

1 Introduction
This review article aims to show the research developed by the current group of scientists at the School of Chemistry at Newcastle University in the last five years, focused on artificial photosynthesis and covering various aspects from light harvesting and electron transfer to catalysis and devices. The researchers in Newcastle are currently involved in this field and the work hereafter showcased are of Professors Anthony Harriman and Andrew Benniston, founders of the Molecular Photonics Laboratory in 2001, who recently were joined by Dr Pau Farras (Materials Science Institute of Barcelona and Institute of Chemical Research of Catalonia), Dr Fabio Cucinotta (University of Muenster and University of Eastern Piedmont), Dr Anna Reynal (Institute of Chemical Research of Catalonia and Imperial College London) and Dr Elizabeth Gibson (University of Nottingham). Although, some of the studies showcased below from the latter four researchers have not been carried out in Newcastle, the authors would like to stress that the experience and knowledge in the field is well represented by the current researchers in Newcastle and will be demonstrated in the following sections.
Artificial photosynthesis (AP) is an old term to describe the area of science that studies the natural processes of photosynthesis and tries to mimic them in artificial systems. Early texts on the topic can be found in Japan and the USSR but were mainly related to plant biochemistry [1]. In the 70s, the oil crisis prompted a huge demand for new and alternative energy sources, and AP was already thought to be a viable and promising solution to help solve the energy issue in a sustainable manner. Most of the work done was summarized in the report of Archer et al. [2]. AP became the hot topic from the 70s on and the early studies of Fujishima and co-workers on molecular water splitting using TiO2 [3a] are seen to be seminal contributions to the field. Since then, most notable scientists, including Graetzel, Lehn, Balzani, Wasielewski, Meyer, Porter and Harriman [3b–g], have been working on this topic. Harriman's early report on AP was published in 1978 in Nature [3h], and almost 40 years later, there has not yet been a practical demonstration of economically viable technology.
In Fig. 1 the effect of the oil crisis in 1973 on the number of publications related to AP in the following years is shown. After the first series of preliminary results achieved worldwide with a generally steady publication rate, there has been a steep increase of papers from 2009 onwards. The timescale coincides with the publication of the first single-site molecular system for light-driven water oxidation, which is well known to be the bottleneck for light-driven water splitting [4a], and photochemical-assisted water splitting using a photoelectrochemical cell [4b]. At the same time, the high resolution of the crystal structure of the natural Photosystem II (PSII) helped to improve the understanding of the mechanism of water oxidation in the natural photosystems [5]. Overall, along with the observed increase in the number of scientific publications, the amount of investments worldwide from both the public and private sectors has ramped up. An example of this was the establishment of the UK Solar Fuels Network in which Prof. Harriman, as the Royal Society of Chemistry’s Solar Champion, is still actively involved.
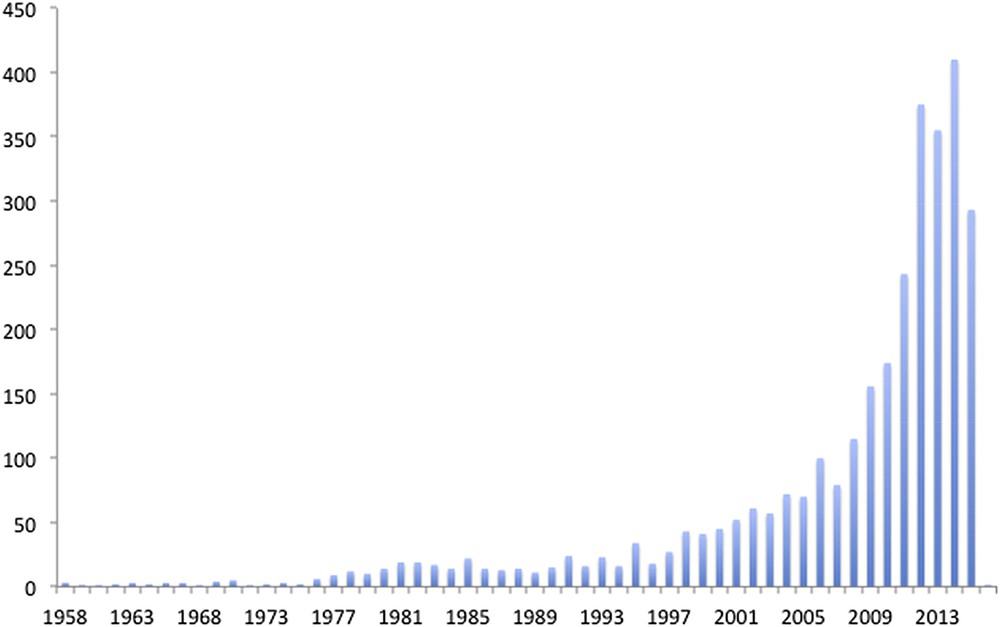
Evolution of publications related to Artificial Photosynthesis in the last 50 years.
Source: Scifinder 2015 (accessed September 9, 2015).
In the following sections, the most remarkable work involving current research studies at Newcastle University in the topics of light harvesting, photocatalysis and devices will be summarised, in order to give an overview of the expertise acquired in recent years both in molecular systems and solid state materials.
2 Light harvesting systems
2.1 Light harvesting studies using molecular systems
The study of light harvesting and electron and energy transfer has been the main focus of research within the photochemistry community in Newcastle. The supramolecular assembly found in natural photosynthesis is a very complex machinery that absorbs light and transfers it to a reaction centre located in the membrane of the plant chloroplasts, where the catalysis takes place. At the same time, the light-driven process is able to redirect some of the energy absorbed to other sections of the membrane in order to protect it from overexposure to light.
Synthetically, one of the main interests has been the preparation of highly fluorescent dyes with the aim of minimizing non-radiative energy losses while maximizing the efficiency of light absorption over the visible spectrum by the dyes. Simple molecules have been studied using standard photophysical techniques including 1H,3H-isochromeno[6,5,4-mna]xanthene-1,3-dione (CXD) [6].
In the natural system, multiple donor-acceptor units are present with uniquely high spatial organization and, taking inspiration from the design of these photosystems, a vast range of multi-chromophoric complexes have been prepared with the aim of studying and manipulating the charge-transfer processes occurring at both intramolecular and intermolecular levels. Boron dipyrromethene (bodipy) dyes have been extensively studied in Newcastle by Harriman and Benniston and are described in the following sections [7].
