

1 Introduction
Amlodipine (A-1) ((R,S)-3-ethyl 5-methyl 2-[(2-aminoethoxy)methyl]-4-(2-chlorophenyl)-6-methyl-1,4-dihydropyridine-3,5-dicarboxylate) (Fig. 1) is a 1,4-dihydropyridine-based long-acting calcium channel blocker extensively used for the treatment of hypertension, angina pectoris, and other cardiovascular diseases [1]. Like other calcium channel blockers, it acts by relaxing the smooth muscle cells of arterial wall by decreasing total peripheral resistance, thus reduces blood pressure; in angina, it increases blood flow to the heart muscle [2]. Essential hypertension results in an increased oxidative stress level, which requires the use of potent antiatherosclerotic drugs that not only directly affect the arterial wall, but also demonstrate antioxidant and free radical scavenging properties. The ability of dihydropyridine as calcium channel blockers to protect against oxidative endothelial cell injury in comparison with vitamin E (Trolox) is well established [3–7].
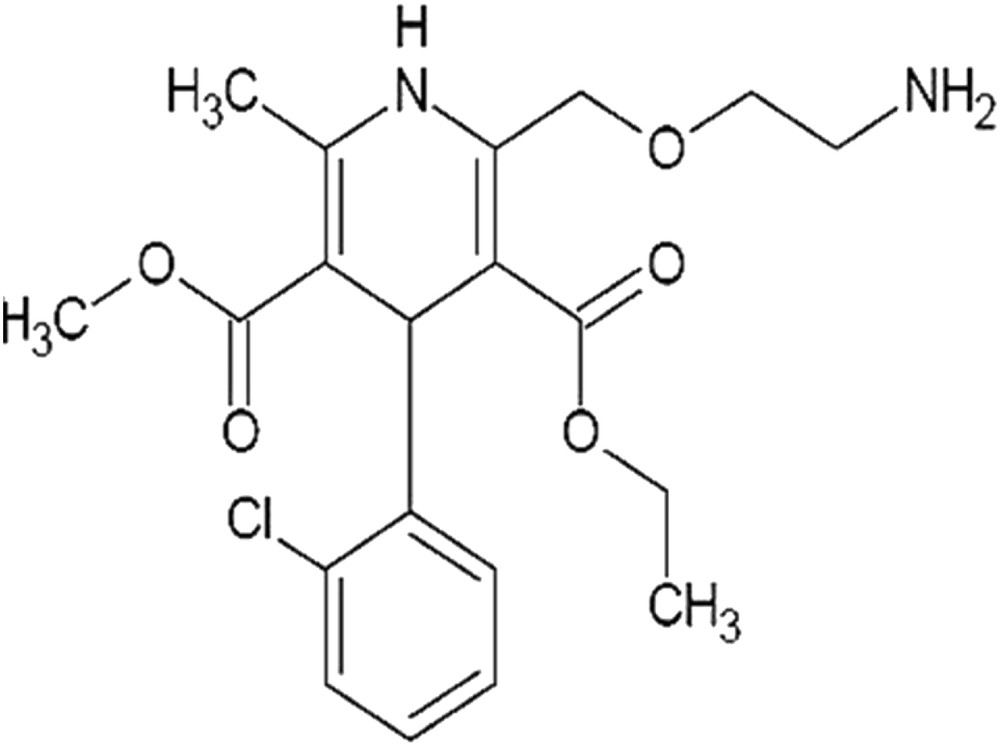
Chemical structure of A-1.
Herein, we report synthesis, crystal structure, thermal properties, in vitro antioxidant potential, metal chelating ability, and antiurease activity of 2-[2-(4-chloro-benzenesulfonylamino)-ethoxymethyl]-4-(2-chloro-phenyl)-5-hydroperoxycarbonyl-6-methyl-1,4-dihydro-pyridine-3-carboxylic acid 3-ethyl ester 5-methyl ester dehydrate (A-2), 2-[2-(4-chloro-benzoylamino)-ethoxymethyl]-4-(2-chloro-phenyl)-6-methyl-1,4-dihydro-pyridine-3,5-dicarboxylic acid 3-ethyl ester 5-methyl ester (A-3), and 4-(2-chloro-phenyl)-2-[2-(2,5-dichloro-benzenesulfonylamino)-ethoxymethyl]-6-methyl-1,4-dihydro-pyridine-3,5-dicarboxylic acid 3-ethyl ester 5-methyl ester (A-4) (Fig. 2). This work was a successful effort to enhance the therapeutic effect of amlodipine by preparing new more effective analogues. To achieve the task, sulfonamide and benzoyl ester groups were incorporated in A-1. These groups impart antimicrobial, anti-inflammatory, anticancer, antiprotozoal, antiviral, and carbonic anhydrase inhibitory activities [8–10]. All the synthesized compounds A-2–A-4 were screened for their in vitro antioxidant, pro-oxidant chelation, and antiurease activities.
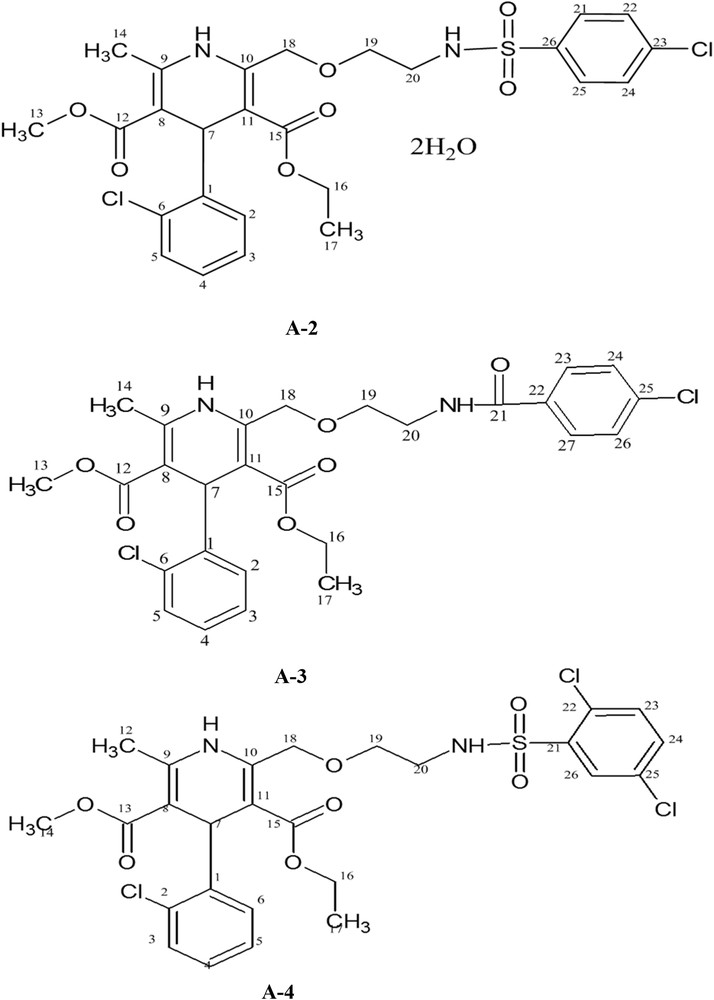
Chemical structures of A-2, A-3, and A-4.
