent as the internal lock frequency. The chemical shifts for 1H and 13C{1H} NMR are reported in parts per million (δ) relative to TMS (Me4Si). For 19F{1H} and 31P{1H} NMR spectroscopy, the chemical shifts were measured in parts per million relative to external perfluoromethylbenzene (δ = −63.73) and 85% H3PO4 in a sealed capillary, respectively. GC–MS was performed using an HP 6890 gas chromatograph equipped with an HP 5973 MSD and an HP-Ultra 1 column (Crosslinked Methyl Silicone Gum, 50 m × 0.2 mm, 0.33 μm film thickness). The injection temperature was 250 °C, and the column temperature ramped from 40 to 250 °C at 10°/min. The conductivity measurements were taken using a TOA conductivity meter (CM-40S). Nitromethane was used as the solvent in a cell containing platinized electrodes (cell constant = 1.014 cm−1). Elemental analyses were performed at the Korea Basic Science Institute in Seoul, Korea.
4.3 Synthesis
4.3.1 anti-[Pd(PEt3)(Ph)(μ-NH2)]2 (1)
In a glovebox under an argon atmosphere, a mixture of trans-[Pd(PEt3)2(Ph)(NH3)]OTf (500 mg, 0.85 mmol) and NaNH2 (150 mg, 3.8 mmol) was stirred in THF (20 mL) for 6 h at ambient temperature. The color of the reaction suspension was changed slowly from light gray to deep gray during the course of the reaction. The resulting suspension was filtered under vacuum to give a pale yellow solution. The removal of all volatiles from the filtrate under high vacuum resulted in yellow residues, which were extracted with n-pentane (4 × 10 mL) to give a pale yellow solution. The volume of solution was reduced to ca. 10 mL to slowly give colorless precipitates, which were filtered, washed with cold n-pentane, and dried in vacuo. Yield 176 mg (65%). IR (KBr): ν(NH) = 3238, 3367 cm−1 (w, br). 1H NMR (C6D6): δ −1.35 (br, 4H, NH2), δ 0.93 (m, 18H, CH3), δ 1.09 (m, 12H, CH2), δ 7.08 (t, 2H, p-CH (Ph), 3J(HH) = 7.3 Hz), δ 7.26 (t, 4H, m-CH (Ph), 3J(HH) = 7.3 Hz), δ 7.78 (d, 4H, o-CH (Ph), 3J(HH) = 7.3 Hz). 13C{1H} NMR (C6D6): δ 8.22 (s, CH3), δ 15.61 (d, CH2, 1J(CP) = 26.7 Hz), δ 122.3, 127.1, 137.6. 31P{1H} NMR (C6D6): δ 20.2 (s). Anal. Calcd for C24H44N2P2Pd2: C, 45.4; H, 6.98; N, 4.41. Found: C, 45.1; H, 6.84; N, 4.20.
4.3.2 [Pd(dppe)(μ-NH2)]2(OTf)2 (2)
NaNH2 (170 mg, 4.36 mmol) was added to a stirred solution of [Pd(dppe)(NH3)2](OTf)2 (1.20 g, 1.43 mmol) in THF at −78 °C (dry ice/acetone). The reaction mixture was stirred for 2 h. The solvent was evaporated to dryness under high vacuum at ca. −20 °C (caution should be taken at this step that the solution temperature should be controlled so as not to exceed more than −20 °C while the solvent was removed, otherwise the color of the solution changed rapidly from colorless to deep-purple, resulting in decomposed species; see Section 2). The residue was extracted with CH2Cl2 (3 × 5 mL) giving a pale yellow solution. The solution volume was reduced to ca. 3 mL. The addition of diethyl ether (10 mL) to the concentrated solution gave colorless crystals, which were washed with diethyl ether and dried in vacuo. Yield: 642 mg (67%). IR (KBr): ν(NH) = 3250, 3360 cm−1 (w, br), ν(SO3) = 1154, 1266 (s, br). 1H NMR (CDCl3): δ −0.19 (br, 4H, NH2), δ 2.52 (m, 8H, CH2), δ 7.4–7.7 (m, 40H, Ph). 31P{1H} NMR (CDCl3): δ 53.2 (s) 19F{1H} NMR (CDCl3): δ −78.9 (s). Anal. Calcd for C54H52F6N2O6P4Pd2S2: C, 48.41; H, 3.91; N, 2.09; S, 4.79. Found: C, 48.10; H, 4.11; N, 1.77; S, 4.73. ΛM = 184 Ω−1 cm2 mol−1 (CH3NO2, [Pd] = 0.50 × 10−3 M).