2.1.1 Small dyad systems
The understanding of charge transfer processes in small systems has been the fundamental for the preparation of more elaborated complexes. Electronic energy transfer strongly depends on the distance between the donor and acceptor and also on their mutual spatial organization. To maximize the transfer efficiencies, cofacial bodipy dyads were prepared with varying lengths of the bridging unit and their transfer rates were measured to give a clear correlation between distance and rates [8]. Nevertheless, the same dye was used as the donor and acceptor; further studies were conducted using others dyes such as anthracene [9], hydroquinone [10,11], julolidine [12] and dicyanovinyl [13] in which the effect of the electrochemical potential on the transfer rates was measured. During those studies, it was found that the bridging unit plays a key role in the transfer rates, in particular in the recombination to the ground state. Using a bodipy dye as the donor and a simple amine as the acceptor, charge transfer rates with or without a naphthalene bridging unit were measured; however, the energy levels of the donor and acceptor did not display an optimal match and no direct effect could be observed [14]. It was found that a bridging unit with a strong coupling between the donor and acceptor promotes a rapid recombination. To prove that, a triazolyl unit, with a weaker coupling capability, was used to join a bodipy acceptor and a pyrene-thiophene (see Fig. 2) or an anthracenyl donor and their transfer rates were measured, giving a good correlation between the Förster energy transfer and the calculated overlap integrals [15,16]. Other systems that have been studied contain the expanded acridinium dye as the donor and dimethylaniline or trialkyloxybenzene as the acceptor with ultra fast charge transfer rates and charge recombinations within the 100–200 ps time domain [17].
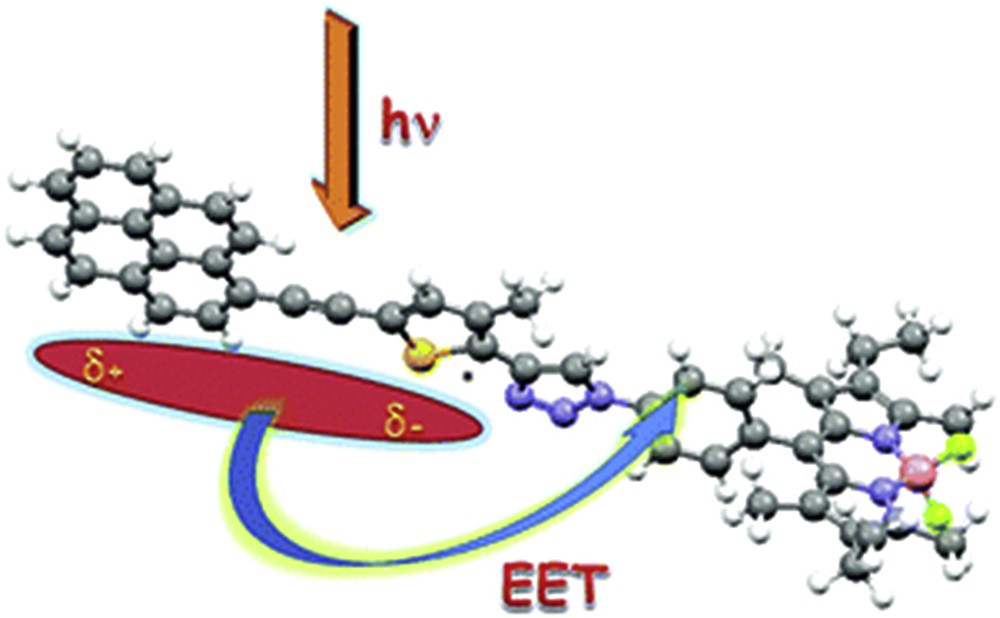
Representation of electronic energy transfer from a pyrene-based excited state to Bodipy [15]. Reproduced with permission of the Royal Society of Chemistry.
The natural system adapts itself towards external perturbations using a seamless of processes occurring in the photosynthetic membrane. It is possible to mimic some of them in artificial systems and understand the effect of external inputs on the rates of charge transfer. Some of the perturbations have been studied by our group and are described below:
2.1.1.1 Solvent polarity
A rigid molecular dyad, comprising two disparate boron dipyrromethene dyes operating as terminals and being separated by a spirobifluorene connector, was selected as the model to study the influence of solvent polarity on the rate of intramolecular electronic energy transfer. Förster theory is widely used to describe energy transfer processes and is of crucial significance in biological studies. However, the theory lacks a proper description of molecular dyads where the separation distance does not exceed the dipole length, and also fails to account for solvent molecules that might affect the parameters that govern the electronic energy transfer processes (EET). In this work, Harriman et al. have found that the effect of the solvent on EET increases in non-polar solvents and it is due to small perturbations of the optical absorption spectrum of the acceptor, which affects its transition dipole moment [18].
2.1.1.2 Interaction with ions
Ion channels control natural membranes; likewise, the photosynthetic system is largely governed by the presence of ions that can redirect or switch on and off the charge transfer processes. A dyad system bearing a bodipy unit as the acceptor and a dimethylamino moiety as the donor was bridged using 2,2′-biphenol [19]. The rate constants for forward and back electron transfer were measured in the presence and absence of a Li+ cation, and an enhanced excited state quenching was found with Li+.
Similar to the previous case, the complexation of a bodipy dyad bridged by a polyethylene glycol chain using ammonium hexafluorophosphate promotes the intramolecular charge transfer, while in the absence of cations each dye acts independently [20].
Hablot et al. [21] presented another example of ionic effect using a triad system comprising two disparate bodipy units connected with a central pyrrolyl derivative core that absorbs and emits at a higher energy than either termini. The design principle is based on the possibility to switch the intramolecular EET flow by the protonation at the amine sites of one of the terminals, as shown in Fig. 3. Remarkably, the strategy works using a photoacid generator in both solution and poly(methyl methacrylate) (PMMA) films.
