

1 Introduction
La qualité des médicaments traditionnels africains a un impact direct sur leur innocuité et leur efficacité. Des rapports ont montré des effets secondaires associés aux falsifications et à la présence de contaminants (métaux lourds, pesticides et micro-organismes) [1].
L'évaluation de la qualité des médicaments traditionnels africains est difficile, car ce sont des mélanges complexes de plusieurs drogues végétales ou de préparations à base de drogues végétales auxquels peuvent être associées des drogues minérales et/ou animales. Aussi la source, la récolte et la préparation des matières premières sont très variables [2, 3].
Pour l’évaluation de la qualité de ces médicaments complexes, une approche multi-technique est nécessaire afin d'authentifier le lien entre les composants et l'usage traditionnel [4,5].
Le profil chromatographique des métabolites polaires, apolaires ou globaux, modèle des constituants pharmacologiquement actifs et/ou chimiquement caractéristiques présents, constitue un bon outil pour l’évaluation qualitative et quantitative des médicaments traditionnels africains [4–7].
Les énormes volumes de données contenus dans les profils chromatographiques exigent de puissants outils de traitement des données, tels que la chimiométrie, qui permet l'extraction de l'information multivariée utile pour comprendre les changements et les variations des profils chromatographiques [8–10].
Parmi ces techniques, l’HPLC est une méthode populaire et couramment utilisée pour l’analyse des médicaments traditionnels à base de plantes, car elle est facile à utiliser et n'est pas limitée par la volatilité ou l’instabilité thermique des constituants des échantillons, contrairement à la GC. Aussi plusieurs types de phase stationnaire sont utilisés et cette méthode chromatographique peut être associée à de nombreux détecteurs (UV, DEDL, Corona-CAD, IR-TF, SM et RMN), offrant ainsi beaucoup de possibilités pour l’analyse de différentes classes de composés chimiques [5–7,11,12].
Notre première étude sur les profils chromatographiques des composés phytochimiques porte sur la fraction peu polaire ou apolaire des drogues végétales et elle a pour objectif de proposer une technique performante d’établissement de profils en chromatographique liquide haute performance des métabolites apolaires pour l’évaluation de la qualité des phytomédicaments africains.
Pour l'évaluation qualitative des profils métaboliques durant l’étude expérimentale, nous avons sélectionné deux drogues végétales largement utilisées en Afrique de l'Ouest, notamment en Côte d'Ivoire [13], et dans lesquelles plusieurs composés polaires et apolaires ont été identifiés (feuilles de kinkéliba Combretum micranthum G. Don, Combretaceae, et feuilles de Mitracarpus scaber Zucc., Rubiaceae).
Elles diffèrent également par rapport aux fractions utilisées en thérapeutique: pour C. micranthum l’activité thérapeutique traditionnelle est liée aux composés polaires (extrait aqueux) [14–16] ; en revanche, pour M. scaber, l’activité thérapeutique traditionnelle est basée sur les composés peu polaires ou apolaires [17–23].
2 Matériel et méthodes
2.1 Produits chimiques
2.1.1 Tous les produits utilisés sont de qualité analytique
Substances de référence : azaanthraquinone (benz-[γ]-isoquinoléine-5,10-dione), α-amyrine, artémisinine, carotène, campestérol, trans-caryophyllène, ginkgolide A, linalol, lupéol, 4-methoxyacetophenone 1,4-naphtoquinone, acide oléanolique, acide palmitique, α-pinène, psoralène, réserpine, β-sitostérol, squalène, α-terpinéol, thymol, α-tocophérol, α-tocophérol acétate, tripalmitoléine, acide ursolique et acide formique ont été obtenus auprès de Sigma-Aldrich (Saint-Quentin-Fallavier France), eugénol d'extrasynthèse (Genay, France), charbon actif de Roth (Lauterbourg, France).
Solvants de grade HPLC: chloroforme, dichlorométhane, éthanol 96%, heptane, méthanol et propanol-2 fournis par VWR© International (Fontenay-sous-Bois, France).
2.2 Appareillage
- - La chaîne HPLC comprend : un dégazeur (Degasys D6-1310 Eurosep, Cergy-Pontoise, France), une pompe ternaire (Jasco 980 système gradient GPL, Kyoto, Japon), un passeur automatique d'échantillon (Kontron 360) avec une boucle de 20 μl, un four à colonne (Cluzeau 560-CIL, Saint Foy Le Grande, France) et un détecteur d’aérosol chargé (Corona® CAD™ ESA Biosciences, Chelmsford, MA, USA). Les chromatogrammes ont été enregistrés en utilisant le logiciel KromaSystem 2000 de Bio-Tek Kontron Instruments (Milan, Italie).
- - Pour la HPLC/MS, le système chromatographique comprend : un dégazeur, une pompe quaternaire, un échantillonneur automatique HP 1050 (Hewlett-Packard® GmbH, Böblingen, Allemagne) et un four à colonne (Cluzeau 560-CIL, Sainte-Foy-la-Grande, France) couplé à un spectromètre de masse API3000 triple quadripôle (TQ-MS ABSciex, Allemagne) équipé d'une source ESI et APCI. Le système est contrôlé par un ordinateur avec le logiciel d’acquisition de données Analyst 1.4.1 (Applied Biosystems/MDS-SIEX).
