

1 Introduction
The transition from a non-renewable, carbon-based energy economy to the one that is sustainable in the long-term will require a concerted global effort heavily dependent on solar energy, as it is the only renewable energy source with the potential to meet current and projected demands [1]. Importantly, variations in the solar flux with time (daily and seasonally) and geography necessitate the conversion of solar energy to chemical energy that can be stored and transported. Bacteria and green plants perform solar energy conversion through natural photosynthesis and the structural features which enable this process are well understood. However, despite the remarkable ability of natural photosynthetic organisms to operate with near unity quantum efficiency, their total efficiency is inherently limited by other demands on the organism including respiration and reproduction [2]. Artificial photosynthetic architectures with targeted functionality have the potential to surpass the performance efficiency of plants by integrating relevant structural themes of natural photosynthesis with known chemical principles and appropriate synthetic chemistry. Sustained fundamental research with these goals in mind is imperative to foster further technological development and ultimately, enable the wide-spread use of artificial photosynthetic technologies.
Effective conversion of solar energy to chemical energy spans multiple time and length scales: an artificial photosynthetic system must manage photons, electrons, and protons from initial photon absorption and excitation on the femtosecond time scale, through intra- and intermolecular charge transfer and separation, to diffusionally controlled substrate binding and bond formation on the nano- to microsecond time scale (Fig. 1). Identification of the light-generated transient species is crucial to understand the key steps in a photocatalytic cycle and exactly how system parameters influence their structure and stability, and ultimately influence system optimization. Ex situ mechanistic information has been conventionally obtained by either trapping these intermediates (i.e., by thermal [3] or steric means [4]) or generating the proposed intermediates (i.e., by pulse radiolysis [5–7]) and probing their structure using appropriate steady-state characterization techniques. This has undoubtedly supplied important and useful information regarding light-activated catalysis. However, in situ mechanistic information on a photocatalytic cycle is now quite readily obtained by pump-probe characterization techniques which combine a light trigger with a probe relevant to the dynamic structure of interest: for example a light probe for transient optical spectroscopy which spans the UV to IR, an EPR probe to track radical formation and decay within a photocatalytic cycle, or an energy-tunable X-ray probe to monitor changes in metal oxidation state and coordination geometry. Of particular interest to research on artificial photosynthesis, the advent of free-electron laser X-ray sources has reduced the X-ray pulse width to as low as 10 fs and now routinely used X-ray techniques such as X-ray absorption spectroscopy, scattering, or diffraction can be synced to ultrafast laser pulses to interrogate the very earliest steps following excitation, in situ, with extraordinary resolution [8]. These new capabilities have already hosted pioneering experiments and yielded remarkable insight into light-induced structural dynamics [9–12] which will likely stimulate breakthroughs in photocatalyst development.

Representation of photophysical and photochemical steps relevant to artificial photosynthesis in an idealized bimetallic linked chromophore—catalyst dyad, Mn = metal center of either the chromophore or catalyst in ground oxidation state (n), D = sacrificial electron donor. For simplicity, only the oxidative quenching mechanism for photoinduced electron transfer and heterolytic H2 bond formation are depicted. Depending on the specific chromophore—catalyst system and conditions, photocatalysis may also proceed via reductive quenching of the chromophore excited state by the sacrificial electron donor and homolytic H2 bond formation.
In recognition of the International Year of Light, this perspective will focus on our research in using light sources to probe newly developed supramolecular architectures towards understanding the fundamental interactions necessary to achieve artificial photosynthesis. We will specifically discuss our work on linked cobaloxime-based H2 photocatalysts as an example of how physical characterization by light sources has been used to guide new designs for next-generation photocatalyst architectures. The applicability of emerging light source characterization methods will be discussed in the context of the outlook for advanced supramolecular photocatalyst development.