2 Experimental
2.1 Material and methods
All chemicals and materials used were of analytical grade. All melting points were obtained on an Electrothermal (Griffin 1090) melting point apparatus and reported without correction. The IR spectra of the compounds were scanned through Perkin Elmer 1600 FTIR (USA) and MIDAC-M 2000 (USA) by using KBr pellets over the range of 4000–400 cm−1. Thermogravimetric analysis (TGA) (25–600 °C) profiles were recorded under an inert atmosphere (N2) on an SDT Q600 (TA instruments, USA) at a ramp rate of 10 °C/min. Antioxidant capacity was evaluated using a UV-1700 Pharma Spec UV–Visible Spectrophotometer, Shimadzu, Japan, equipped with Peltier temperature-controlled device. Elemental analysis (CHNS) was performed using a Vario Micro Cube (Elementar, Germany). Mass spectra were recorded on a JEOL MS Route with ionization mode EI+. 1H NMR spectra were recorded on Bruker AVANCE AV 600 and DPXQ 400 spectrometers. The IUPAC name of each compound was derived using Chem sketch (freeware version) and atomic numbering scheme was assigned to interpret 1H NMR spectra. The single-crystal data were collected using a Bruker Kappa APEX II CCD diffractometer (graphite monochromated Mo Kα radiation, λ = 0.71073 Å) at room temperature, and data reductions were performed using SAINT [11]. The structures were solved by direct methods with SHELXS, the resulting atomic models were developed and refined against |F|2 using SHELXL [12]. The “observed data” threshold for calculating the R(F) residuals was set as I > 2σ(I). The C and N bound H atoms were placed at idealized locations (C–H = 0.93–0.97 Å and N–H = 0.86 Å) and refined as riding atoms. The O-bound H atoms were located in the difference Fourier maps and refined as riding on their relative atoms. All non-hydrogen atoms were refined with anisotropic parameters. The structural models were analyzed and validated with PLATON [13] and full refinement details using the crystal information file. Mercury program [14] software WinGX was used to prepare molecular graphics for publication [15]. Supramolecular analyses were performed, and the diagrams were prepared with the aid of PLATON and CrystalMaker [16].
2.2 Syntheses
Aqueous sodium carbonate solution (3–5%) containing A-1 (286 mg, 0.7 mmol) was treated with equimolar ratio of 4-chlorobenzenesulfonyl chloride, 4-chlorobenzoyl chloride, and 2,5-dichlorobenzenesulfonyl chloride separately for 3–6 h. The execution and completion of reactions were indicated by thin-layer chromatography. White precipitates were formed by adding 3 N HCl dropwise up to 2.0 pH. The products were filtered and recrystallized in MeOH/EtOAc (50:50 v/v) to obtain colorless blocks.
2.2.1 2-[2-(4-Chloro-benzenesulfonylamino)-ethoxymethyl]-4-(2-chloro-phenyl)-5-hydroperoxycarbonyl-6-methyl-1,4-dihydro-pyridine-3-carboxylic acid ethyl ester dehydrate (A-2)
Yield: 67%, mp 148–150 °C, 1H NMR (400 MHz, CDCl3, δ, ppm): 7.9 (d, J = 8.1 Hz, 2H, H-23, H-25), 7.5 (d, J = 8.1 Hz, 2H, H-22, H-26), 7.4–7.1 (m, 4H, H-2–H-6), 5.4 (s, 1H, H-7), 4.9 (s, 2H, H-18), 4.07 (m, 2H, H-16), 3.7 (m, 2H, H-19), 3.6 (s, 3H, H-13), 3.2 (m, 2H, H-20), 2.3 (s, 3H, H-14), 1.2 (t, 3H, H-17). IR (KBr) νmax (cm−1) 3377 (N–H stretch), 3267 (sec. sulfonamide), 1686, 1662 (CO stretch), 1645, 1603 (CC stretch), 1480, 1429 (aliphatic C–H, bending), 1281 (SO, asymmetric stretch), 1202 (OC–O ester, stretching), 1186 (O–C–C of ester, stretching), 1082 (SO symmetric stretch), 1035 (aromatic C–Cl), 757, 737 (aromatic C–H, bending). MS m/z (%): 618 (2, M+), 585 (45), 583 (100), 581 (94), 318 (6), 286 (8). Anal. Calcd for C26H32Cl2N2O9S (619.51): C, 50.41; H, 5.21; N, 4.52; S, 5.18. Found: C, 50.24; H, 5.33; N, 4.60; S, 5.43%.
2.2.2 2-[2-(4-Chloro-benzoylamino)-ethoxymethyl]-4-(2-chloro-phenyl)-5-hydroperoxy carbonyl-6-methyl-1,4-dihydropyridine-3-carboxylic acid ethyl ester (A-3)
Colorless crystals. Yield: 71%, mp 79–82 °C. 1H NMR (400 MHz, CDCl3, δ, ppm): 7.9 (d, J = 8.1 Hz, 2H, H-23, H-27), 7.5 (d, J = 8.2 Hz, 2H, H-24, H-26), 7.4–7.1 (m, 4H, H-2–H-5), 5.4 (s, 1H, H-7), 4.9 (s, 2H, H-18), 4.07 (m, 2H, H-16), 3.7 (m, 2H, H-19), 3.6 (s, 3H, H-13), 3.2 (m, 2H, H-20), 2.3 (s, 3H, H-14), 1.2 (m, 3H, H-17). IR (KBr) νmax (cm−1) 3327 (N–H stretch), 1700, 1612 (CO stretch), 1535 (CC stretch), 1482, 1443 (aliphatic C–H, bending), 1348, 1203 (OC–O of ester, stretching), 1169 (O–C–C of ester, stretching), 1037 (aromatic C–Cl), 826, 740 (aromatic C–H, bending). MS m/z (%): 547 (10, M+), 531 (35), 529 (100), 527 (60), 362(7), 318(6). Anal. Calcd for C27H28Cl2N2O6 (547.43): C, 59.24; H, 5.15; N, 5.12. Found: C, 58.92; H, 5.35; N, 4.87%.