- - Colonne PolyVinyl Alcool (PVA-Sil YMC ; 5 μm ; 150 × 2,1 mm avec précolonne 10 × 2,1 mm) fournie par Interchim, Montluçon, France).
- - Colonne de carbone graphite poreux (PGC Hypercab 5 μm ; 100 × 2,1 mm avec précolonne 10 × 2,1 mm) obtenue chez Thermo Fisher Scientific (Courtabœuf, France).
2.3 Matériel végétal
Cette étude a été réalisée sur les échantillons de deux drogues végétales :
- • Cinq lots de feuilles de C. micranthum G. Don. (CM1 à CM5) :
- - CM1 (septembre 2009), CM4 (octobre 2010) obtenus au marché aux plantes dans la commune d'Adjamé (Côte d'Ivoire) ;
- - CM2 (avril 2009) et CM3 (octobre 2010) (matières premières d’une industrie de phytomédicament, fournies par le laboratoire de chimie analytique, faculté de pharmacie, université de Cocody-Abidjan) ;
- - CM5 (octobre 2010) provenant du Sénégal.
- • Quatre lots de feuilles de M. scaber Zucc. (MS1 à MS4) :
- - MS1 (octobre 2008), MS2 (avril 2009), MS3 (octobre 2010) récoltés au jardin botanique de l'université d'Abidjan Cocody ;
- - MS4 (octobre 2010) récolté dans la commune de Bingerville (Côte d'Ivoire).
2.4 Préparation des échantillons
2.4.1 Extraction
Deux grammes de poudre de plante ont été extraits deux fois par chaufage à reflux avec 20 ml de dichlorométhane pendant 30 min et ensuite filtrés à travers un papier filtre Whatman n° 42. Les filtrats ont été réunis et évaporés sous pression réduite à 40–45° C. Le résidu sec a été repris avec 5 ml du même solvant, puis filtré sur filtre seringue en téflon (0,22 μm). Les extraits ont été dilués à 10 mg/ml dans le dichlorométhane.
2.4.2 Élimination de la chlorophylle
L'élimination de la chlorophylle a été réalisée par adsorption sur charbon actif (6 mg pour 1 mg de résidu sec), en secouant 2 min, puis filtration à travers un papier filtre (Whatman 42). Le filtrat a été utilisé pour l'analyse HPLC.
2.5 Optimisation de la séparation chromatographique
L’optimisation de la séparation chromatographique a été réalisée sur les colonnes (PVA-Sil et PGC Hypercab) à 25 °C (température ambiante).
Les solvants ont été dégazés avant utilisation et la phase mobile est dégazée en continu à un débit de 0,4 ml/min. La détection est réalisée avec un détecteur d’aérosol chargé (CAD) à 35 psi de pression de gaz et à une température de 35 °C.
2.6 Couplage et détection en spectrométrie de masse
Après séparation chromatographique de 10 μl d’extrait à un débit de 0,4 ml/mn, l’éluat est introduit dans le spectromètre de masse avec source d’ionisation chimique à pression atmosphérique (APCI) et un analyseur triple quadripôle.
Les paramètres d’ionisation sont les suivants : gaz de nébulisation 6 (unité arbitraire), gaz auxiliaire 9 (u.a.), température de la source (350° C), potentiel de declustering 56 V, potentiel de focalisation 370 V et potentiel d'entrée 11 V. Une méthode d’acquisition en balayage « full scan » en ionisation positive entre m/z 100 et m/z 1200 a été utilisée.
2.7 Identification des composés
Les composés ont été caractérisés par leur temps de rétention et leur spectre de masse et si possible identifiés par comparaison à des composés purs analysés à la concentration de 25 μg/ml dans les mêmes conditions.
2.8 Analyse chimiométrique
Avant l'analyse chimiométrique, les données ont été converties au format mzData avec le logiciel Wiff to mzData (v 1.0.0.4) fourni par ABSciex. Les données brutes ont ensuite été traitées par le logiciel Mzmine 2.2 pour la détection, l'alignement des pics et le remplissage des lacunes, la création et l'exportation des données traitées sous la forme d'une matrice.
La matrice de données a été utilisée pour l'analyse chimiométrique par le logiciel SIMCA P-11 (Umetrics AB, Umeå, Suède).
Nous avons effectué une analyse multivariée non supervisée (analyse en composantes principales : ACP) afin d'étudier la similarité entre les différents lots de plantes.
L'impact de l’élimination de la chlorophylle des extraits a été évalué par une analyse multivariée supervisée (régression partielle des moindres carrés : PLS) après correction orthogonale du signal (OSC).