2 Supramolecular architectures for artificial photosynthesis
A complete system for artificial photosynthesis will ideally functionally mimic natural photosynthesis: the absorption of light energy to oxidize water and provide highly reducing equivalents to reduce protons to H2 or CO2 to CO, formate, or possibly higher hydrocarbon fuels. As structural mimics of natural photosynthesis, a photocatalyst minimally requires a chromophore module and a catalyst module, and some means to link the two modules to support electron transfer. We favor a supramolecular approach to link functional modules using non-covalent but specific interactions such as hydrogen bonding, metal coordination bonding, or electrostatic interactions. These interactions can be used to construct self-assembling and self-repairing structures that would be prohibitively difficult to obtain using traditional synthetic routes. For example, even the most “simple” transformation of artificial photosynthesis, the reduction of protons to hydrogen, requires two electrons and two protons which must be precisely manipulated over several orders of magnitude in time and space by the chromophore excited states and catalyst oxidation states. Water oxidation and CO2 reduction require four and six electron processes, and accordingly introduce a substantial degree of additional complexity to photocatalyst design. Therefore, the integration of multiple light-absorbing sites into photocatalyst architectures will facilitate the distribution of the redox equivalents necessary for multi-electron photocatalysis across multiple chromophores, and avoid the requirement for fast regeneration of the chromophore ground state to transfer the second and subsequent electrons. Additionally, molecular modules which facilitate sequential and accumulative proton or electron transfer, or modules which anchor homogeneous supramolecular photocatalysts to electrode surfaces for interfacial electron transfer to electron sources or sinks are readily incorporated by this approach. The enormous potential to design and realize complex and hierarchical structures coupled with their responsiveness to traditional and emerging high-resolution structural characterization techniques position homogeneous supramolecular photocatalysts to drive a breakthrough in artificial photosynthesis.
Bis(glyoximato)cobalt(II) macrocycles, or cobaloximes (Fig. 2) were originally developed as vitamin B12 mimics [13] and later found to reduce protons to H2 in the presence of a sacrificial electron donor [14]. More recently cobaloximes experienced a resurgence in interest following the discovery that they behave as efficient electrocatalysts for proton reduction with a very low overpotential [15–18]. In the interest of using solar energy to initiate H2 catalysis, seminal work from Hawecker, Lehn, and Ziessel recognized that the cobalt reduction potentials are aligned well with the energetics of common chromophores and demonstrated that H2 photocatalysis by 1 can be initiated by [Ru(bpy)3]+, formed following reductive quenching of [Ru(bpy)3]2+∗ by triethylamine [19]. Numerous chromophores have now been used to initiate H2 photocatalysis by cobaloximes in multimolecular systems [20–23], even reaching over 9000 TON using water as a proton source and chromophores based on earth-abundant elements [24]. Elegant experimental [25,26] and computational [27,28] studies have provided insight into the mechanism for H2 catalysis which is proposed to proceed from reduction of Co(II) to Co(I) followed by protonation which yields Co(III) hydride. The Co(III)—H species is then reduced and protonated to release H2 and return the catalyst to the Co(II) resting state. The mechanism for H2 photocatalysis by cobaloximes is fairly well understood at this point and therefore renders cobaloximes an ideal platform by which new chromophores for photocatalysis can be studied. Significant shortcomings of cobaloxime-based catalysis, however, are their structural instability and solubility under acidic or neutral aqueous conditions, particularly as compared to other first–row transition metal based water reduction catalysts. Nevertheless, among first-row transition metal catalysts, cobaloximes have been used most extensively in linked photocatalyst designs. In 2008 Fihri et al. were the first to describe a supramolecular cobaloxime photocatalyst by coordination of a pyridyl-functionalized [Ru(bpy)3]2+-based chromophore to the Co(II) center of 3 [29]. The ability to link a chromophore by simple pyridyl coordination to the cobalt center and still observe H2 photocatalysis inspired others to test this strategy by coordination of a number of molecular chromophores including several variations on the original [Ru(bpy)3]2+ motif [30,31], as well as Ir(III) [32], porphyrin [33–35], bodipy [36–38], perylene [39], and fluorescein chromophores [40], and our group's approach to the design and investigation of linked cobaloxime photocatalysts will be discussed in detail in the following section. In recognition of the limitations of linking a single molecular chromophore to support multi-electron catalysis, the same pyridyl coordination was used to anchor cobaloximes to the surface of TiO2 and CdSe/ZnS nanoparticles, both of which act as an electron reservoir to quickly supply multiple electrons to the catalyst site [41,42]. Pyridyl coordination has also been used to link cobaloxime catalyst modules to semiconductor surfaces via polymer [43] and molecular bridges [44]. A related strategy links molecular cobaloxime catalysts to a protein scaffold, either by attractive van der Waals interactions [45] or via histidine coordination [46,47]. The complex electron transfer mechanism of the protein structures involved in natural photosynthesis can support extremely long-lived charge separation and facilitate H2 catalysis under near-neutral aqueous conditions.