2.2.3 2-[2-(2,5-Dichloro-benzenesulfonylamino)-ethoxymethyl]-4-(2-chloro-phenyl)-5-hydroperoxycarbonyl-6-methyl-1,4-dihydro-pyridine-3-carboxylic acid ethyl ester (A-4)
Colorless blocks. Yield: 81%, mp 150–153 °C. 1H NMR (600 MHz, CD3OD, δ, ppm): 7.84 (dd, 1H, J = 8.1 Hz, 2.1 Hz, H-24), 7.82 (d, 1H, J = 8.1 Hz, H-23), 7.81 (d, 1H, J = 2.0 Hz, H-26), 7.39–7.05 (m, 4H, H-3–H-6), 5.39 (s, 1H, H-7), 4.05 (m, 2H, H-16), 3.62–3.58 (m, 4H, H-19, H-20), 3.57 (s, 3H, H-14), 2.33 (s, 3H, H-12), 1.15 (t, J = 6.6 Hz, 3H, H-17). IR (KBr) νmax (cm−1) 3389 (N–H stretch), 3258 (sec. sulfonamide), 1692 (CO stretch), 1604 (CC stretch), 1479 (aliphatic C–H, bending), 1295 (SO, asymmetric stretch), 1203 (OC–O of ester, stretching), 1103 (SO symmetric stretch), 1037 (aromatic C–Cl), 748 (aromatic C–H, bending). MS m/z (%): 617 (0.2, M+), 610 (2), 528 (2), 418 (32), 417 (100), 347 (20), 323 (29), 297 (18), 254 (17), 208 (61) 164 (12), 44 (12). Anal. Calcd for (C26H27Cl3N2O7S) (617.92): C, 50.54; H, 4.40; N, 4.53; S, 5.19. Found: C, 50.33; H, 4.58; N, 4.71; S, 5.03%.
2.3 Antioxidant activity
2.3.1 Preparation of solutions
Standard solutions of antioxidants were prepared in ethanol or methanol depending on the solubility. All sample solutions (2 mg/mL) were prepared in a 1:1 mixture of acetone and water and then stored at 4 °C.
2.3.2 α,α-Diphenyl-1-picrylhydrazyl radical scavenging capacity assay
For α,α-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging capacity assay [17], 3 mL of DPPH solution (25 mg/L) in methanol was mixed with suitable volumes of sample solutions and the absorption was recorded at 515 nm after an interval of every 5 min for a total duration of 30 min. The color of the solution fades on reduction of DPPH free radical. The percentage inhibition of the DPPH was calculated using the formula:
[DPPH]T = 0 is the DPPH free radical concentration without samples
[DPPH]T = t is the DPPH concentration with sample at time t.
Kinetic curve showing the scavenging of DPPH free radical in terms of decrease in its absorbance at 515.0 nm as a function of time (in minutes) was plotted for every sample and compared with the standard antioxidants, BHA and Trolox.
2.3.3 2,2-Azinobis(3-ethylbenzothiazoline-6-sulfonic acid) assay protocol
2,2-Azinobis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS•+) (A = 0.700 ± 0.020) solution (2.99 mL) was added to10 μL sample [18] and the absorbance values were recorded at 30 °C after every 1 min interval for a total duration of 8 min along with blanks. All determinations were carried out in triplicates and the percentage inhibition at 734.0 nm was calculated by the following equation:
2.3.4 Ferric reducing antioxidant power assay
Ferric reducing antioxidant power (FRAP) assay was used to measure the reducing capacity of novel derivatives according to the method by Benzie and Strain [19]. Aliquot sample (100 μL) was mixed with 3 mL of FRAP reagent and 300 μL of doubly distilled water. Absorbance was measured at 593 nm after every minute for a total duration of 4 min. Results were compared with standard curve of FeSO4 and FRAP values were calculated.
2.3.5 Metal chelating activity
The metal chelating activity was estimated on the basis of decrease in the absorbance of the ferrous–ferrozine complex [20]. An aliquot of 100 μL of sample was added to FeSO4 solution (2 mM, 50 μL) and the reaction was initiated by the addition of 200 μL of ferrozine (5.0 mM) and total reaction volume was adjusted to 4.0 mL with ethanol. The absorbance at 562 nm was recorded after 10 min. Fe2+ chelating activity of the test compounds was calculated using the following formula and is compared with a positive control EDTA.
2.4 Antiurease activity
Urease activity was estimated by measuring the ammonia production by the indophenol method [21]. The reaction mixture having 25 μL of urease enzyme solution and 55 μL of phosphate buffer containing 100 mM urea were incubated with 5 μL of test compound at 30 °C for a period of 15 min in a 96-well plates spectrophotometer. The increase in absorbance at 630 nm was measured after 50 min. All reactions were carried out in triplicate and percentage inhibitions were calculated with the formula:
3 Results and discussion
3.1 Crystallography
3.1.1 Crystallographic description of A-2
The molecular structure and atom-labeling scheme are shown in Fig. 3 (see Tables 1 and 2).