3 Résultats et Discussion
3.1 Optimisation de la séparation chromatographique
L’optimisation des conditions chromatographiques est fondée sur l’exploitation des informations complémentaires des deux types de méthodes chromatographiques liquides : polarité de phases normales (élution par ordre de polarité croissante) et polarité de phases inversées (élution par ordre de polarité décroissante).
Le développement de la méthode de séparation chromatographique liquide des métabolites apolaires (composés lipophiles) était basé sur les travaux de Deschamps et al. [24, 25], travaux portant sur le profilage chromatographique de différentes classes de lipides sur des phases stationnaires PVA (phase normale) et PGC (phase inverse).
L’optimisation des conditions chromatographiques s’est effectuée en utilisant un détecteur d’aérosol chargé (CAD), détecteur adapté à l’analyse des composés sans chromophores (composes lipophiles) et dont le principe fondé sur l’ionisation des composés [26] se rapproche de l’ionisation en spectrométrie de masse.
Le choix des différents solvants a été fonction des caractéristiques physicochimiques (force éluante et miscibilité).
Les premiers essais ont été effectués en testant les gradients ternaires de Deschamps et al. [24], sur colonne PVA en utilisant comme solvant faible l’heptane, solvant intermédiaire mélange propanol-2/chloroforme et solvant fort le méthanol ou l’éthanol 96% et en additionnant aux phases mobiles uniquement de l’acide formique à 0,1%.
Les profils chromatographiques obtenus (Fig. 1A, B) des gradients ternaires proposés par Deschamps et al. [24] avec le méthanol ou l’éthanol à 96% sont similaires ; on note également une similarité des ces profils chromatographiques avec le profil chromatographique obtenu (Fig. 1C) avec un gradient binaire de 100% heptane à 100% éthanol (solvant miscible) à 0,4 ml/mn. Il n’existe donc pas une grande différence de sélectivité (nombre de pics) entre les différentes phases mobiles, et particulièrement entre le méthanol et l’éthanol 96%, mais avec des pertes de charge plus élevées pour l’éthanol. Cette similarité de sélectivité entre le méthanol et l’éthanol a été indiquée par divers auteurs [27, 28], ce qui nous a amenés à proposer le gradient binaire entre l’heptane et l’éthanol à 96% pour la suite de nos essais sur les phases stationnaires polyvinyle alcool (PVA) et carbone graphite poreux (PGC).
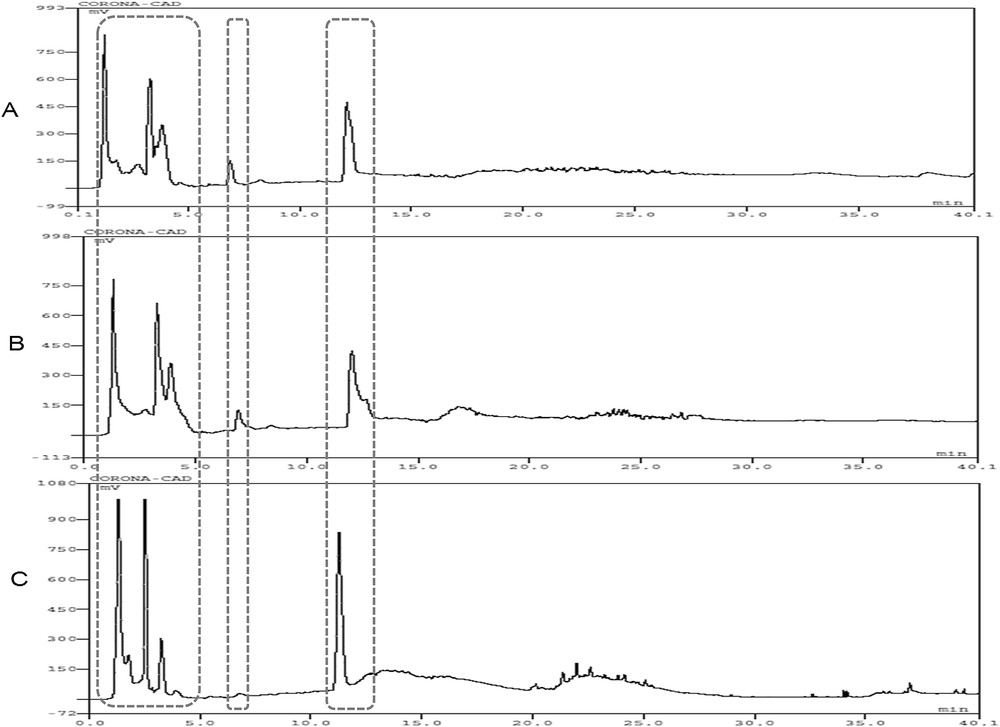
Chromatogrammes du mélange de 23 témoins apolaires (100 μg/ml) sur colonne PVA des gradients ternaires de Deschamps et al. [24] avec heptane/chloroforme-isopropanol/méthanol (A) ; heptane/chloroforme-isopropanol/éthanol (B) et du gradient binaire 100% heptane à 100% éthanol à 0,4 ml/mn (C). Toutes les phases mobiles contiennent 0,1% d’acide formique.