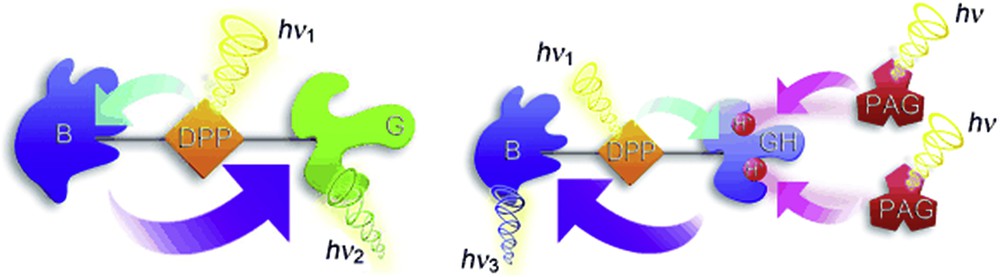
Representation of the chemically modified switching process, in which conversion of G into GH+ causes a change in the direction of EET from the central donor to the peripheral acceptors [21]. Reproduced with permission of John Wiley & Sons Ltd.
2.1.1.3 Supramolecular assemblies
Ionic self-assembly is a powerful technique that employs attractive electrostatic forces to organize functionalized molecular modules into materials. This strategy was used by Olivier et al., utilizing charged bodipy dyes to form cassettes that display intracomplex EET [22]. Red- and blue-absorbing bodipy dyes, bearing opposite charges, associate in solution in a 1:2 complex to promote rapid charge transfer kinetics. In addition, the complex shows liquid crystalline properties when heated and the mesomorphic state retains the efficient EET.
2.1.1.4 External pressure
Alamiry et al. have done an extensive study on how subtle changes in the molecular conformation affect the fluorescence properties and, in turn, charge transfer kinetics using high pressures [23]. Using the same cofacial bodipy dyes described above, it was possible to observe the formation of excimer emission at ambient temperature under high pressures. Using a more rigid system, a bodipy dye is appended by ancillary pyrene or perylene units. The structures are shown in Fig. 4. The net effect of high pressure on this molecular system is to curtail EET from pyrene to bodipy by 5% while EET from perylene to bodipy suffers a 26% decrease in efficacy [24]. The same authors have used a prototypic donor-acceptor dyad featuring a bodipy dye linked to a dicyanovinyl unit. In this case, under moderate applied pressure, the formation of the exciplex is hindered by restricted rotation around the semirigid meso-phenylene ring, whereas at high pressures the molecule undergoes reversible distortion that severely quenches exciplex fluorescence [25].
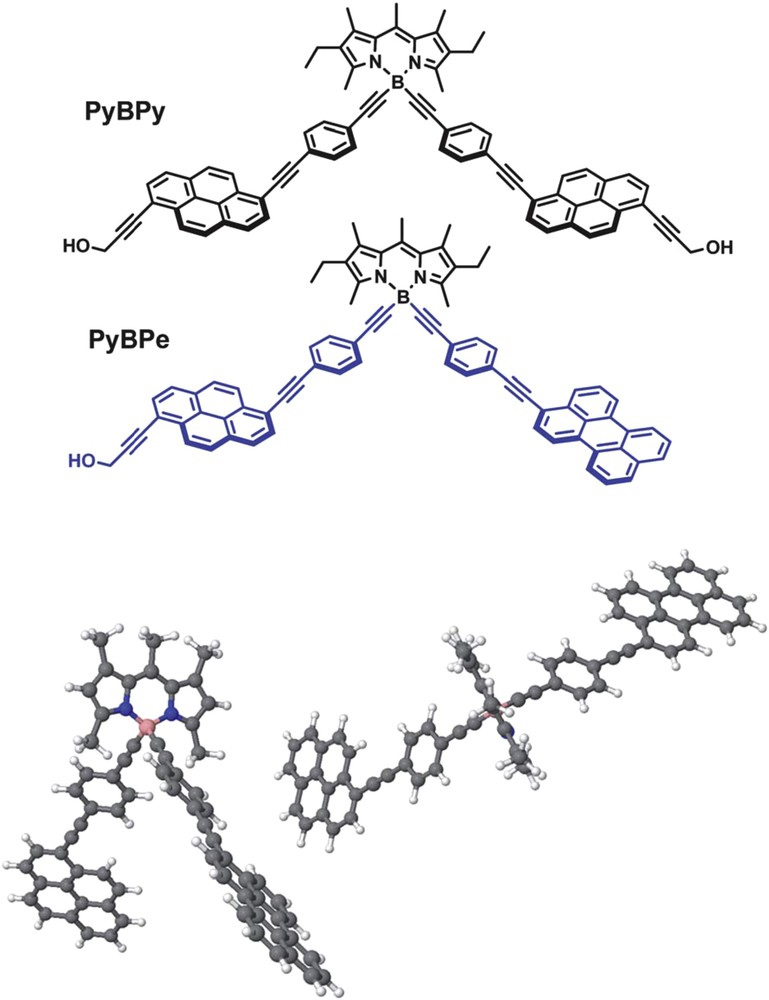
Molecular formulae for PyBPy and PyBPe (upper panel). Two orientations of the energy-minimized structure computed for PyBPe with the ancillary ethyne function removed for clarity of presentation (lower panel) [24]. Reproduced with permission of the Royal Society of Chemistry.
A better control on the molecular conformation has been achieved using a slightly more rigid bridging unit, namely carborane. The combination of bodipy dyes and o- and p-carborane bridging units allowed unprecedented information about the effect of temperature and pressure in long-range EET along the molecular axis [26] and through-space across distances from 36 to 16 Å [27]. The experimental data obtained in these studies have been compared with theoretical protocols for expressing the EET between closely spaced but weakly coupled reactants with good to excellent agreement.
2.1.2 Multidyad systems
Numerous molecular-scale arrays have been built and studied to fully understand light-induced events between well-defined dyes. These multicomponent systems have been very successful as bioinspired mimics and as vehicles for cascade effects leading to long-lived charge separated states. The control of directionality and flux of transfer events still remains a challenge although recent advances in the topic have opened new avenues towards designing better systems. A good example can be found in the work by Benniston et al. [28], in which a molecular tetrad constructed from a porphyrin, a ruthenium terpyridyl moiety and two terminal quinones presents the properties of bidirectional electron transfer. The excitation of the molecule with two different wavelengths, corresponding to either the porphyrin or ruthenium unit, leads to directing EET in opposite directions along the molecular axis.
A multicomponent dendrimeric system prepared solely from porphyrins has been studied in which there exists a logical gradient for either energy or electron transfer between the porphyrin units. When using a central free-base porphyrin with peripheral zinc(II) porphyrins, energy transfer steps occur at the triplet level. In contrast, when using a gold(III) porphyrin in the core of the dendrimer equipped with the zinc porphyrins, light-induced electron transfer is possible. All possible forward and back transfer kinetics has been measured by time-resolved spectroscopy, and the final charge-shift state of the gold analogue survives for some milliseconds in liquid solution at room temperature [29].