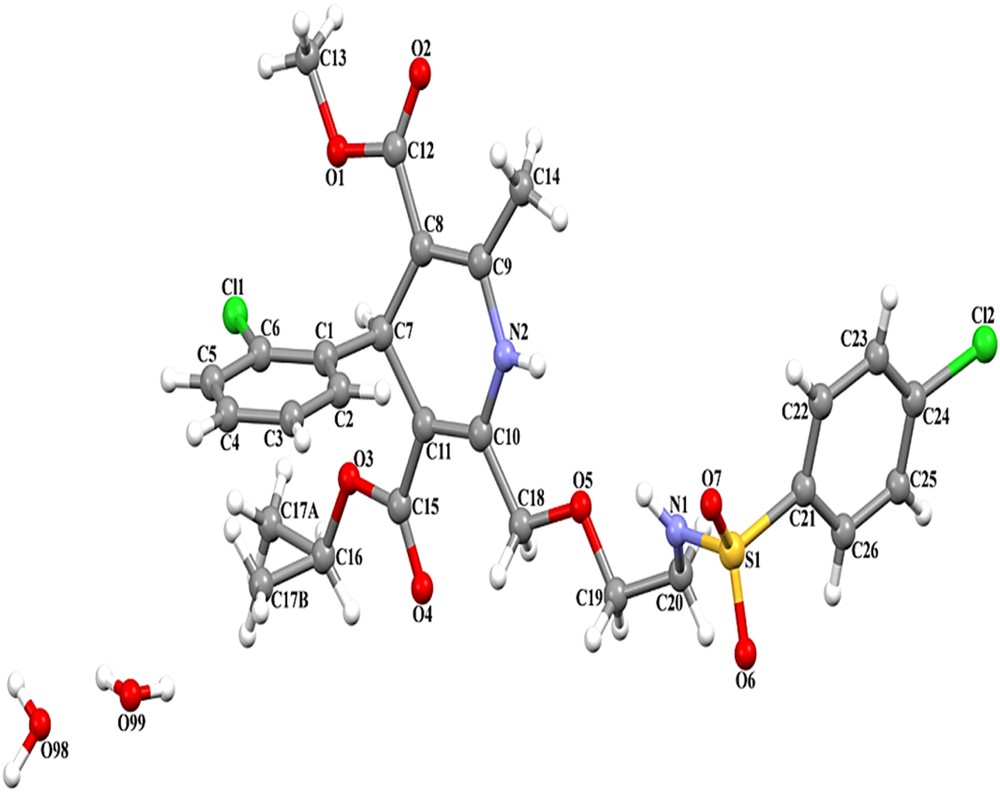
The structure of A-2 presentation with atom numbering scheme.
Selected geometric parameters (Å, °) for A-2 and A-3.
| A-2 | A-3 | ||
| Bond distance | |||
| Bond | Distance | Bond | Distance |
| S1–O7 | 1.428(4) | C15–O4 | 1.214(4) |
| S1–O6 | 1.427(4) | C12–O2 | 1.196(5) |
| C12–O2 | 1.201(7) | C21–O6 | 1.224(4) |
| C15–O4 | 1.213(7) | C6–Cl1 | 1.716(5) |
| C6–Cl1 | 1.742(7) | C25–Cl2 | 1.725(4) |
| C24–Cl2 | 1.720(9) | ||
| Bond angle | |||
| Angle | (°) | Angle | (°) |
| O6–S1–O7 | 120.0(3) | O1–C12–O2 | 121.4(3) |
| N2–C10–C18 | 115.7(4) | O3–C15–O4 | 120.9(3) |
| C1–C7–C8 | 113.4(4) | C1–C7–C8 | 110.1(3) |
| S1–N1–C20–C19 | 147.6(4) | C10–C18–O5–C19 | −156.5(3) |
| C10–C18–O5–C19 | 174.5(4) | C20–N1–C21–C22 | −176.4(3) |
| C21–S1–N1–C20 | 69.7(5) | O5–C19–C20–N1 | −58.9(4) |
Crystal data and structure refinement parameters for A-2 and A-3.
| Parameters | A-2 | A-3 |
| Empirical formula | C26H32Cl2N2O9S | C27H28Cl2N2O6 |
| Formula weight (g/mol) | 619.50 | 547.41 |
| Crystal size (mm3) | 0.42 × 0.36 × 0.28 | 0.37 × 0.33 × 0.32 |
| Crystal system | Monoclinic | Triclinic |
| Space group | C2/c | P |
| a (Å) | 23.8754(9) | 8.2968(3) |
| b (Å) | 8.6725(3) | 9.3112(4) |
| c (Å) | 30.5777(12) | 18.1359(7) |
| α (°) | 90 | 100.692(2) |
| β (°) | 90.673(2) | 98.316(3) |
| γ (°) | 90 | 102.747(2) |
| V (Å3) | 6331.0(4) | 1317.39(9) |
| Z | 8 | 2 |
| Calculated density (g/cm3) | 1.300 | 1.380 |
| λ(Mo Kα) (Å) | 0.71073 | 0.71073 |
| μ(Mo Kα) (mm−1) | 0.32 | 0.29 |
| Tmin, Tmax | 0.631, 0.730 | 0.685, 0.873 |
| F(000) | 2592 | 572 |
| θ range for data collection (°) | 1.3–6.5 | 1.2–26.5 |
| h, k, and l ranges | −29 ≤ h ≤ 28, −10 ≤ k ≤ 8, −38 ≤ l ≤ 38 | −10 ≤ h ≤ 8, −11 ≤ k ≤ 11, −22 ≤ l ≤ 22 |
| Reflections measured | 26,524 | 21,631 |
| Independent reflections | 6533 | 5434 |
| Observed reflection (I > 2σ(I)) | 3322 | 3384 |
| Rint | 0.060 | 0.035 |
| Final R* indices (I > 2σ(I)) | R = 0.094, wR = 0.348 | R = 0.073, wR = 0.253 |
| Goodness-of-fit | 1.11 | 1.07 |
| Max./min. Δρ (e/Å3) | 1.04 and −1.03 | 1.16 and −0.53 |
The 1,4-dihydropyridine ring exhibits a puckered conformation, with puckering parameters [22] q2 = 0.215(5) Å, q3 = −0.069(5) Å, Q = 0.226(5) Å, φ = 4.5(14)°, and θ = 107.9(13)°. The largest deviations are −0.1014(35) Å for C11 and 0.1441(34) Å for C7, and the ring revealed a dihedral angle of 84.78(18)° with the C1–C6 ring (Table 1). The Phenyl moiety is approximately planar, with maximum deviations of 0.0099(40) Å for C1 atom and 0.0214(58) Å for C26 atom, respectively. The crystal packing is stabilized by intermolecular C–H⋯O and N–H⋯O hydrogen bonds (Table 3). Atoms N1 and N2 in the reference molecule at (x, y, z) act as hydrogen bond donors to atom O7 in the molecule at (−x + 1, −y + 1, −z + 1), thus forming centrosymmetric R22(8) and R22(20) rings [23] centered at (1/2, 1/2, 1/2). The combination of the N–H⋯O hydrogen bonds produces R21(10) ring. Atom C16 in the reference molecule at (x, y, z) acts as a hydrogen bond donor to atom O6 in the molecule at (−x + 3/2, −y + 3/2, −z + 1), thus making a centrosymmetric R22(26) ring. Similarly, atom C23 in the reference molecule at (x, y, z) acts as a hydrogen bond donor to atom O2 in the molecule at (−x + 1, −y, −z + 1), thus shaping a centrosymmetric R22(32) ring centered at (1/2, 0, 1/2) (Table 2). The combination of the C–H⋯O hydrogen bonds along [110] generates a chain of edge-fused R22(26) and R22(32) rings (Fig. 4).