Les profils chromatographiques (Fig. 2A, B) des essais de gradients binaires entre l’heptane et l’éthanol 96% sur des phases stationnaires PVA et PGC montrent que la plupart des composés sont élués pratiquement avec le front du solvant sur la PVA ; en revanche, la PGC, plus rétentive [29], permet de différencier davantage de composés. La phase stationnaire PGC, présentant une meilleure sélectivité (nombre de pics) par rapport à la PVA, a été choisie pour le profilage des composés phytochimiques apolaires.
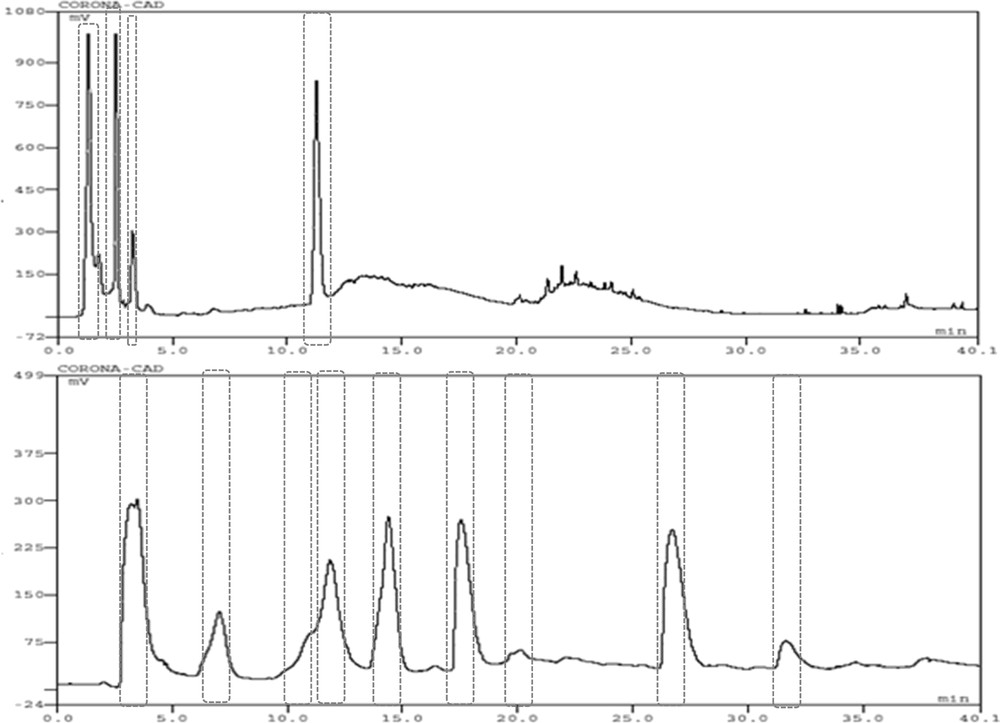
Chromatogrammes du mélange de 23 témoins apolaires (100 μg/ml) des gradients binaires à 0,4 ml/mn entre 100% heptane à 100% éthanol 96 sur colonne PVA (A) et 100% éthanol à 100% heptane sur colonne PGC (B). Toutes les phases mobiles contiennent 0,1% d’acide formique.
Des essais ont été ensuite réalisés sur la phase stationnaire PGC pour le choix du solvant fort entre l’heptane, le dichlorométhane et le chloroforme. Les profils chromatographiques (Fig. 3A, B, C) indiquent une meilleure sélectivité (nombre de pics) avec l’heptane durant les 25 premières minutes ; en revanche, après 25 min, on observe une meilleure efficacité (pic plus fin) avec les deux autres solvants et surtout avec le chloroforme. L’heptane possède une force éluante intermédiaire par rapport au dichlorométhane et au chloroforme, qui sont des solvants forts sur la phase stationnaire carbone graphite poreux (PGC) [29, 30] Tableau 1
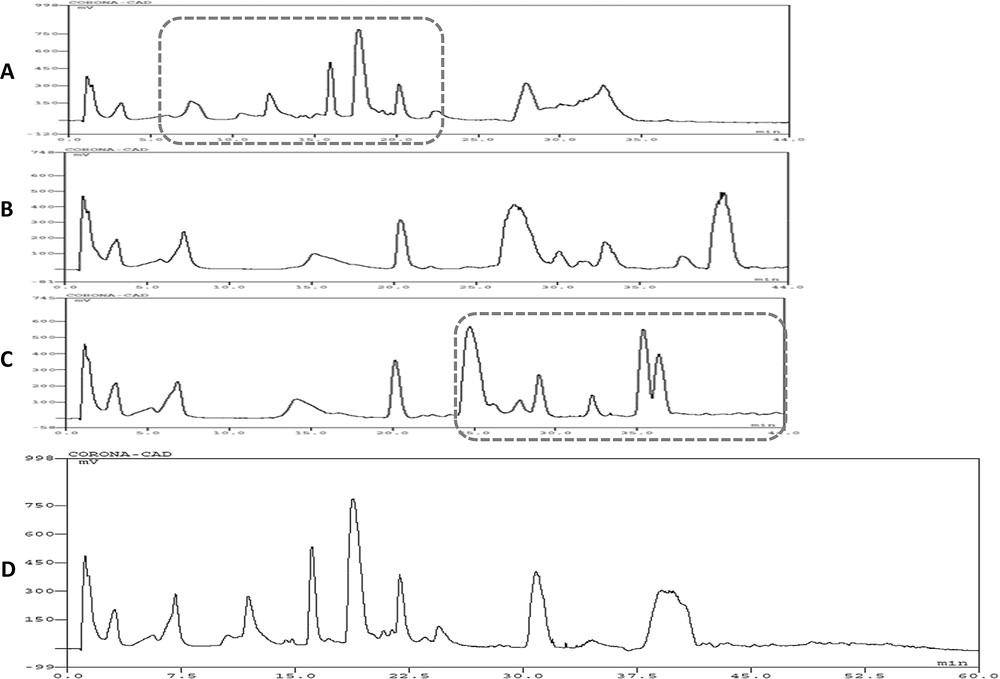
Chromatogrammes du mélange de 23 témoins apolaires (100 μg/ml) des gradients binaires à 0,4 ml/mn sur colonne PGC entre 100% éthanol à 100% heptane (A), 100% éthanol à 100% dichlorométhane (B), 100% éthanol à 100% chloroforme (C) et du gradient ternaire (Tableau 2) (D). Toutes les phases mobiles contiennent 0,1% d’acide formique.
Liste de 23 composés apolaires appartenant à différentes classes chimiques.
| Classe chimique | Composés |
| Monoterpène | linalol |
| Monoterpène | α-terpinéol |
| Monoterpène | α-pinène |
| Monoterpène | thymol |
| Sesquiterpène | artémisinine |
| Sesquiterpène | trans-caryophyllène |
| Diterpène | ginkgolide A |
| Triterpène | acide ursolique |