4.3.3 Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (3)
Under similar reaction conditions as for complex 1, a mixture of [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)]OTf (500 mg, 0.65 mmol) and NaNH2 (100 mg, 2.6 mmol) was stirred in THF (30 mL). The color of the reaction mixture was changed slowly from light gray to pale yellow during the course of the reaction. After 4 h, the resulting mixture was filtered under vacuum to give a yellow solution. The removal of all volatiles from the filtrate under high vacuum resulted in orange residues, which were extracted with n-hexane (4 × 5 mL) to give a pale yellow solution. The removal of solvent from the extracted solution under high vacuum gave a spectroscopically pure compound of Pd(2,6-(Cy2PCH2)2C6H3)(NH2). Yield 270 mg (68%). Spectral data for Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (4): 1H NMR (C6D6): δ −0.20 (br, 2H, NH2), δ 1.0–2.4 (m, 44H, Cy), δ 3.10 (vt, 4H, CH2, ∣2J(PH) + 4J(PH)∣ = 8.4 Hz), δ 7.08 (m, 3H, CH(aryl)). 31P{1H} NMR (C6D6): δ 50.7 (s).
4.4 Stoichiometric reactions of 3 with various electrophiles
The NMR sample preparation for the stoichiometric reactions of complex 3 with various electrophiles was performed in a glovebox under an argon atmosphere. An NMR sample was prepared by the addition of reactant into a d6-benzene solution of 3 using a 5 mm screw-capped NMR tube (Wilmad, 528-TR) or a 5 mm vacuum NMR tube (Wilmad, 507-LPV). The reaction products were analyzed by NMR (1H, 13C, 19F, and 31P) spectroscopy and GC–MS.
4.4.1 Reaction of 3 with H2O to yield Pd(2,6-(Cy2PCH2)2C6H3)(OH) (4)
A d6-benzene (0.3 mL) solution of 3 (ca. 10 mg) in a 5 mm screw-capped NMR tube prepared in a glovebox was removed and stored at ambient temperature. The reaction was monitored by 1H and 31P NMR spectroscopy, showing that complex 3 reacts slowly (over a period of days) with trace amounts of H2O in solution to give Pd(2,6-(Cy2PCH2)2C6H3)(OH) (4). The hydroxo complex Pd(2,6-(Cy2PCH2)2C6H3)(OH) can be prepared independently by a reaction of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) with KOH in THF. The prepared hydroxo complex was characterized by 1H and 31P NMR spectroscopy. For the microanalytical data, the hydrogen content was in good agreement with the calculated values but the carbon value was deviated slightly from an acceptable range because of its instability in air. The data is included as a reference. Treatment of the d6-benzene solution of 3 with an excess of water resulted in two species, exhibiting the 31P NMR resonances at δ 52.7 (minor) and 54.7 (predominant). The former can be attributed to a cationic ammine species [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)]+ by the observation of identical 31P{1H} NMR to that of [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)](OTf) at δ 52.7 in d6-benzene. The latter was attributed to cationic aqua complex [Pd(2,6-(Cy2PCH2)2C6H3)(OH2)]+, as evidenced by the identical 31P{1H} NMR at δ 54.7 to a complex prepared from Pd(2,6-(Cy2PCH2)2C6H3)(BF4) and H2O in d6-benzene. For Pd(2,6-(Cy2PCH2)2C6H3)(OH), 1H NMR (C6D6): −1.22 (t, 1H, OH, 3J(PH) = 3.3 Hz), 0.8–2.5 (m, 44H, Cy), 3.00 (vt, 4H, CH2, ∣2J(PH) + 4J(PH)∣ = 8.2 Hz), 7.1 (m, 3H, CH(aryl)). 31P{1H} NMR (C6D6): δ 48.7 (s). Anal. Calcd for C32H52O1P2Pd: C, 61.9; H, 8.44. Found: C, 59.3; H, 8.64. For [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)](OTf): 31P{1H} NMR: 52.7 (s, C6D6), 52.4 (s, CDCl3).