As mentioned above, bodipy dyes have been the object of numerous studies in Newcastle. In this section, we focus on the design of multichromophoric molecules bearing several bodipy units attached with themselves or in combination with other subunits. The first example is constructed combining C60 decorated by yellow and/or blue bodipy dyes and the main feature of the system is that the fullerene core is a poor electron acceptor and does not participate into the light-induced transfer processes. On the other hand, intramolecular electronic energy transfer from the yellow to the blue dyes can be followed by steady-state and time-resolved spectroscopy [30]. Ziessel and Harriman summarized the most important features to design and construct efficient molecular funnels using multiple chromophores and ensure a directed flow of light-induced transfer processes. They also compared some of the properties of the artificial systems with the biological machinery. In particular, they highlighted the application of bodipy molecules since selective illumination of these dyes leads to a multitude of energy and electron transfer events, involving both singlet and triplet excited states [31]. To conclude with the section, one of the most elegant examples of cascade energy transfer has been published in which an artificial light harvesting array comprising 21 discrete bodipy chromophores is arranged in order to act as a molecular funnel that is able to direct the excitation energy from the peripheral units to a focal point. The transfer kinetics slows down as the exciton moves toward the central target. The measured rates from the rim to the focal point of the molecule, abbreviated as PMEX, are shown in Fig. 5. In addition, the system is able to adapt depending on the photon flux and, as such, it appears to provide effective means for self-protection by junctions where photon annihilation might take place [32].
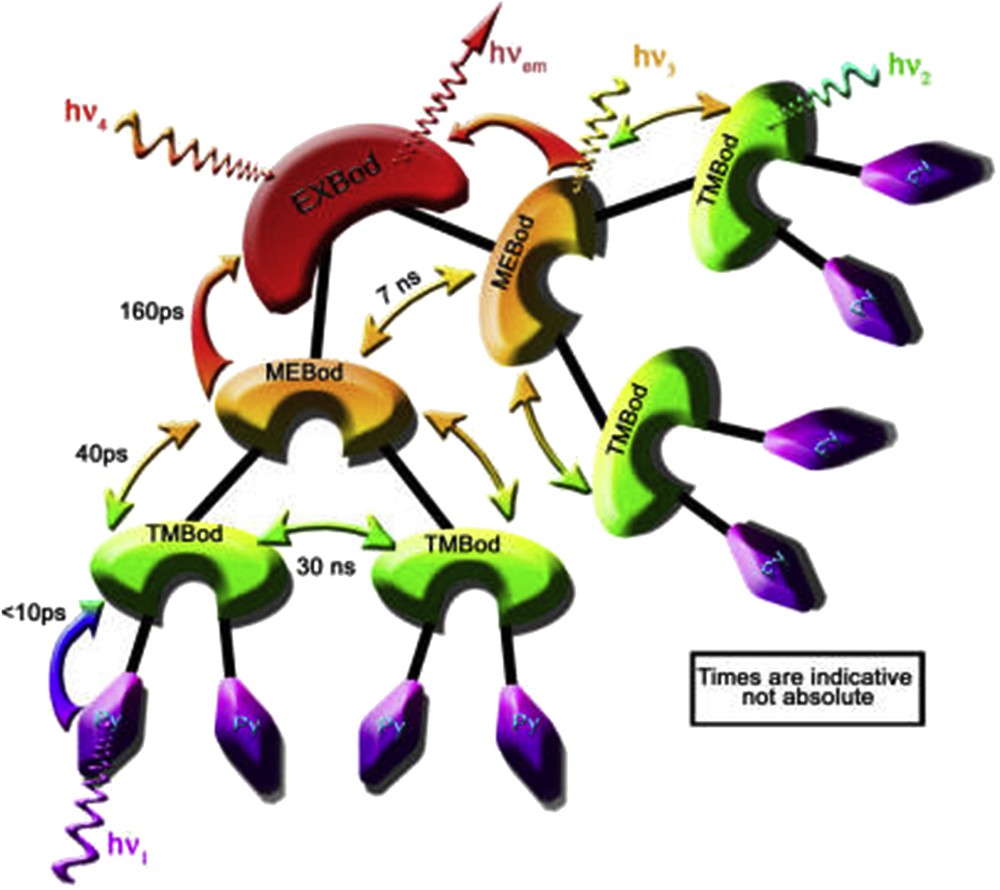
Pictorial representation of the EET flow in the PMEX array, allowing for excitation into any of the main chromophores. Reprinted with permission from reference [32]. Copyright 2013 American Chemical Society.
2.2 Light-harvesting studies using solid state materials
The search for light harvesting and converting systems has inspired great efforts in designing artificial antennae based on a wide variety of supramolecular systems made up from small chromophores, as described above. In most of them, the molecular units are organized in a very primitive level and, although molecularly dispersed, still show drawbacks related to photostability and aggregation phenomena that might quench photo-generated excited states. To circumvent these problems, host-guest systems have been developed that take advantage of non-covalent interactions and self-assembly to impose specific spatial organizations of chromophoric units, mimicking the natural photosystems [33]. This is of fundamental importance as it enables to control the dimensions of space, energy and time within which the antenna effect is realized and the processes of unidirectional energy and electron transfer are driven.
Among various multifunctional materials, nano-containers are promising candidates as controlled devices because of their ability to enclose molecules. They can also be used as chambers for controlled chemical reactions, as already well demonstrated in catalysis. In this regard, channel-forming microporous host-guest compounds display unique properties due their uni-dimensional character [34]. In particular, alumino-silicates like zeolite L and porous silica such as MCM and SBA have been intensively investigated, owing to the versatility and stability of the inorganic host system, and a recent work by Cucinotta and colleagues is reported below.
2.2.1 Zeolites
Photoactive dye molecules can be entrapped within the channels of zeolite L and, according to their size, are mostly present as monomers, although aggregation might occur under particular conditions that can partially quench the fluorescence. The resulting host-guest systems exhibit high internal ordering, owing to the strict orientation of the entrapped molecules in the narrow channels. This leads to new functionalities, such as optical anisotropy and efficient unidirectional Förster resonance energy transfer (FRET). Several photonic antenna materials have been reported that contain up to three different kinds of dyes [33,34]. Recently, a FRET antenna was studied, for which light emission from the inserted dye molecules was obtained by photo-excitation of quantum dots decorating the whole zeolite L surface (see Fig. 6) [35].