| Triterpène | α-amyrine |
| Stérol | β-sitostérol |
| Stérol | campestérol |
| Tétraterpène | carotène |
| Alcaloïde | réserpine |
| Coumarine | psoralène |
| Phénol | 4-méthoxyacetophénone |
| Phénol | eugénol |
| Quinone | 1,4-naphthoquinone |
| Quinone alcaloïde | azaanthraquinone |
| Quinone terpénique | α-tocophérol |
| Quinone terpénique estérifiée | α-tocophérol acétate |
| Alcane | squalène |
| Acide gras | acide palmitique |
| Triglycéride | tripalmitoleine |
Au vu de ces résultats, nous avons proposé à un débit de 0,4 ml/mn [25] sur phase stationnaire carbone graphite poreux, un gradient ternaire de 100% éthanol à 60% heptane en 30 min, puis de 60% heptane à 100% chloroforme en 20 min. On reste à 100% chloroforme pendant 10 min, puis on opère un reconditionnement aux conditions initiales pendant 30 min (Tableau 2), conditions qui permettent d’obtenir un meilleur (plus de pics fins) profil chromatographique (Fig. 3D).
Gradient ternaire à un débit de 0,4 ml/min.
| Temps (min) | Éthanol à 96% (0,1% d'acide formique) (%) | Heptane (0,1% d'acide formique) (%) | Chloroforme (0,1% d'acide formique) (%) |
| 0 | 100 | 0 | 0 |
| 30 | 40 | 60 | 0 |
| 50 | 0 | 0 | 100 |
| 60 | 0 | 0 | 100 |
| 61 | 100 | 0 | 0 |
| 90 | 100 | 0 | 0 |
3.2 Couplage et détection en spectrométrie de masse
Pour la méthode de spectrométrie de masse, nous avons choisi la source d’ionisation APCI, plus adaptée à l’analyse des métabolites apolaires [12].
Après optimisation des paramètres du spectromètre de masse par la méthode d’infusion des standards (20 μg/ml), les meilleurs résultats ont été obtenus avec le mode d’ionisation positive, ceci étant en effet lié à présence d’acide formique dans la phase mobile.
De nombreuses classes (Fig. 4) de composés apolaires (terpènes, dérivés phénoliques, alcaloïdes, quinones, alcanes et dérivés estérifiés) ont été détectées, avec en général comme fragments majoritaires [M+H–H2O]+ et/ou [M+H]+. Ceci est en accord avec les travaux de divers auteurs sur les composés lipophiles [31–33]. En revanche, les composés monoterpéniques très volatils, ceux de faible poids moléculaire (m/z < 150) et les acides gras n’ont pu être détectés.