4.4.2 Reaction of 3 with [Ph2I](SO3CF3) or [Ph2I]Cl
A slight excess amount of [Ph2I]OTf or [Ph2I]Cl was added to a d6-benzene (0.3 mL) solution of 3 (15 mg, 0.024 mmol) in a 5 mm screw-capped NMR tube. The reaction of 3 with [Ph2I]OTf produced a cationic aniline complex [Pd(2,6-(Cy2PCH2)2C6H3)(NH2Ph)]OTf, which was confirmed by its independent preparation from the reaction of Pd(2,6-(Cy2PCH2)2C6H3)OTf and NH2Ph, showing an identical 31P NMR peak at δ 52.1 in d6-benzene. On the other hand, attempts at isolating the cationic aniline species were unsuccessful because of dissociation of the coordinated aniline converting to Pd(2,6-(Cy2PCH2)2C6H3)(OTf), which displays a single 31P{1H} NMR at δ 54.5 in d6-benzene. The reaction of 3 with [Ph2I]Cl produced Pd(2,6-(Cy2PCH2)2C6H3)Cl (31P{1H} NMR (C6D6): δ 52.3) along with the liberated NH2Ph (GC–MS: m/z = 93, 66, 39).
4.4.3 Reaction of 3 with phenylacetylene to yield Pd(2,6-(Cy2PCH2)2C6H3)(CCPh) (5)
A slight excess of phenylacetylene (HCCPh) was added to a d6-benzene (0.3 mL) solution of 3 (15 mg, 0.024 mmol) in a 5 mm screw-capped NMR tube. The reaction proceeded readily, in 30 min, to yield the palladium(II) acetylenide Pd(2,6-(Cy2PCH2)2C6H3)(CCPh) (5), quantitatively. The acetylenido complex is extremely soluble in conventional organic solvents including n-pentane. Therefore, the removal of all volatiles from the reaction mixture resulted in a pale yellow solid, which was characterized by IR and NMR (1H, 13C{1H}, 31P{1H}) spectroscopy. A preparative scale experiment for this reaction was conducted in a glovebox as follows: an excess amount of phenylacetylene (0.02 g, 0.38 mmol) was added to a benzene solution of 3 (150 mg, 0.24 mmol). The reaction mixture was stirred for 1 h. All volatiles were removed under high vacuum to give yellowish residues. The residues were dissolved in n-pentane. The n-pentane solution was filtered through a Celite column to give a pale yellow solution. The resulting solution was dried completely under high vacuum to remove all volatiles, yielding an analytically pure compound of Pd(2,6-(Cy2PCH2)2C6H3)(CCPh). Spectral data for Pd(2,6-(Cy2PCH2)2C6H3)(CCPh) (5): IR (KBr): ν(CC) = 2096 cm−1 1H NMR (C6D6): δ 1.0–2.4 (m, 44H, Cy), δ 3.20 (vt, 4H, CH2, ∣2J(PH) + 4J(PH)∣ = 8.1 Hz), δ 7.00 (t, 1H, p-H(Ph), 3J(HH) = 7.4 Hz), δ 7.2 (m, 3H, CH (aryl)), δ 7.21 (m, 2H, m-H(Ph)), δ 7.74 (d, 2H, o-H(Ph), 3J(HH) = 7.0 Hz). 13C{1H} NMR (C6D6): δ 113.1 (t, Pd–CCPh, 2J(CP) = 12 Hz), δ 118.3 (Pd–CCPh). 31P{1H} NMR (C6D6): δ 56.4 (s). Anal. Calcd for C40H56P2Pd: C, 68.1; H, 8.00. Found: C, 68.5; H, 7.89.