Hydrogen-bond geometry (Å, °) for A-2 and A-3.
| D-H⋯A | D–H | H⋯A | D⋯A | D–H⋯A |
| A-2 | ||||
| N1–H1⋯O7i | 0.86 | 2.19 | 2.912 (6) | 141 |
| N2–H2A⋯O7i | 0.86 | 2.61 | 3.427 (6) | 159 |
| C16–H16B⋯O6ii | 0.97 | 2.45 | 3.121 (9) | 126 |
| C23–H23⋯O2iii | 0.93 | 2.45 | 3.270 (10) | 148 |
| A-3 | ||||
| N1–H1⋯O4 | 0.85 (5) | 2.26 (5) | 3.082 (5) | 162 (4) |
| N2–H2A⋯O6iii | 0.91 (5) | 2.15 (5) | 3.015 (4) | 158 (4) |
| C4–H4⋯O2iv | 0.93 | 2.57 | 3.258 (6) | 131 |
| C20–H20A⋯O5iii | 0.97 | 2.50 | 3.390 (5) | 152 |
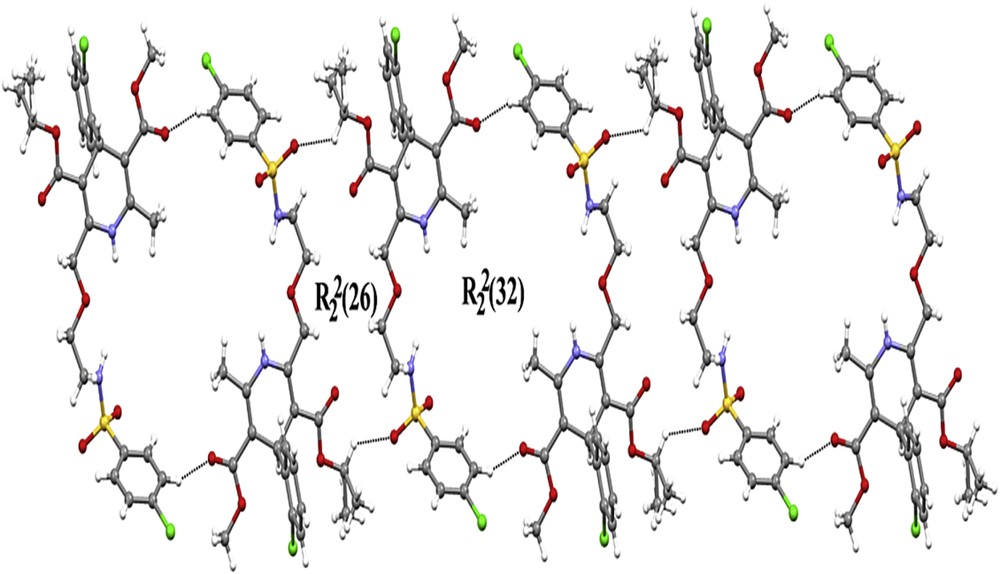
Part of the crystal structure of A-2 showing the formation of a chain edge-fused R22(26) and R22(32) rings along [110].
3.1.2 Crystallographic description of A-3
The molecular structure is illustrated in Fig. 5. The C12–O2, C15–O4, and C21–O6 bond lengths of 1.196(5), 1.214(4), and 1.224(4) Å are indicative of significant double-bond character (Table 1). The 1,4-dihydropyridine ring exhibits a puckered conformation, with puckering parameters q2 = 0.190(4) Å, q3 = 0.044(4) Å, Q = 0.195(4) Å, φ = 172.5(12)°, and θ = 76.9(12)°. The largest deviations from the best plane are −0.1206(23) Å for C7 and 0.0874(24) Å for C8, and the ring makes a dihedral angle of 89.01(11) with the C1–C6 ring. The maximum deviations of 0.0101(27) Å for C2 atom and 0.0116(28) Å for C22 atom, respectively, show approximate planarity of three phenyl rings (Table 2).
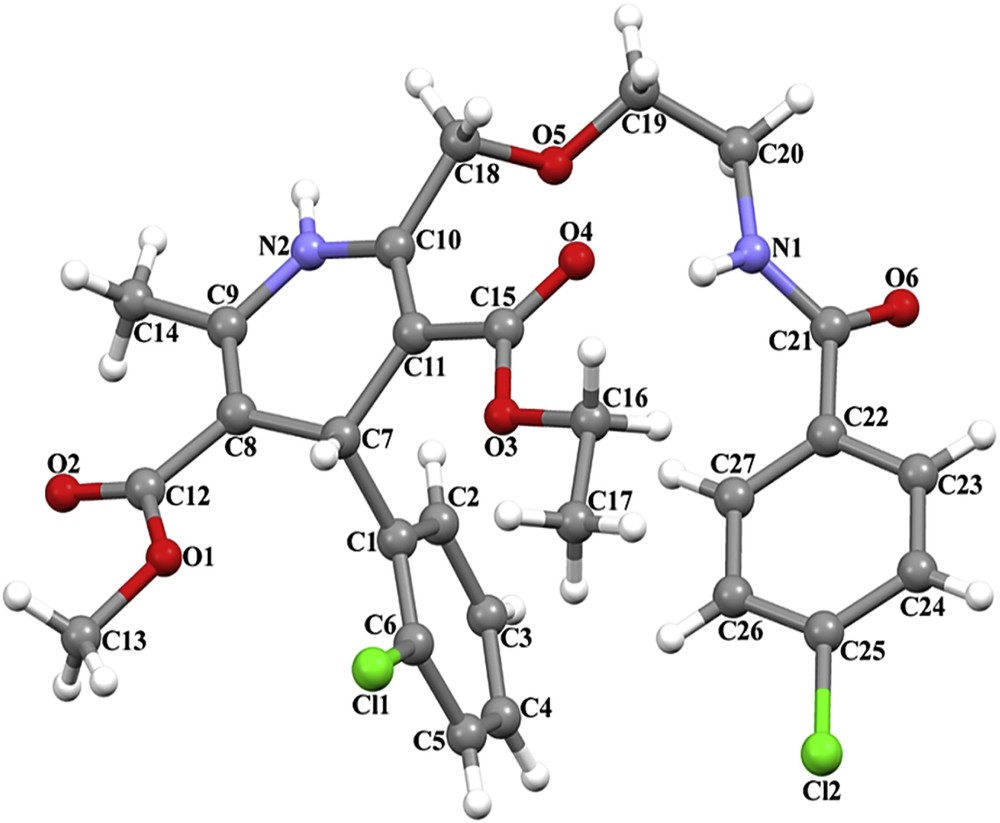
The structure of A-3 demonstrating the atom numbering scheme.