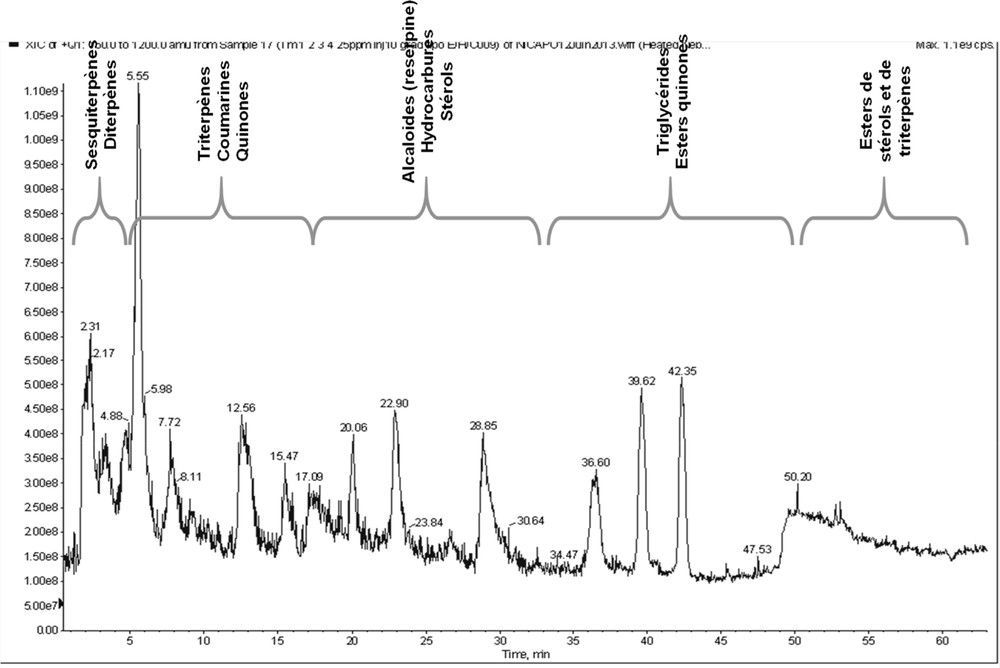
Chromatogramme TIC LC/MS du mélange de 23 témoins apolaires (100 μg/ml) du gradient ternaire (Tableau 2) sur la phase stationnaire carbone graphite poreux (PGC).
3.3 Identification des composés
Dans le cadre de notre étude, l’identification a porté principalement sur les composés phytochimiques apolaires, déja identifiés dans les deux drogues végétales, afin de vérifier que l’on retrouve tous ces composés dans les profils chromatographiques.
Les différents composés de C. micranthum et de M. scaber dont les spectres de masse étaient exploitables sont caractérisés par leur temps de rétention et leur spectre de masse. Ils sont distingués selon les différentes catégories proposées par Sumner et al. [9] : identifés si le temps de rétention et le spectre de masse sont similaires à ceux d’un composé pur ; appartenant à une classe chimique si la zone de temps de rétention et les fragments m/z sont caractéristiques de cette classe ; les autres composés sont considérés comme «inconnu».
L'analyse par NARP-LC/MS sur colonne PGC des extraits dichlorométhaniques traités au charbon actif de C. micranthum et M. scaber (Figs. 5 et 6) a permis respectivement de détecter environ 40 et 70 composés phytochimiques.
- - Pour C. micranthum (Tableau 3), 44 composés ont été trouvés : 5 ont été identifiés et correspondent au lupéol (Rt 11.90), à l'α-amyrine (Rt 12.23), α-tocophérol (Rt 13.04), squalène (Rt 17.75), au β-sitostérol (Rt 20,48) ; 14 appartiennent à différentes classes chimiques : 1 dérivé α -tocophérol, 5 triterpènes, 3 esters de stérol et 5 triglycérides ; 25 composés inconnus.

Chromatogramme TIC LC/MS d'un extrait traité au charbon actif de C. micranthum (CM1).

Chromatogramme TIC LC/MS d'un extrait de M. scaber (MS1) traité au charbon actif.
Composés phytochimiques détectés par LC-MS (APCI) d'un extrait de C. micranthum (CM1) traité au charbon actif.