4.4.4 Reaction of 3 with dialkyl acetylenedicarboxylate (RCCR; R = CO2Me, CO2Et) to yield (Z)-Pd(2,6-(Cy2PCH2)2C6H3)(CRCR(NH2)) (R = CO2Me (6a), CO2Et (6b))
To a d6-benzene (0.3 mL) solution of 3 (15 mg, 0.024 mmol) in a 5 mm screw-capped NMR tube was added a slight excess of dialkyl acetylenedicarboxylate (RCCR; R = CO2Me, CO2Et: 0.032 mmol; 0.1 mL of a diluted d6-benzene solution, which was prepared by the addition of dmad (46 mg) or dead (54 mg) into 1.0 mL of d6-benzene). The insertion product Pd(2,6-(Cy2PCH2)2C6H3)(CRCR(NH2)) was formed quantitatively from the reaction, which was monitored by NMR spectroscopy. The product was barely isolated from the solution because of its high solubility in most organic solvents. Therefore, the removal of all volatiles from the solution under high vacuum resulted in yellow solids, which afforded satisfactory FABMS data. For Pd(2,6-(Cy2PCH2)2C6H3)((MeO2C)CC(CO2Me)(NH2)) (6a): 1H NMR (C6D6): δ 1.0–2.4 (m, 44H, Cy), δ 3.49 (s, 3H, CH3), δ 3.73 (s, 3H, CH3), δ 3.10 (dt, Ha, CH2, 2J(HaHb) = 17.6 Hz,∣2J(PH) + 4J(PH)∣ = 7.9 Hz), δ 3.31 (dt, Hb, CH2, 2J(HaHb) = 17.6 Hz,∣2J(PH) + 4J(PH)∣ = 7.9 Hz), δ 3.89 (br, 2H, NH2), δ 7.21 (m, 3H, CH (aryl)). 13C{1H} NMR (C6D6): δ 50.53, 51.45 (s, CO2CH3), δ 162.08, 176.40 (CO2CH3). 31P{1H} NMR (C6D6): δ 51.7 (s). FABMS (observed m/z 761.22): calcd for C38H59NO4P2Pd, 761.30. For Pd(2,6-(Cy2PCH2)2C6H3)((EtO2C)CC(CO2Et)(NH2)) (6b): 1H NMR (C6D6): δ 1.0–2.4 (m, 44H, Cy), δ 1.08 (t, 3H, CH3, 3J(HH) = 7.1 Hz), δ 1.34 (t, 3H, CH3, 3J(HH) = 7.1 Hz), δ 4.12 (q, 2H, CH2, 3J(HH) = 7.1 Hz), δ 4.31 (q, 2H, CH2, 3J(HH) = 7.1 Hz), δ 3.10 (dt, Ha, CH2, 2J(HaHb) = 17.6 Hz,∣2J(PH) + 4J(PH)∣ = 8.3 Hz), δ 3.30 (dt, Hb, CH2, 2J(HaHb) = 17.6 Hz,∣2J(PH) + 4J(PH)∣ = 8.3 Hz), δ 3.94 (br, 2H, NH2), δ 7.21 (m, 3H, CH(aryl)). 13C{1H} NMR (C6D6): δ 14.47, 15.15 (CO2CH2CH3), δ 58.98, 60.30 (CO2CH2CH3), δ 162.08, 176.09 (CO2CH2CH3). 31P{1H} NMR (C6D6): δ 51.9 (s). FABMS (observed m/z 789.25): calcd for C40H63NO4P2Pd, 789.33.