The crystal packing became stable by intermolecular C–H⋯O and N–H⋯O hydrogen bonds (Table 3). The intramolecular N–H⋯O hydrogen bond defines S (10) motif. Atom N2 in the orientation molecule at (x, y, z) acts as a hydrogen bond source to O6 in the molecule at (−x + 1, −y, −z + 1), thus forming centrosymmetric R22(20) ring centered at (1/2, 0, 1/2). Atom C20 in the reference molecule at (x, y, z) acts as a hydrogen bond donor to atom O5 in the molecule at (−x + 1, −y, −z + 1), thus forming centrosymmetric R22(8) ring centered at (1/2, 0, 1/2). Atom C4 in the reference molecule at (x, y, z) acts as a hydrogen bond donor to atom O2 in the molecule at (x, y − 1, z), thus forming a C(9) chain running parallel to the [010] direction (Fig. 6). The combination of the hydrogen bonds along [010] generates a chain of edge-fused R44(42) rings.
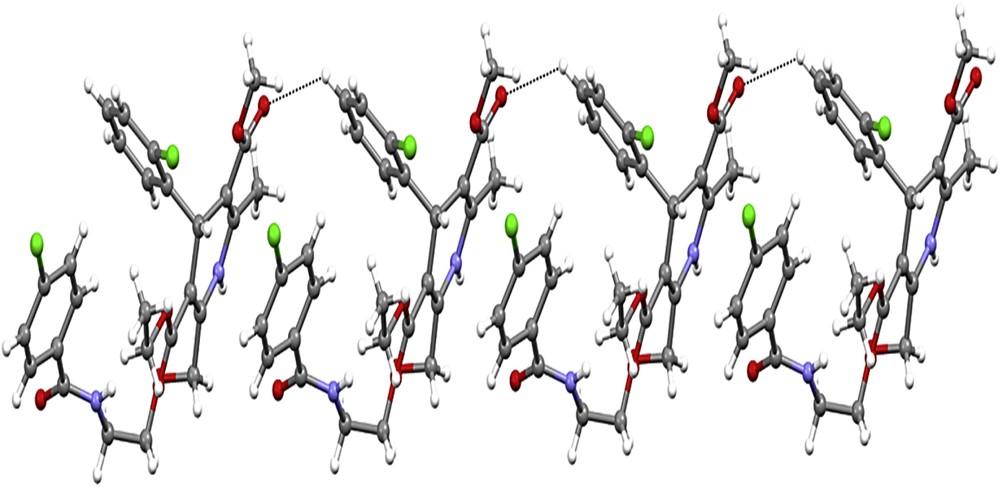
Crystal structure of A-3 showing the formation of a chain along [010] generated by the C–H⋯O hydrogen bonds. H atoms have been omitted from the motif for clarity.
The crystals of A-4 were not refined enough to obtain crystallographic data; its confirmation was achieved with the help of 1H NMR spectrum. The successful incorporation of 2,5-dichloro-benzenesulfonyl amide was evident by a double doublet at δ 7.84 which was attributed to H-24. Two protons resonated as doublets at δ 7.82 and 7.81 were corresponding to H-23 and H-26, respectively. The multiplet at δ 7.39–7.05 was attributed to H-3–H-6 phenyl protons of A-1, the rest of the signals in 1H NMR were in complete agreement with those observed for parent molecule.
3.2 Thermal analysis
The results of thermal analysis of A-2 were found consistent with the proposed stoichiometry Fig. 7. According to the pattern of decomposition loss of water molecules of crystallization referred at 60–150 °C with weight loss of about 5–6% (calculated value 5.49% for two lattice water molecules). Compound was then thermally stable up to 200 °C and afterward started decomposing slowly between 200 and 275 °C, followed by rapid decomposition up to 380 °C. Total weight loss of 94% occurred during these steps because of successive release of organic compounds (calculated value 94.50%). The differential scanning calorimetry (DSC) plot showed a broad endothermic peak near 75 °C, because of loss of water of crystallization which is followed by a sharp endothermic peak attributed to melting temperature of the compound. Several small exothermic peaks were observed in the temperature range between 310 and 550 °C because of phase changes.

TGA/DSC curve of A-2.
Thermogram of A-3 showed a principle sharp endothermic peak around 100 °C attributed to the melting of compound (Fig. 8). It was thermally stable up to 180 °C and sharply decomposed at 185 °C owing to the loss of carbonyl, followed by a rapid decomposition process between 190 and 280 °C.
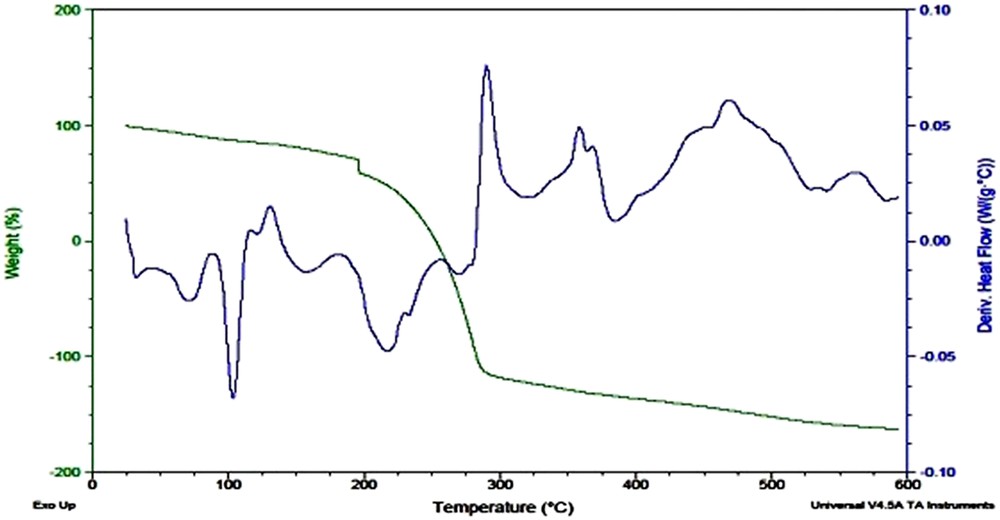
TGA/DSC curve of A-3.