| Tr (min) | Composés | m/z | |
| 1.09 | Inconnu | 197.1 | 181.1 |
| 2.85 | Inconnu | 278.7 | |
| 5.94 | Inconnu | 353.5 | |
| 6.37 | Triterpèneb | 439.5 | 463.5 |
| 7.64 | Triterpèneb | 391.5 | 463.5 |
| 8.82 | Triterpèneb | 425.5 | 443.5 |
| 10.78 | Triterpèneb | 409.5 | |
| 11.90 | Lupéola | 409.4 | |
| 12.23 | α-Amyrinea | 427.4 | 338.6 |
| 13.04 | α-Tocophérola | 429.3 | |
| 13.68 | Triterpèneb | 427.2 | 435 |
| 14.91 | Inconnu | 445.4 | |
| 15.18 | Inconnu | 444.9 | |
| 16.78 | Inconnu | 445.4 | 425.4 |
| 17.75 | Squalènea | 411.3 | |
| 18.33 | Inconnu | 613.5 | |
| 20.48 | β-Sitostérola | 397.4 | |
| 21.90 | Inconnu | 617.7 | 591.5 |
| 22.20 | Inconnu | 615.5 | 597.7 |
| 23.78 | Inconnu | 557.7 | |
| 25.05 | Dérivé α-tocophérolb | 428.8 | |
| 25.94 | Inconnu | 575.8 | 747.2 |
| 27.20 | Inconnu | 603.6 | 585.6 |
| 28.37 | Inconnu | 577.6 | 585.6 |
| 29.66 | Inconnu | 409.5 | |
| 36.53 | Inconnu | 413.6 | |
| 37.11 | Inconnu | 613.7 | 423.4 747.2 870.9 |
| 38.77 | Inconnu | 409.7 | |
| 40.32 | Inconnu | 579.8 | 613.5 |
| 41.60 | Triglycérideb | 874.7 | 595.8 |
| 41.87 | Triglycérideb | 872.8 | 595.8 |
| 42.77 | Triglycérideb | 876.6 | |
| 44.00 | Triglycérideb | 882.7 | 555.7 607.8 |
| 44.40 | Triglycérideb | 853.1 | 597.8 575.7 663.8 |
| 45.87 | Inconnu | 423.6 | 601.7 577.7 854.8 |
| 46.86 | Inconnu | 409.6 | 881.0 |
| 47.26 | Inconnu | 577.8 | 603.7 |
| 48.29 | Inconnu | 589.7 | 575.6 882.8 |
| 49.14 | Inconnu | 409.7 | 533.8 |
| 49.69 | Inconnu | 577.7 | 603.6 359.1 |
| 52.28 | Inconnu | 631.9 | 561.7 359.5 |
| 54.18 | Ester de stérolb | 397.4 | |
| 54.94 | Ester de stérolb | 589.8 | |
| 56.52 | Ester de stérolb | 397.5 | 749.2 |
a Composés identifiés.
b Composés appartenant à une classe chimique (classification Summer et al. [9]).
Tous les métabolites secondaires apolaires rapportés par plusieurs auteurs [15,16] ont été identifiés (sitostérol, lupeol, α-amyrine). Comme cela était prévisible, les composés polaires présents dans les feuilles de C. micranthum [15,16] n'ont pas été détectés, mais le profil chromatographique obtenu par notre méthode chromatographique liquide en phase inverse des métabolites apolaires peut être proposé pour distinguer des produits à base des feuilles de C. micranthum.
- - En ce qui concerne M. scaber (Tableau 4), 68 composés ont été détectés : 6 composés ont été identifiés : acide oléanolique (Rt 6,77), acide ursolique (Rt 7.42), α-amyrine (Rt 12.38), α-tocophérol (Rt 12.97), squalène (Rt 18.31), sitostérol (Rt 21,32) ; 19 appartiennent à différentes classes chimiques : 1 dérivé α -tocophérol, 8 triterpènes, 4 esters de stérol et 6 triglycérides ; 43 composés inconnus.
Composés phytochimiques détectés par LC-MS (APCI) d'un extrait de M. scaber (MS1) traité au charbon actif.
| Tr (min) | Composés | m/z |
| 1.07 | Inconnu | 196.9 208.8 243.2 179.0 |
| 2.30 | Inconnu | 279.0 |
| 3.10 | Inconnu | 343.1 279.2 329.2 |
| 6.02 | Inconnu | 353.3 277.1 |
| 6.77 | Acide Oléanoliquea | 439.1 457.5 411.5 503.8 |
| 7.20 | Triterpèneb | 457.4 411.3 |
| 7.42 | Acide Ursoliquea | 438.8 411.5 457.3 |
| 9.30 | Triterpèneb | 439.3 425.4 327.2 |
| 9.78 | Triterpèneb | 439.3 425.4 359.1 327.1 |
| 10.91 | Triterpèneb | 439.3 327.3 371.1 |
| 12.38 | α-Amyrinea | 409.6 |
| 12.75 | Triterpèneb | 427.4 338.4 |
| 12.97 | α-Tocophérola | 429.3 475.7 |
| 13.44 | Triterpèneb | 427.3 310.5 409.5 |
| 14.22 | Triterpèneb | 427.3 409.5 435.5 |
| 14.75 | Triterpèneb | 409.6 261.2 427.4 |
| 15.26 | Inconnu | 445.4 |
| 16.24 | Inconnu | 611.6 |
| 17.13 | Inconnu | 425.4 445.5 407.4 |
| 17.54 | Inconnu | 611.8 445.8 |
| 18.31 | Squalènea | 411.3 |
| 18.95 | Inconnu | 613.7 589.4 |
| 19.55 | Inconnu | 555.6 |
| 20.48 | Inconnu | 615.6 557.7 |
| 20.72 | Inconnu | 613.8 |
| 21.32 | β-Sitostérola | 383.4 397.3 |
| 21.61 | Inconnu | 573.6 591.4 311.3 337.0 |
| 21.94 | Inconnu | 599.6 589.8 |
| 22.14 | Inconnu | 615.8 597.8 |
| 22.27 | Inconnu | 615.6 597.8 |
| 22.64 | Inconnu | 573.6 613.6 |
| 22.96 | Inconnu | 613.5 595.6 |
| 23.45 | Inconnu | 609.7 |