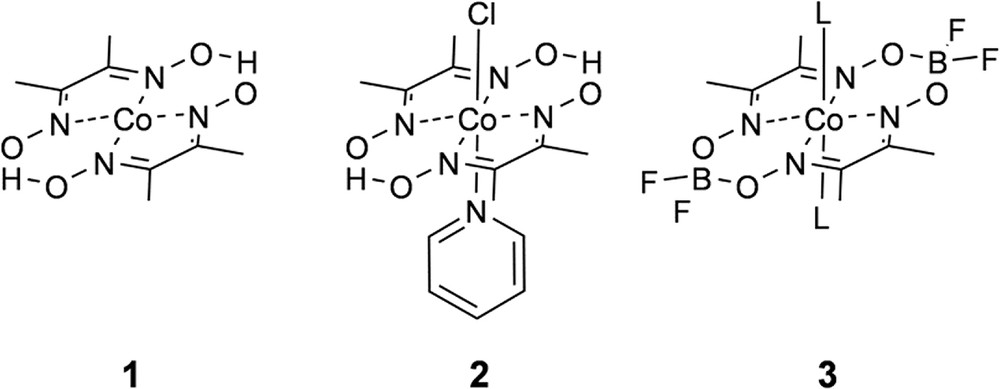
Chemical structures of cobaloxime macrocycles used in H2 electrocatalysis and photocatalysis.
As mentioned above, cobaloximes have so far dominated the research space with respect to the diversity of linked photocatalyst systems, but there are notable examples of other earth-abundant H2 catalyst modules linked to chromophores. Iron hydrogenase mimics linked to chromophores including [Ru(bpy)3]2+ [48], porphyrins [49], and peptide scaffolds [50] have shown modest H2 production following visible excitation. The self-assembly of DuBois-type [Ni(P2N2)2]2+ H2 catalysts [51] with Photosystem I [52] and perylene-diimide based nanostructures [53] have both shown very high activity for H2 photocatalysis under near-neutral aqueous conditions. Several other promising molecular electrocatalysts for H2 production have been recently described and represent prospective new catalyst modules for linked photocatalyst architectures [54].