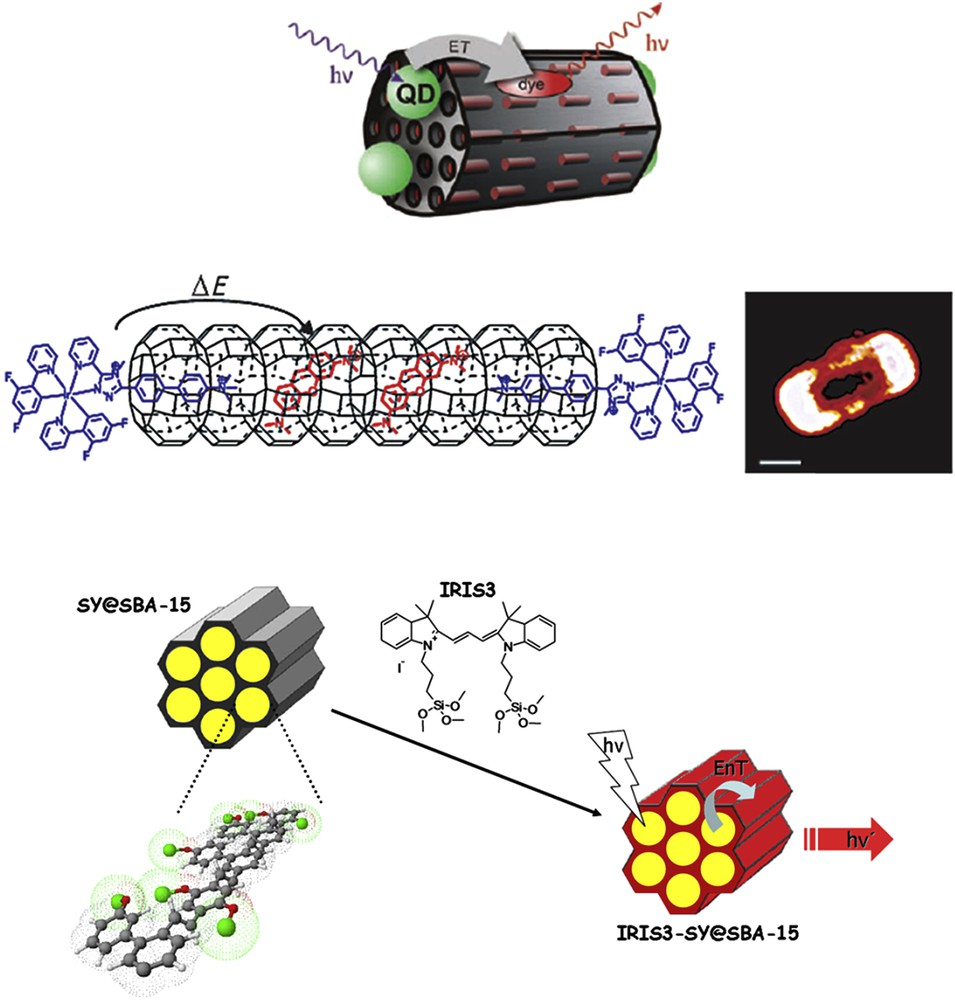
Top: Schematic representation of the assembly and of the photoinduced energy transfer from the excited QDs to the dye molecules. Middle: Schematic representation and fluorescence microscopy images of a zeolite L crystal, loaded with an iridium(III) complex stopper at the channel entrances and an acceptor phenoxazine dye. Bottom: Schematic preparation route for the silica-based hybrid antenna: a) impregnation of SBA-15 pores with a semiconductive polymer followed by b) surface functionalization of the silica particles with a cyanine dye [35–36].
Furthermore, to interface the guests to the outer environment, stopper molecules, that is, molecules that are able to enter partly into the channels, are found to be a powerful tool, especially for injecting excitation energy into the channels or, alternatively, collecting it from inserted dye molecules [34]. In this regard, transition-metal complexes, particularly triplet emitters, fulfill the requisites of effective closure molecules and energy-transfer units. By using octahedral complexes, it is possible to design stopper systems made of a head moiety, a coordinated metal ion, which is too big to enter the pore openings of a zeolite channel, and a tail moiety, such as an extension of one of the coordinated ligands, which is narrow enough to penetrate the channel. By using iridium(III) stopper molecules, which also display a tuneable light emission over the whole visible range, it has been possible to design and study hybrid antennae where the excitation energy is harvested by the complex and transferred to a small organic dye loaded into the zeolite channels and possessing a suitable orientation to enhance the transfer efficiency (see Fig. 6) [36].
2.2.2 Silica
Photonic antenna arrays using silica and organosilica nanoparticles have also been recently reported and their applications as possible light-harvesting antennae have been extensively researched. It has been shown that energy transfer occurs from excited polymer molecules inserted into a mesoporous material (SBA-15) to cyanine dyes covalently linked to the surface of the silica [37]. As displayed in Fig. 6, a photostable antenna system was made, in which an encapsulated PPV derivative acts as the energy donor to an acceptor stably linked outside the SBA-15 framework.
A step forward is represented by a modification of the micelle-templated sol-gel mechanism for the synthesis of mesoporous silica, by which it is possible to prepare ordered materials with built-in photoactivity in the network, enabling energy transfer processes from the host to the guest molecules and vice versa. The advantage of such systems is that the structural and morphological properties can be tuned by using specific micellar templates, typically constituted by organic amphiphilic mesophases that are able to model the pore size of the material. Also, porous frameworks may lead to stabilisation of the guest dyes by shielding them against photo-oxidative degradation, commonly induced over time by oxygen diffusion in solution. In a recently developed approach for the construction of host-guest architectures, a new class of photoactive amphiphilic molecules was designed that, upon micelle formation, can be used as suitable template for porous materials [38]. The study showed that it is possible to modulate the photoactivity of the materials, depending on the aggregation properties of the chromophoric templates.