A principle sharp endothermic peak A-4 was observed around 150 °C attributed to the melting of compound (Fig. 9). It was thermally stable up to 190 °C and then started decomposing first because of elimination of sulfonyl group, followed by a rapid degradation process between 275 and 350 °C.
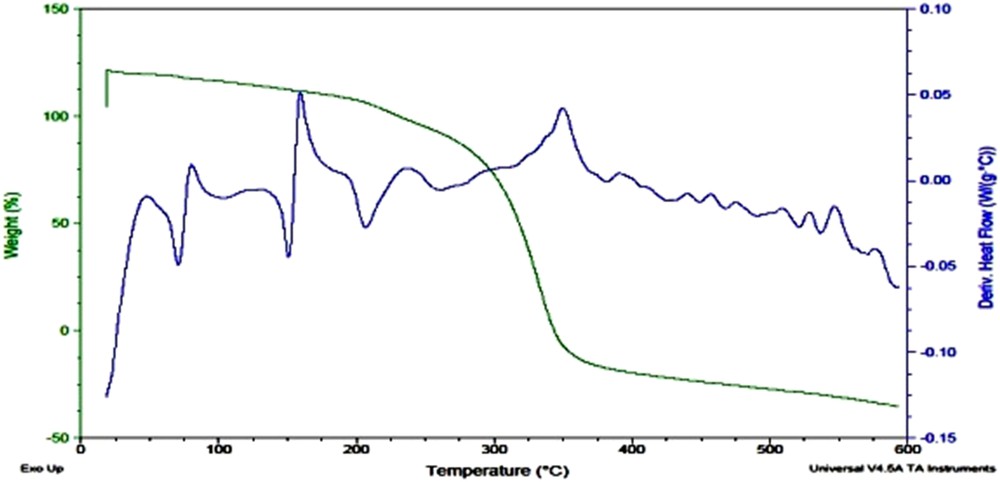
TGA/DSC curve of A-4.
3.3 Antioxidant potential
Antioxidant potential of a compound refers to the capability of a compound to decelerate the oxidative stress [24]. To evaluate the antioxidant potential of these novel derivatives, three major mechanisms of action of antioxidants are exploited which include free radical scavenging mechanism, pro-oxidant reduction mechanism, and pro-oxidant chelating mechanism.
3.3.1 Free radical scavenging activity
To evaluate in vitro free radical scavenging ability ABTS and DPPH assays were used. The basic working mechanism of these assays is as follows:
| Probe (oxidant) + e− (from antioxidant) → reduced probe + oxidized antioxidant |
The probe (radical cation) is an oxidant that snatches an electron from the antioxidant, causing color changes of the probe. The extent of the color reduction is proportional to the antioxidant concentrations. The reaction end point is reached when color change stops.
3.3.1.1 DPPH assay
DPPH is a stable nitrogen-centered free radical owing to the delocalization of the auxiliary electron over the molecule as a whole. The delocalization also gives rise to the deep violet color that gives an absorption band in methanol solution at about 517 nm [25]. In the present study, DPPH assay is used to evaluate the ability of the investigated compounds acting as hydrogen atom donors and transformation of DPPH radical into its reduced form DPPH-H. This method computes the scavenging capacity by measuring an absorption index of the test compound at different concentrations with respect to time (Fig. 10). The radical scavenging activity was found to be in the decreasing order as follows: A-2 > A-3 > A-4 > A-1.
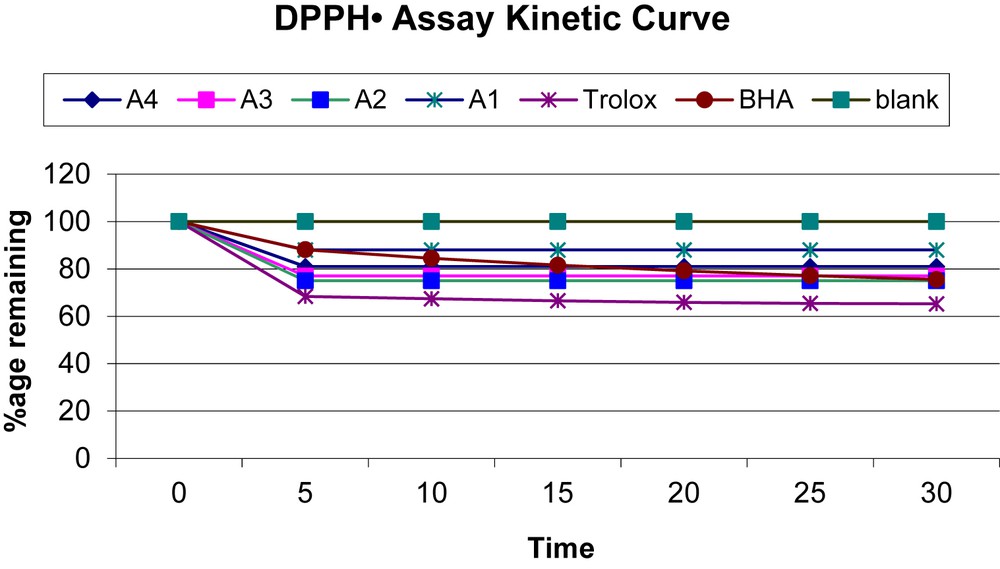
Kinetic curves of amlodipine and its derivatives.
3.3.1.2 ABTS assay
The ABTS•+ assay is a decolorization assay that measures the capacity of antioxidants to scavenge free radicals. ABTS is a phenothiazine drug, which on reacting with an oxidizing agent, potassium persulfate, forms a green radical cation [26]. ABTS•+ is a nitrogen-centered radical cation having a redox potential of 0.68 V which is equal to the redox potential of peroxyl radical at physiological pH [27]. ABTS•+ has a characteristic blue green color with a λmax of 734 nm. ABTS•+ gets reduced to its colorless form ABTS when antioxidant compound reacts with this radical cation. Results are generally expressed in terms of TEAC value which describes the antioxidant capacity of the compound relative to a standard antioxidant Trolox [28]. All synthetic derivatives were assayed against ABTS and decreases in the following order: A-1 > A-4 > A-3 > A-2. The results revealed that free radical scavenging power of A-1 is significantly higher than the novel derivatives (Fig. 11).