3 A case study on the evolution of linked cobaloxime photocatalysts
Our research has focused largely on using the cobaloxime catalyst module as a platform on which various supramolecular linking strategies for photocatalyst development can be explored. Motivated by the original supramolecular work of Artero, we expanded the design portfolio to include Ru(II)bis(terpyridyl)-based and perylene-diimide chromophore modules linked to [Co(dmgBF2)2] (3) (Fig. 3) [55]. Importantly, for each assembly we observed a slight positive shift in the Co(II/I) reduction potential, indicative of nitrogen coordination to the Co(II) site. Note, as is typical for supramolecular assemblies, characterization techniques such as NMR and mass spectrometry provided no useful information regarding the assembly structure [56,57]. Therefore, solution phase X-ray scattering was used to quantitatively characterize the structure of the photocatalyst architectures at millimolar concentrations in dimethylformamide and acetonitrile. Furthermore, solution phase X-ray scattering can be performed under a variety of conditions (i.e. temperature, solvent, and pH) to monitor structural changes in response to environment or follow an assembly structure throughout the photocatalytic cycle [58]. In this study, X-ray scattering data was collected from 0.1 < q < 2.8 Å−1 at millimolar concentrations in acetonitrile and dimethylformamide which enabled analysis of both the wide-angle (WAXS) and small-angle (SAXS) regimes to obtain information on the global structure and intra-assembly interactions, where q is the scattering angle defined as q = (4π/λ)sinθ (λ = X-ray wavelength and θ = half scattering angle). A fragment analysis of the WAXS data was used to overcome difficulties related to the strong solvent background and slight perturbations of the solvent shell in response to the charged chromophore module. After subtracting the sum of the scattering from each module from that of the chromophore-catalyst supramolecular assembly, we observed new scattering interactions as a result of assembly formation. The Co(II)—Ru(II) distance appeared as a distinct peak in the transformation of the difference scattering into real space for assemblies 4 and 5, and matched extremely well with the metal–metal distance obtained from energy-minimized models (Fig. 4). Therefore, the WAXS data yielded confirmation of assembly formation, but not quantitative information regarding the completion of assembly formation. The SAXS response was analyzed according to the Guinier relationship [59] (Equations 1, 2) to estimate the assembly radius of gyration, Rg, a common method for macromolecular structural characterization [60–66]:
| (1) |
| (2) |
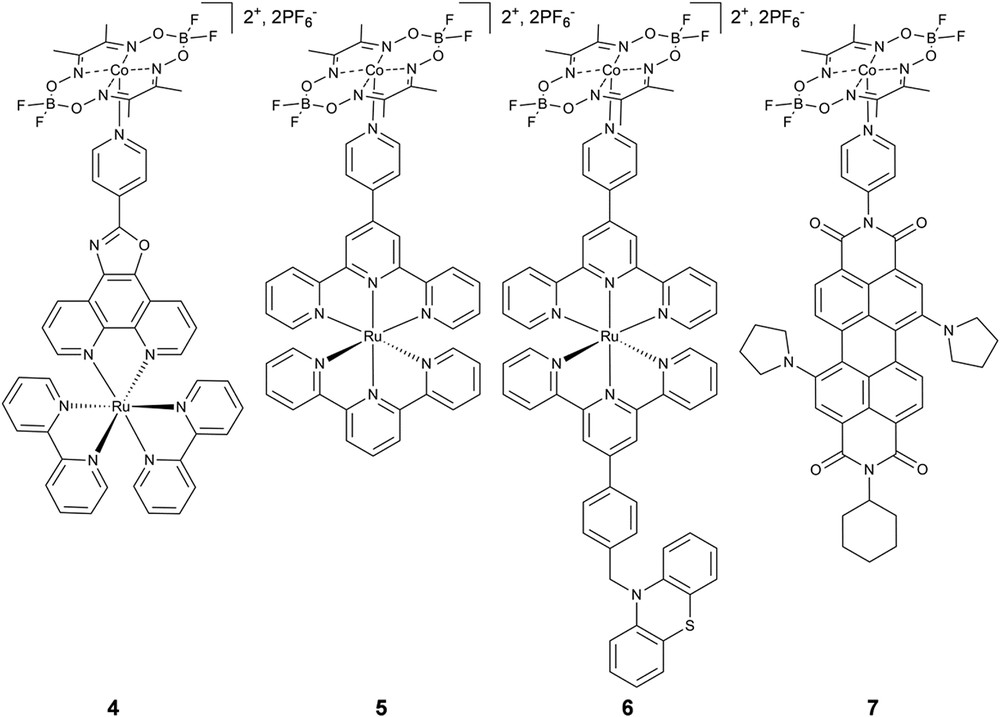
Chemical structures of axially-linked cobaloxime photocatalyst assemblies.