3 Photocatalysis
In the previous section we have summarised the studies on light harvesting systems and how to obtain charge separation in a unidirectional manner. Once we understand how to do this, the next logical step is to use the separated charges to drive chemical reactions. As a charge-separated state causes the formation of a positive and a negative charge, one can envisage the possibility of performing both oxidation and reduction reactions [39]. Therefore, this section will cover the work done on photocatalysis using sacrificial reagents to study separately the oxidation and reduction reactions occurring during water splitting.
3.1 Photochemical oxidation reactions
Photochemical oxidation reactions require the use of a sacrificial electron acceptor to drive the reaction. In the case studies reported by Farràs and co-workers, persulfate or [Co(NH3)5Cl]2+ has been used, although other reagents are employed by other research groups. As described in the literature, both sacrificial agents have advantages and disadvantages: for persulfate, the generation of a radical species SO4−· with a very high oxidation potential might cause unwanted side reactions that can mask the efficiency of the catalyst. On the other hand, Co2+ is coloured and cannot be used when studying reaction kinetics using UV-vis spectroscopical methods. In conclusion, depending on the information that one wants to gather, it is better to use one agent or the other, so that unwanted reactions are avoided.
The photochemical oxidation of water was studied using a mononuclear Ru complex (RuC) in the presence of [Ru(bpy)3]2+ as the sensitizer (RuP) [40]. This is one of the first examples in which a well-defined mononuclear water oxidation catalyst works when activated by light. This study paved the way to design new and more efficient oxidation catalysts; indeed, several papers have appeared since then showing higher oxidation activity. The working principle is that the sensitizer is excited by light and the photo-induced charge-transfer state is quenched by the sacrificial acceptor, leaving an oxidized sensitizer. The latter has enough oxidation potential to oxidize the catalyst to a higher oxidation state, and the step is repeated 4 times to reach the necessary active state of the catalyst to oxidize a water molecule to oxygen (Eq. 1). The electron transfer from the catalyst to the sensitizer must be faster than the charge recombination process to prevent back-electron transfer and reduction of the catalyst. For this reason, many research groups worldwide have explored the possibility of preparing multinuclear complexes whereby a light-harvesting antenna is connected to a catalytic unit, so that a more efficient intramolecular charge transfer could occur.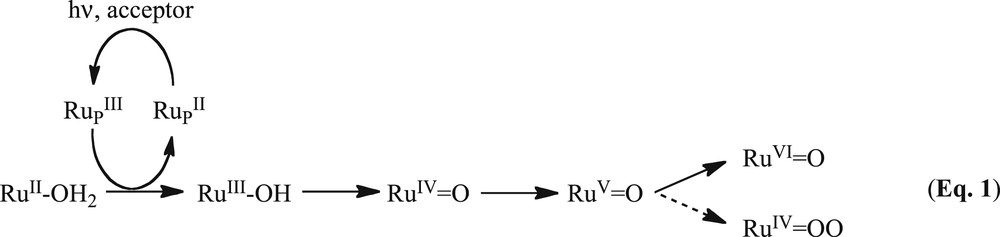
In our case, a series of dyads based on ruthenium complexes were prepared and studied for the photo-oxidation of water, having tetrakis(2-pyridyl)pyrazine (tppz) as a bridging ligand, and with a general formula RuP(tppz)RuC. The latter is able to efficiently couple the metal centres with both sides and to tweak the oxidation potentials of the catalytic unit, making it harder to oxidize. The result is that the RuP is unable to oxidize 4 times the RuC to its active state and is only capable to generate the RuIV=O species (see Eq. 1). It is known that RuIV=O is a good oxidizing agent for organic substrates such as alcohols, sulfides and alkenes. Therefore, the dyad molecules were tested for the oxidation of a series of substrates in the presence of a sacrificial agent and they showed the desired activity. Furthermore, a full photophysical and electrochemical characterization of the molecules was done to understand the charge transfer events happening within the dyad [40].
The sensitizer used in most of the photo-oxidation reactions studied to date is the [Ru(bpy)3]2+. New sensitizers are needed to be prepared and studied to increase the oxidation potential of the sensitizer unit, and also to improve the light-absorbing properties, by covering a wider range of the visible spectrum and increasing the extinction coefficients to absorb more photons. New light-harvesting complexes were prepared with a ruthenium and a bodipy unit, connected through a triazole bridging ligand that decouples both centres with the aim to generate a charge-separated state. As shown in Fig. 7, upon light excitation, energy transfer from the bodipy to the Ru centre occurs. In the presence of a quencher (Q), an electron is removed leaving a RuIII centre that either can oxidize directly the catalyst or oxidize the bodipy sub-unit, which will finally oxidize the catalyst. With this system, a series of organic substrates were oxidized with moderate to good yields but the sensitizers were not able to oxidize a catalyst to higher oxidation states and generate oxygen [41].
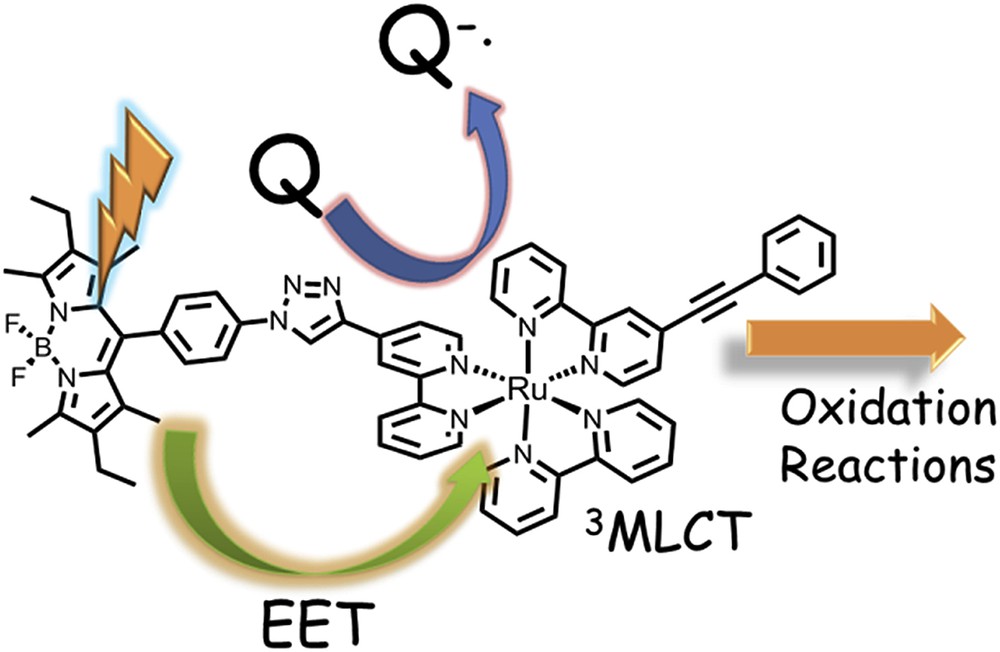
Light induced processes for the oxidation of organic substrates using a bodipy-Ru dyad [41].