4.4.5 Reaction of (Z)-Pd(2,6-(Cy2PCH2)2C6H3)(CRCR(NH2)) (R = CO2Me (6a), CO2Et (6b)) with HOC6H4-p-NO2 to yield cis-CHRCR(NH2) (R = CO2Me, CO2Et) and Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4-p-NO2) (7)
HOC6H4-p-NO2 (5 mg) was added to a d6-benzene (0.3 mL) solution of 6a or 6b (ca. 15 mg) in a 5 mm screw-capped NMR tube. The reaction proceeded quantitatively to produce cis-CHRCR(NH2) (R = CO2Me, CO2Et) and Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4-p-NO2) (7). All products were analyzed by 1H and 31P NMR spectroscopy, and GC–MS. The p-nitrophenoxide complex Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4-p-NO2) (7) was prepared independently from an equimolar reaction of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) and NaOC6H4-p-NO2 in THF. For cis-((MeO2C)CHC(CO2Me)(NH2)): 1H NMR (C6D6): δ 3.15 (s, 3H, CH3), δ 3.44 (s, 3H, CH3), δ 5.77 (s, 1H, CH). GC–MS: m/z = 159, 128, 100, 68, 59. For cis-((EtO2C)CHC(CO2Et)(NH2)): 1H NMR (C6D6): δ 0.78 (t, 3H, CH3, 3J(HH) = 7.13 Hz), δ 1.02 (t, 3H, CH3, 3J(HH) = 7.13 Hz), δ 3.79 (q, 2H, CH2, 3J(HH) = 7.13 Hz), δ 4.07 (q, 2H, CH2, 3J(HH) = 7.13 Hz), δ 5.84 (s, 1H, CH). GC–MS: m/z = 187, 142, 114, 86, 68. For Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4-p-NO2) (7): IR (KBr): ν(NO) = 1583, 1303 cm−1 (sh, s). 1H NMR (C6D6): δ 0.9–2.1 (m, 44H, Cy), δ 2.84 (vt, 4H, CH2, ∣2J(PH) + 4J(PH)∣ = 8.4 Hz), δ 6.88 (d, 2H, 3J(HH) = 9.2 Hz), δ 8.49 (d, 2H, 3J(HH) = 9.2 Hz). 31P{1H} NMR (C6D6): δ 50.7 (s). Anal. Calcd for C38H55NO3P2Pd: C, 61.5; H, 7.47; N, 1.89. Found: C, 61.1; H, 7.52; N, 1.58.
4.4.6 Reaction of dialkyl acetylenedicarboxylate with ammonia
Anhydrous gaseous ammonia was bubbled into a d6-benzene (0.3 mL) solution of dialkyl acetylenedicarboxylate (dmad, dead; ca. 20 mg, respectively) for ca. 30 s in a 5 mm screw-capped NMR tube. The reaction produced an isomeric mixture of cis- and trans-(CHRCR(NH2)) in a trans/cis ratio of ca. 1.3. For trans-((MeO2C)CHC(CO2Me)(NH2)): 1H NMR (C6D6): δ 3.42 (s, 3H, CH3), δ 3.59 (s, 3H, CH3), δ 4.80 (s, 1H, CH). For trans-((EtO2C)CHC(CO2Et)(NH2)): 1H NMR (C6D6): δ 0.75 (t, 3H, CH3, 3J(HH) = 7.13 Hz), δ 1.01 (t, 3H, CH3, 3J(HH) = 7.13 Hz), δ 3.37 (q, 2H, CH2), δ 4.08 (q, 2H, CH2), δ 4.90 (s, 1H, CH). For cis-((MeO2C)CHC(CO2Me)(NH2)) and cis-((EtO2C)CHC(CO2Et)(NH2)) refer to the preceding experiment.