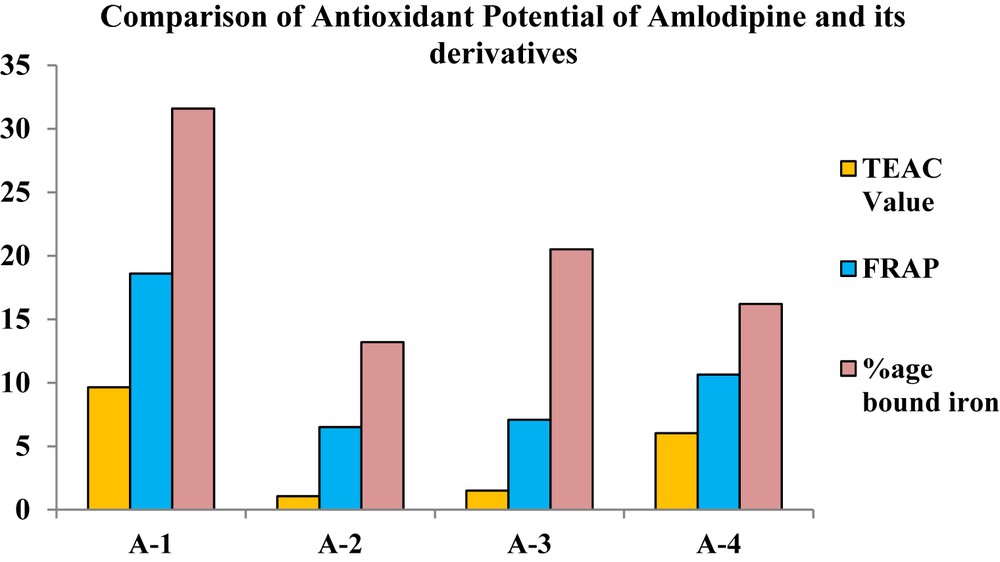
Comparative results of antioxidant activity assays of A-1, A-2, A-3, and A-4.
3.3.2 Ferric reducing antioxidant power
This assay measures the ability of antioxidants to reduce the Fe(III) which serves as a pro-oxidant in our body system. For evaluating the reducing power of antioxidant, a ferric salt, Fe(III)-(TPTZ)2Cl3 (TPTZ), is used as an oxidant. When this oxidant came into contact with antioxidant it gets reduced to Fe(II)-(TPTZ)2Cl3 [29]. The redox potential of Fe(III) salt is 0.70 V which is comparable to that of ABTS radical cation but at acidic pH. The antioxidant activity of the amlodipine and its derivatives decreases in the following order: A-1 > A-2 > A-3 > A-4 (Fig. 11). The results revealed that pro-oxidant reduction capacity of A-1 is higher than the derivatives.
3.3.3 Pro-oxidant chelating activity
The transition metal ion Fe2+ has the talent to furnish an electron, thus allowing the formation and propagation of many chain reactions, even starting with relatively nonreactive radicals [30]; hence, to decrease the availability of these pro-oxidants some specific proteins in the body form chelate. To study this antioxidant mechanism a synthetic compound ferrozine is used which quantitatively chelates iron(II). However, in the presence of other chelating agents, the complex formation is disrupted with the result that the red color of the complex is decreased. Measurement of color diminution, therefore, allows the estimation of the chelating activity of the concomitant chelator [31]. The results of this assay are expressed as percent bound iron and A-1 has the highest pro-oxidant chelating potential than other derivatives (Table 4).
Results of antioxidant activity.
| Sr. no. | Sample | TEAC value (mM) | FRAP value (mM) | %age bound iron |
| 1 | A-1 | 9.631 ± 0.865 | 18.6 ± 0.679 | 31.6 ± 0.520 |
| 2 | A-2 | 1.062 ± 0.570 | 6.5 ± 0.703 | 13.2 ± 0.898 |
| 3 | A-3 | 1.498 ± 0.346 | 7.08 ± 0.528 | 20.5 ± 0.262 |
| 4 | A-4 | 6.02 ± 0.820 | 10.64 ± 0.962 | 16.2 ± 0.133 |
3.4 Antiurease activity
Many microorganisms use urease (EC: 3.5.1.5) enzyme to obtain nitrogen for growth and in plant nitrogen metabolism during the germination process [32]. Urease is a virulence feature in certain human and animal pathogens; it contributes in the development of kidney stones, pyelonephritis, peptic ulcers, and other disease states. The results revealed that sulfonamide derivatives A-2 and A-4 showed an excellent antiurease activity (Table 5). The percent antiurease activity is not correlated with sample concentration [33].
Results of antiurease activity.
| Sr. no. | Sample | 50 μg/mL (%age) | 250 μg/mL (%age) |
| 1 | A-2 | 73.37 ± 0.05 | 76.92 ± 0.01 |
| 2 | A-4 | 66.37 ± 0.00 | 75.44 ± 0.08 |
4 Conclusion
The projected structures of the synthesized compounds A-2 and A-3 were well supported by X-ray diffraction, whereas A-4 was confirmed by 1H NMR spectroscopic data. From the results of antioxidant activity, it might be concluded that the compounds have potent radical scavenging activity against ABTS and DPPH radical cation along with good reducing and chelating abilities. Hence, on the basis of the aforementioned results, these synthesized derivatives provide an overall obligatory basis to introduce newfangled drugs for the management of hypertension and other associated diseases.
Acknowledgment
We would like to thank Professor Khalid M. Khan, HEJ Institute of Chemistry, University of Karachi for his kind assistance in 1H NMR and mass spectral measurements, Dr. Faiz-ul-Hasan Nasim, Department of Biotechnology, Islamia University Bahawalpur for antiurease activity measurements. We are also thankful to Mr. Naeem Razzaq (Schazoo Laboratories) for providing working standards of amlodipine. The article has been vetted by Dr. Naeem Lodhi, Department of Chemistry, York University, Canada and Mr. M. Siddique Awan, Assistant Professor of English, GC University Lahore, so as to ascertain language accuracy. We strongly acknowledge the financial support of the Higher Education Commission of Pakistan to undertake this research work under its Indigenous Ph.D. Program (Batch III).