3.2 Photochemical reduction reactions
3.2.1 Proton reduction
If oxidation reactions require the use of sacrificial acceptors, photochemical reduction reactions make use of sacrificial donors to drive the reactions. All the examples in this section deal with hybrid materials combining semiconductors and molecular catalysts and, in particular, only proton reduction to hydrogen will be considered.
Developing efficient photocathodes for proton reduction requires a detailed study of injection and recombination processes occurring between the catalyst and the semiconductor. For that reason, Reynal et al. designed a series of cobalt catalysts with anchoring groups to TiO2 at various distances. Forgive the redundancy, as mentioned throughout the text the distance between the donor and acceptor is crucial and limits the transfer kinetics. As shown in Fig. 8, the distances vary from 7 to 9 Å. The photoinduced reduction of the three catalysts anchored on TiO2 is 104 times faster than the reverse recombination and both processes show an exponential dependence on the distance between the semiconductor and the catalytic core [42].
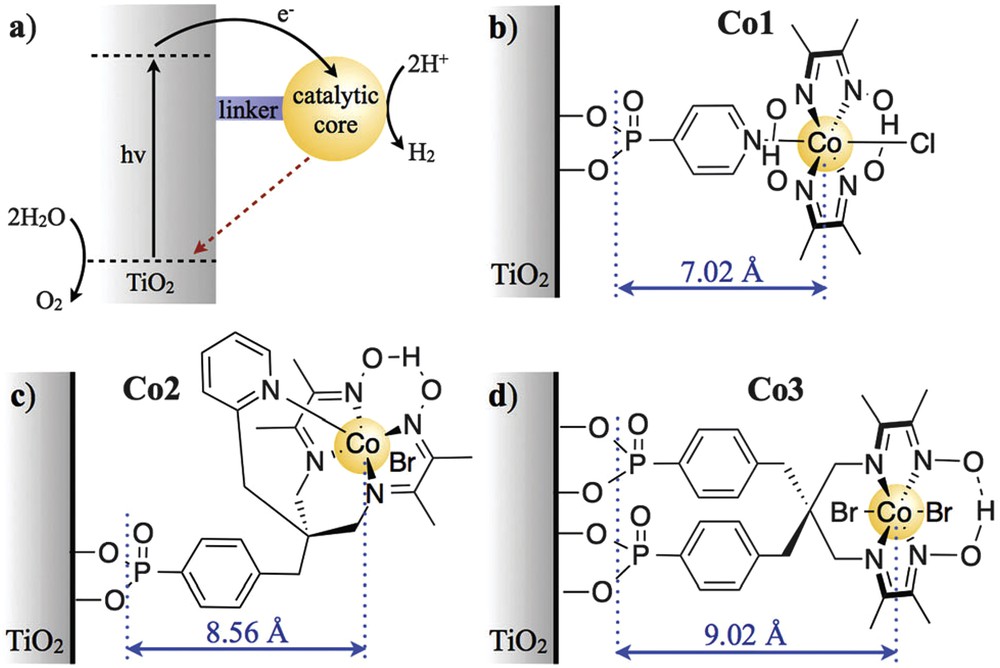
(a) Electron transfer processes in TiO2 functionalised with a molecular catalyst for H2 production after UV-light excitation. The solid black and dashed red arrows indicate charge separation and recombination, respectively. Molecular structures of the catalysts for H+ reduction are shown in (b) for Co1, (c) for Co2 and (d) for Co3 (charges omitted for clarity). The blue arrows indicate the distance between the anchoring groups and the catalyst metal centre (r, Å), as determined by energy minimised DFT calculations [42]. Published by the Royal Society of Chemistry.
Lakadamyali et al. have proposed a hybrid system constructed with TiO2 nanoparticles decorated with phosphonated ruthenium trisbipyridyl (RuP) chromophores and molecular cobaloxime proton reduction catalysts. The reaction conditions were optimized in terms of dye and catalyst loading, type and concentration of the sacrificial donor reagent and electrolyte. Time-resolved spectroscopy showed electron transfer kinetics from the chromophore to TiO2 in approximately 180 ps, and from TiO2 to the catalyst in a 10 μs timescale [43,44].
Even though cobalt catalysts have shown good results towards proton reduction, pendant nickel amines, first reported by DuBois et al. [45], are considered amongst the fastest electrocatalysts for hydrogen generation. Therefore, it is not unusual that a phosphonated derivative of the catalyst has been immobilized on TiO2 and photoreduction of protons has been accomplished using RuP as a sensitizer. Time-resolved spectroscopy confirmed that directed electron transfer from RuP to the nickel catalyst occurred on the nano- to micro-second time scale and the overall system achieved a photon-to-hydrogen quantum yield of approximately 10% [46].
3.2.2 CO2 reduction
A second merit in photochemical reductions is the selective conversion of CO2 to CO; the latter can be used in the Fischer-Tropsch process to produce liquid fuels. Gibson et al. have demonstrated that homogeneous molecular systems can perform this reaction using a zinc porphyrin as the sensitizer and a rhenium complex as the catalyst, as shown in Fig. 9. The importance of the bridging ligand on the performance of the system was discussed in detail, as coupled units showed a reduced efficiency for CO2 reduction [47].
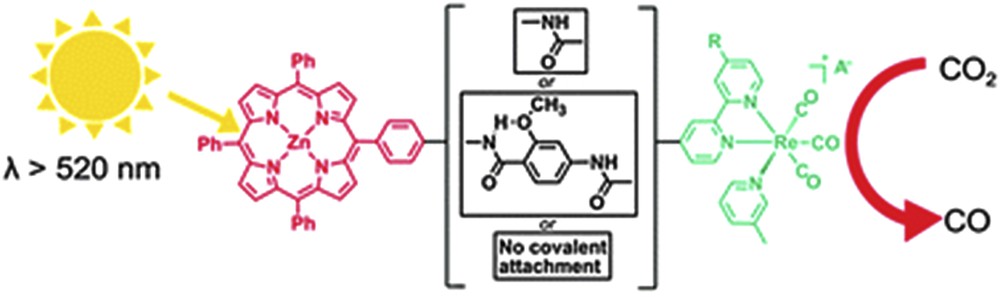
Scheme of the photochemical reduction of CO2 to CO by a molecular dyad consisting of a zinc porphyrin as the sensitizer and [Re(CO)3(picoline)bpy]+ as the catalyst [47]. Reproduced with permission of the Royal Society of Chemistry.