4.4.7 Reaction of 3 with dialkyl maleate (cis-(CO2R)CHCH(CO2R)) (R = CH3, CH2CH3)
An excess of dialkyl maleate (cis-(CO2R)CHCH(CO2R), R = CH3, CH2CH3) was added to a d6-benzene (0.3 mL) solution of 3 (15 mg, 0.024 mmol) in a 5 mm screw-capped NMR tube. The dialkyl maleate slowly isomerizes to dialkyl fumarate (trans-(CO2R)CHCH(CO2R), R = CH3, CH2CH3) in the presence of catalytic amounts of 3 at ambient temperature, as evidenced by 1H NMR spectroscopy. After 4 h, the observed trans/cis ratio was 0.8 (for R = Me) and 0.7 (for R = Et), respectively. After 24 h, the trans/cis ratio was 8.9 (for R = Me). The conversion of dialkyl maleate (cis-isomer) into dialkyl fumarate (trans-isomer) was complete in 5 days. For dimethyl maleate (cis-(CO2CH3)CHCH(CO2CH3)): 1H NMR (C6D6): δ 3.35 (s, 6H, CH3), δ 5.71 (s, 2H, CH). For dimethyl fumarate (trans-(CO2CH3)CHCH(CO2CH3)): 1H NMR (C6D6): δ 3.25 (s, 6H, CH3), δ 6.87 (s, 2H, CH). For diethyl maleate (cis-(CO2CH2CH3)CHCH(CO2CH2CH3)): 1H NMR (C6D6): δ 0.96 (t, 6H, CH3, 3J(HH) = 9.2 Hz), δ 3.98 (q, 4H, CH2, 3J(HH) = 9.2 Hz), δ 5.77 (s, 2H, CH). For diethyl fumarate (trans-(CO2CH2CH3)CHCH(CO2CH2CH3)): 1H NMR (C6D6): δ 0.89 (t, 6H, CH3, 3J(HH) = 7.6 Hz), δ 3.88 (q, 4H, CH2, 3J(HH) = 7.6 Hz), δ 6.91 (s, 2H, CH).
4.4.8 Reaction of 3 with cis-stilbene
An excess of cis-stilbene (cis-CHPhCHPh) was added to a d6-benzene (0.3 mL) solution of 3 (15 mg, 0.024 mmol) in a 5 mm screw-capped NMR tube. No reaction occurred at ambient temperature for 10 h. At an increased temperature of 50 °C (in a silicone oil bath), however, cis-stilbene isomerized catalytically to trans-stilbene in the presence of 3: trans/cis = 2.0 for 2 h. For cis-stilbene (cis-CHPhCHPh), 1H NMR (C6D6): δ 6.49 (s, (2H, CH). For trans-stilbene (trans-CHPhCHPh), 1H NMR (C6D6): δ 7.02 (s, 2H, CH).
4.4.9 Reaction of 3 with acrylonitrile
A slight excess of acrylonitrile (ca. 1.5 μL) was added to a d6-benzene (0.3 mL) solution of 3 (15 mg, 0.024 mmol) in a 5 mm screw-capped NMR tube via a microsyringe. The reaction proceeded readily to generate a metastable addition product Pd(2,6-(Cy2PCH2)2C6H3)(CH(CN)CH2NH2). Attempts at isolating the addition product from the solution were unsuccessful because of decomposition to a couple of unidentified species. On the other hand, in the reaction of 3 with a large excess of acrylonitrile, the addition product gradually disappeared to give a couple of new complexes, as evidenced by 31P NMR spectroscopy, along with the generation of insoluble polymeric species of acrylonitrile in solution. After filtration of the precipitates, the 31P NMR spectrum of the solution exhibited a couple of new resonances, one at δ 59.2 corresponding to a cationic acrylonitrile complex [Pd(2,6-(Cy2PCH2)2C6H3)(CH2CHCN)]+, which can be verified by the reaction of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) with CH2CHCN (excess) in d6-benzene. Spectral data for Pd(2,6-(Cy2PCH2)2C6H3)(CH(CN)CH2NH2): 1H NMR (C6D6): δ 1.0–2.4 (m, 44H, Cy), ca. δ 2.2 (m, CH), ca. δ 3.1 (m, CHa), ca. δ 3.5 (m, CHb), δ 3.16 (vt, 4H, CH2, ∣2J(PH) + 4J(PH)∣ = 8.1 Hz). 31P{1H} NMR (C6D6): δ 49.0 (s). For polymeric species: IR (KBr): ν(CN) = 2245, 2204 cm−1, ν(NH2) = 3300, 3360 cm−1. For [Pd(2,6-(Cy2PCH2)2C6H3)(CH2CHCN)](OTf), 31P{1H} NMR: δ 59.2 (C6D6), δ 59.4 (CDCl3).