Left: energy-minimized model of assembly 5, metal–metal distance indicated. Right: Comparison of Fourier transform of difference scattering of 5, model and experimental data, 5 mM in CH3CN.
Following structural characterization of the axially-linked assemblies, as a first test of photocatalyst function, we surveyed 4–7 for electrocatalytic activity in acetonitrile and found no significant decrease in activity following coordination, an important control for photocatalyst activity. Ultrafast transient optical spectroscopy was then used to monitor the transient species following illumination, as well as provide a sensitive functional marker for photocatalyst activity. Following 527 nm excitation in acetonitrile, we did not observe formation of the reduced catalyst module species, [Co(I)(dmgBF2)2], which would indicate oxidative quenching of the chromophore excited state by the catalyst module. Rather, the transient spectra of 4–7 closely resemble that of the chromophore module alone. However, the kinetics follows a biexponential decay consisting of a very fast component on the picosecond time scale followed by a component that decays on the time scale of the chromophore alone (Fig. 5). Interestingly, the fast component slightly increases in amplitude as the catalyst to chromophore ratio increases, but saturates between five- and ten-fold excess of 3. With the X-ray scattering structural analysis in mind, we interpret the multi-exponential kinetics as a combination of 1) photoinduced electron transfer from the chromophore to [Co(II)(dmgBF2)2] which is immediately followed by fast charge recombination so that there is no observable accumulation of the charge-separated state, and 2) unperturbed decay of the chromophore in its unassembled state. These conclusions are supported by a number of other studies. A detailed EPR analysis of cobaloximes in a variety of solvent and coordination environments indicates that axial ligation occurs directly though the dz2 orbital, which is likely the pathway for fast back electron transfer [67]. In studies of axially-linked porphyrin and corrole chromophores, fast charge recombination is also proposed as the reason for the lack of observation of the charge-separated state crucial to H2 photocatalysis [35,68]. Notably, an axially-linked chromophore-bridge-cobaloxime triad did undergo stepwise charge transfer and formation of the Co(I) state in approximately 15 ps following photoexcitation in dichloromethane, but also suffered from structural lability in the charge-separated state [39]. Taken all together, we conclude that the major contributing mechanism to H2 photocatalysis by axially-linked cobaloxime photocatalysts is not oxidative quenching enabled by photocatalyst assembly, but more likely the reductive quenching of the chromophore excited state by the sacrificial electron donor followed by reduction of the cobaloxime ground state to initiate the catalytic cycle.

Transient spectra of chromophore alone (left) and assembly 6 (right) in CH3CN following ultrafast photoexcitation at 527 nm.