A similar Re catalyst bearing a phosphonate anchoring group has been used in a hybrid system using TiO2 as a semiconductor. The immobilization on the metal surface provides more stability than its homogeneous counterpart, and their photochemical kinetics improve showing an enhanced catalytic activity. This work reported one of the highest turnover number (TON) for photochemical reduction of CO2 using a Re catalyst and visible light irradiation [48].
It has been recently reported that nickel catalysts behave as good carbon dioxide reduction catalysts, and a nickel cyclam complex covalently attached to TiO2 and Zr-doped TiO2 has been tested as photocatalyst. As neither the semiconductor nor the catalyst absorb in the visible region, kinetic studies have been performed with 355 nm light excitation showing that the catalyst is able to accept photoelectrons from the metal oxide in aprotic solvents. Further investigations are underway to solve stability in protic solvents [49].
With the aim to construct a full photoelectrochemical device for water splitting, efficient photocathodes made of p-type semiconductors need to be developed. Pastor et al. have studied Cu2O as a semiconductor for CO2 reduction and the addition of ruthenium oxide nanoparticles results in a six-fold increase in the yield of CO formation. This result correlates with an increase of long-lived electrons on the semiconductor and a partial suppression of electron/hole recombination loss through the transfer of photo-generated holes from Cu2O to RuOx [50].
4 Photoelectrochemical devices
In the previous sections we have shown the recent studies in oxidation and reduction reactions using sunlight. However, all these examples and the majority of studies in the literature are based on sacrificial systems using either sacrificial electron acceptors such as persulfate and Co(III) salts or sacrificial electron donors such as triethylamine, triethanolamine, etc. In these, the irreversible reactions allow faster reaction kinetics and simplify the mechanisms by avoiding some of the recombination reactions. In addition, there is always the need to add more reagents to keep up with the production of the fuel. Therefore, such systems are unsustainable and one can argue that full devices without the need of sacrificial reagents need to be developed, in order to develop technology that is able to provide enough fuel to sustain current and future energy demand. Harriman compiled a summary of the implications of a solar fuel industry together with the fuels that can be produced. Additionally, it was pointed out that nitrogen fixation by photochemical means to produce ammonia still remains a challenge for future research [51].
A variety of options have been proposed to avoid the use of sacrificial reagents such as particulate catalysts and electrochemical cells to take advantage of the inherent redox nature of the abovementioned reactions. The latter consists of photoelectrochemical cells that can either be made of semiconductors or, inspired by the dye-sensitized solar cells, sensitized by using light-absorbing antennae to give dye-sensitized photoelectrosynthetic cells (DSPECs). One of the possible solutions envisaged by Harriman was the use of organic substrates as a means to produce a solar fuel. This option has been explored by Farràs et al. using methyl phenyl sulphide and a water-soluble styrene as substrates that can be oxidized in water and produce hydrogen as solar fuel, as shown in Eq. 2 and Fig. 10. In these studies, a complete thermodynamic and kinetic characterization of all the reactions (favourable and unfavourable) occurring at the DSPEC has been done, and the main deactivation pathways have been identified. In addition, the DSPEC uses a Ru-based molecular oxidation catalyst in solution giving the possibility to use different catalysts depending on the substrate [52,53].
| (Eq. 2) |
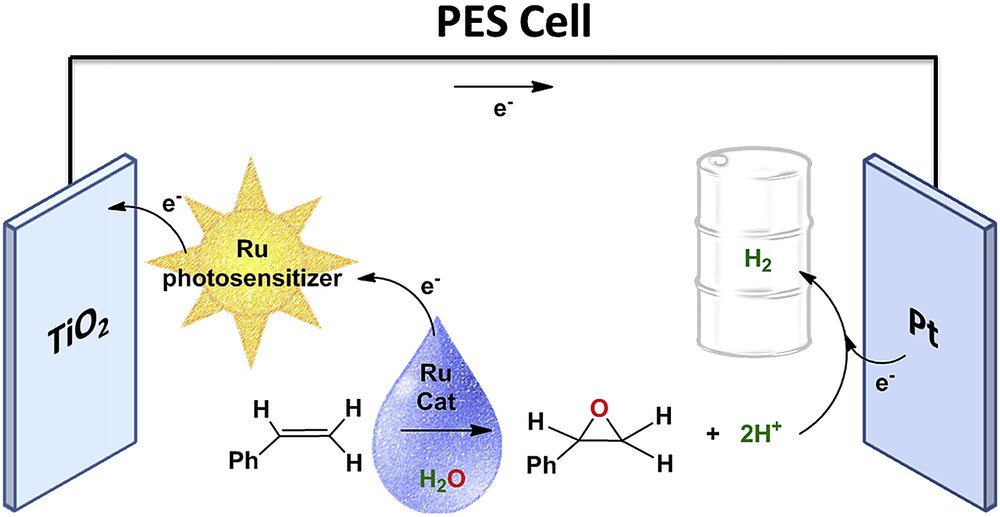
Schematic representation of the photoelectrosynthesis cell for the oxidation of a water-soluble alkene and the production of hydrogen, using a molecular ruthenium sensitizer and a molecular ruthenium catalyst in solution [53].
5 Conclusions
This review intends to give the reader an overview of the expertise developed in Newcastle in the past few years in the area of artificial photosynthesis. The group of researchers has covered most of the aspects needed to construct an efficient and functional photoelectrochemical device for water splitting, as shown in the previous sections. From the synthesis of catalysts and sensitizers to the characterization by electrochemical and photochemical means, there is the intention of leading the research in solar fuels and contributing to the worldwide effort in the pursuit of carbon-neutral or carbon-free fuel products.
Aknowledgements
P.F. gratefully acknowledges the Royal Society (NF120652) for the Newton Fellowship and for financial support.