4.5 A typical procedure for catalytic hydroamination of olefins with ammonia
The catalytic hydroamination of olefins with ammonia in the presence of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) was performed in two ways. Method A using a 5 mm vacuum NMR tube (Wilmad, 507-LPV): anhydrous ammonia was introduced into a d6-benzene (0.5 mL) solution of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) (3 mg, 3.98 × 10−3 mmol) and CH2CHCN (0.026 mL, 3.98 × 10−1 mmol) in a 5 mm vacuum NMR tube, which was precooled and evacuated via several freeze and thaw cycles. The molar ratio of CH2CHCN/NH3 was calculated by integrating the corresponding resonance peaks in the 1H NMR spectrum of the prepared sample. The NMR sample was kept in silicone oil for 12 h at 60 °C. After cooling the sample, the reaction products were analyzed by 1H NMR spectroscopy and GC–MS. For GC–MS analysis, the reaction mixture was transferred to a short glass-column (0.7 × 15 cm) packed with alumina (ca. 1 cm). Eluting the mixture with diethyl ether resulted in a clear pale yellow solution, which was analyzed by GC–MS. Method B using a high pressure reactor (Carl Roth, Model 1, 100 mL): a mixture of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) (3 mg, 3.98 × 10−3 mmol) and CH2CHCN (0.026 mL, 3.98 × 10−1 mmol) in d6-benzene (0.5 mL) was loaded in a glass linear equipped with a high pressure reactor. The reactor was then cooled at −15 °C (ice/NaCl) and evacuated. Anhydrous ammonia gas was introduced into the reactor for 5 min at 0 °C to reach a pressure of 8 atm. When reactor temperature was increased to 60 °C, the pressure increased to 25 atm. The reaction mixture was stirred for 12 h at 60 °C. After cooling the reactor in an ice bath, gaseous ammonia was removed from the reactor in a well ventilating hood. The reaction products were analyzed by 1H NMR spectroscopy and GC–MS in a similar manner. Spectral data for CH2(CN)CH2NH2, 1H NMR (C6D6): δ 1.45 (t, 2H, CH2CN, 3J(HH) = 6.4 Hz), δ 2.13 (t, 2H, CH2N, 3J(HH) = 6.4 Hz). GC–MS: m/z = 69, 42, 30, 28. For (CH2(CN)CH2)2NH, 1H NMR (C6D6): δ 1.50 (t, 4H, CH2CN, 3J(HH) = 6.4 Hz), δ 1.97 (t, 4H, CH2N, 3J(HH) = 6.4 Hz). GC–MS: m/z = 123, 83, 54, 42, 30, 28. For (CH2(CN)CH2)3N, 1H NMR (C6D6): δ 1.59 (t, 6H, CH2CN, 3J(HH) = 6.8 Hz), δ 1.94 (t, 6H, CH2N, 3J(HH) = 6.8 Hz). GC–MS: m/z = 176, 136, 109, 83, 54, 42, 30. For (CH2(CN)CH(CH3))NH2, GC–MS: m/z = 83, 69, 44, 42, 40, 28, 18, 15. For (CH2(CO2CH3)CH2)2NH, GC–MS: m/z = 187, 174, 157, 130, 102, 56. For (CH2(CO2CH3)CH2)3N, GC–MS: m/z = 275, 216, 157, 101, 56.
4.6 X-ray structure determination
All X-ray data collections were performed using Mo Kα radiation (λ = 0.71069 Å) on an Enraf-Nonius CAD4 diffractometer equipped with a graphite crystal, incident beam monochromator. All calculations were carried out using the SHELX-97 programs [31]. All structures were solved by direct methods. All non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were generated in ideal positions and refined in a riding model.
Acknowledgments
This work was supported by the Dongguk University Research Fund. The authors are grateful to Prof. M. S. Lah for the X-ray diffraction studies, and Dr. J. M. Seul and Mr. S. Y. Ryu for the technical assistances.