Our analysis of the axially-linked cobaloxime photocatalyst assemblies, coupled with work from others on similar assemblies, led us to reevaluate the supramolecular assembly geometry and attempt linking the chromophore to the cobaloxime macrocycle through the ligand structure rather than directly to the catalytic site. Toward this goal, we created two new symmetric cobaloxime-based photocatalyst assemblies via functionalization of [Ru(bpy)3]2+-based chromophores with glyoxime groups followed by self-assembly with Co(II) (Fig. 6) [69]. As in the axially-linked assemblies, traditional characterization techniques failed to provide useful structural information and we instead employed synchrotron X-ray based characterization. X-ray absorbance spectroscopy of equatorial assembly 9 at the Co K-edge (7721.5 eV) confirmed the Co(II) oxidation state and the data are consistent with an octahedral local coordination environment including four nitrogen ligands in the equatorial plane and two chloride ligands in the axial positions. Guinier analysis of the SAXS data provided complementary global structural information and confirmed that the Rg of the assemblies was significantly larger than that of the modules alone (6.57 Å vs. 10.65 Å for 8 and 9, respectively) and matched that of energy-minimized models. Again, transient optical spectroscopies were employed to identify and track electron transfer following visible excitation of the [Ru(bpy)3]2+ chromophore modules. The ultrafast transient spectra for 8 following 420 nm excitation show initial ground state bleach and broad excited state formation (Fig. 7), similar to those of common [Ru(bpy)3]2+ chromophore compounds, with an excited state lifetime of 550 ns measured by nanosecond transient optical spectroscopy. In contrast, excitation of equatorial assembly 9 shows an initial ground state bleach coupled to a strong positive feature centered at approximately 570 nm which correlates with the reduced state of the cobaloxime module as determined by spectroelectrochemistry. The kinetic trace of 9 at 570 nm has multiple components, the shortest of which represents decay of the Co(I) state with τ∼26 ps. The multi-exponential decay to ground state following collapse of the Co(I) signal is interpreted as decay through two MLCT states on the phenanthroline-phenazine type bridge, which has been previously proposed for related Ru(II) complexes [70,71]. Despite its short lifetime, the equatorially-linked cobaloxime assembly 9 yielded the first observation of ultrafast formation of the relevant catalytic intermediate in a linked dyad. X-band EPR spectroscopy demonstrated that the Co(II) center of assemblies 9 and 11 are high-spin, where the electronic structure of the Co(II) in most cobaloximes is 3d7 low-spin. Also, no charge separation was observed following 420 nm excitation of assembly 11, which has only one amine bond linking the chromophore and catalyst modules. Therefore, we propose that both the spin state and highly conjugated ligand structure contribute to the fast charge separation observed in assembly 9. To build upon these designs, we are currently exploring modifications to the ligand bridge structure which will continue to facilitate photoinduced electron transfer and also stabilize the Co(I) state long enough for diffusional interaction with protons in solution.
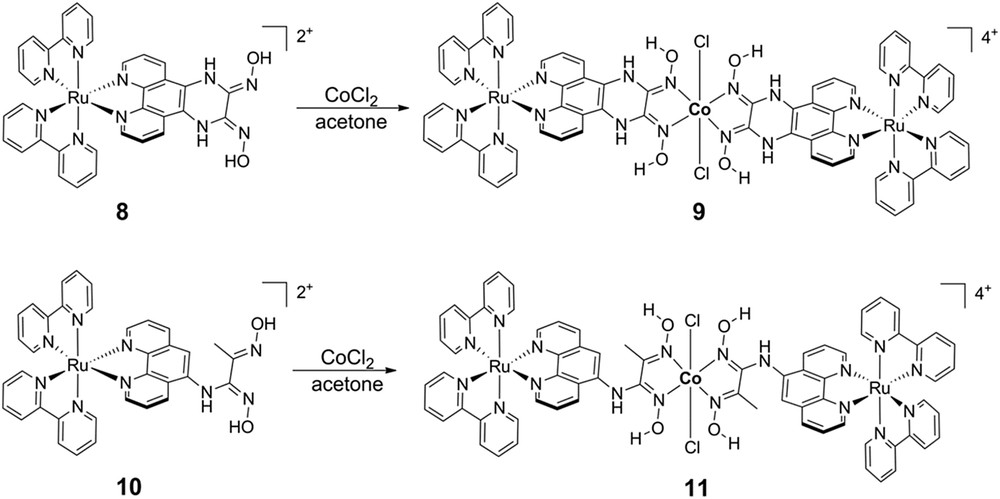
Chemical structures of equatorially-linked cobaloxime photocatalyst assemblies.
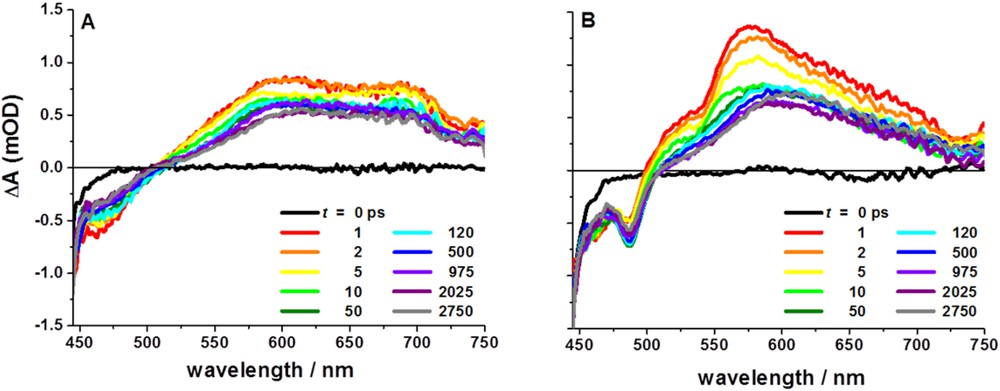
Transient spectra of 8 (A) and 9 (B) following excitation of the Ru(II) chromophore module at 420 nm in CH3CN.
A related strategy to elaborate on the cobaloxime structure is to functionalize the saturated hydrocarbon chain which bridges the imines in cobalt diimine–dioxime structures [72]. As a result of the covalent bond linkage connecting one side of the macrocycle, the cobalt diimine–dioximes enjoy better stability than the more common, symmetric proton-capped or BF2-capped macrocycles, yet maintain the proposed proton binding site at the singularly deprotonated oxime. Although to the best of our knowledge, this approach has not yet been used to attach molecular chromophores but has been successfully used to tether cobaloximes to carbon nanotubes [73] and nanoparticles [74] which quickly supply the reducing equivalents needed for proton reduction and stabilize the molecular species in general.
Recent detailed structural evaluation of the chemical, electrochemical or photochemical reduction of 2 has pointed to the formation of Co(II)—Co(II) dimers in solution [75,76]. This work suggests that a predominant mechanism for H2 catalysis may proceed through reduction and protonation of the ligand, rather than via a Co(III) hydride, which certainly disrupts the prevailing mechanistic understanding for proton reduction catalysis by cobaloximes. This information can be used to envision new photocatalyst designs in two opposing directions. New photocatalyst architectures could be designed so as to either prevent dimer formation by a sterically bulky macrocycle or cobaloxime encapsulation [77,78]. Alternatively, new photocatalysts could capitalize on the proposed non-innocence of the glyoxime macrocycle and extend the typical glyoxime to include those which are engineered to support multi-electron redox chemistry [79].
4 Conclusions and outlook
The imaginative design and thorough study of new supramolecular photocatalysts have provided important mechanistic information applicable to artificial photosynthesis. In particular, because the cobaloxime catalyst module is so well-studied and well understood, cobaloxime-based linked photocatalysts remain an important platform for the development of next generation photosynthetic architectures. As demonstrated by the structural evolution of cobaloxime-based photocatalysts in our own lab, the synergy of new synthesis and targeted and complementary physical characterization by a variety of light sources has enabled the identification of a catalytically important transient intermediate and generated a path forward to extend its stability and photocatalytic efficiency. As a general strategy, the close integration of new synthesis with high-resolution physical characterization will enable a feedback loop for continued evolution in both scientific understanding and technological applicability of artificial photosynthesis. But the need for wide-scale artificial photosynthetic technology is urgent, and the question now is: how do we accelerate this iterative loop and enable the next revolution in solar energy conversion? New photocatalyst designs should be pursued that have the ability to spontaneously capture substrates (i.e., H2O, H+, or CO2), drive multiple and accumulative electron transfers, and effectively couple complementary redox photochemistry. The emergence of faster and brighter X-ray sources will enable extremely high-resolution characterization of these new designs in both time and space, yielding extraordinary insight into light-induced structural dynamics and unprecedented imaging capabilities to directly visualize the mechanism of artificial photosynthesis in action.
Acknowledgments
KM gratefully acknowledges funding from the Division of Chemical Sciences, Geosciences, and Biosciences, and Office of Basic Energy Sciences of the U.S. Department of Energy through Grant DE-AC02-06CH11357.