| 23.63 | Inconnu | 601.7 619.8 |
| 24.16 | Inconnu | 559.9 609.7 |
| 24.39 | Inconnu | 557.6 591.4 |
| 24.48 | Inconnu | 557.4 575.5 383.5 |
| 25.40 | Dérivé α-tocophérolb | 431.4 447.5 |
| 26.08 | Inconnu | 575.5 |
| 27.04 | Inconnu | 573.6 591.5 395.5 |
| 27.65 | Inconnu | 339.5 603.6 549.6 371.2 423.4 |
| 28.41 | Inconnu | 577.8 637.9 409.5 |
| 30.47 | Inconnu | 409.4 |
| 31.16 | Inconnu | 423.4 395.5 651.9 |
| 32.05 | Inconnu | 603.7 409.5 747.1 |
| 34.17 | Inconnu | 591.6 551.8 |
| 34.87 | Inconnu | 409.6 605.6 872.7 619.4 |
| 36.73 | Inconnu | 613.6 423.3 |
| 38.56 | Inconnu | 870.9 545.7 |
| 40.18 | Inconnu | 409.6 613.5 579.6 |
| 41.18 | Inconnu | 872.5 595.9 |
| 43.30 | Inconnu | 876.6 599.9 |
| 44.39 | Inconnu | 601.7 882.7 409.6 |
| 45.05 | Triglycéride | 853.1 598.0 573.7 409.6 |
| 45.90 | Triglycéride | 603.9 341.5 |
| 46.28 | Triglycéride | 601.8 577.8 856.9 |
| 47.45 | Triglycéride | 878.8 597.8 |
| 47.89 | Triglycéride | 577.7 603.6 858.9 |
| 48.73 | Triglycéride | 575.6 601.8 884.8 |
| 49.72 | Inconnu | 908.6 533.8 341.2 |
| 50.24 | Inconnu | 577.8 551.8 605.6 |
| 51.23 | Inconnu | 575.8 579.7 603.6 |
| 52.78 | Inconnu | 577.9 606.0 860.7 |
| 53.49 | Inconnu | 603.9 607.7 |
| 55.04 | Ester de stérolb | 397.5 605.6 |
| 55.64 | Ester de stérolb | 384.5 589.7 |
| 57.59 | Ester de stérolb | 749.1 397.3 |
| 61.04 | Ester de stérolb | 383.5 359.3 395.6 |
a Composés identifiés.
b Composés appartenant à une classe chimique (classification Summer et al. [9]).
La majorité des métabolites secondaires apolaires mentionnés dans la littérature (sitostérol, α-amyrine, acide oléanolique, acide ursolique) ont été identifiés [17–20,34]. Les extraits sont particulièrement riches en composés phytochimiques apolaires tels que quinones, phytostérols, triterpènes et leurs dérivés estérifiés, éventuellement responsables des activités pharmacologiques de M. scaber (antifongique, antimicrobienne, anti-inflammatoire et analgésique) [17–23].
Le profil chromatographique, renfermant de nombreux métabolites secondaires apolaires, avec ou sans activité thérapeutique et/ou pharmacologique, et appartenant à différentes classes chimiques, peut représenter une empreinte chromatographique pour l’évaluation de la qualité des médicaments à base des feuilles de M. scaber.
3.4 Analyse chimiométrique
Les analyses multivariées sont couramment utilisées pour traiter de grandes quantités de données issues de techniques analytiques appliquées aux médicaments à base de plantes [8].
L’analyse en composante principale (PCA) a été déjà utilisée pour renforcer le pouvoir discriminant des techniques spectrales (spectroscopie proche infrarouge, SPIR) afin de comparer des formulations pharmaceutiques et également distinguer des médicaments contrefaits [35] ; et donc permettre de distinguer différentes drogues végétales ou des produits à base de drogues végétales.
L’analyse supervisée (PLS-DA) est utilisée dans le domaine de la métabolomique pour comparer des classes ou des groupes d'observations, comme différents lots d’une drogue végétale [36].
Le filtrage des données, des rayons X par correction orthogonale du signal (OSC) [37–39], éliminant les variations non prédictives qui ne sont pas corrélées avec les variables, renforce la discrimination des classes, facilitant ainsi l’identification des variables les plus importantes, responsables des différences.
L'analyse chimiométrique a été réalisée sur les profils chromatographiques de deux extraits des 5 lots de C. micranthum (CM1 à CM5) et des 4 lots de M. scaber (MC1 à MC4).
3.4.1 Influence de l’élimination de la chlorophylle
Dans notre étude, la propriété d'adsorption des chlorophylles sur charbon actif indiquée par Jones et al. [40] a été utilisée pour l’élimination de la chlorophylle. Cette méthode a été préférée à l'extraction préalable par l'acétone [41] et à l'hydrolyse alcaline [42], en raison de sa facilité de mise en œuvre. Après des essais préliminaires, le rapport 6 mg de charbon actif pour 1 mg de résidu sec correspondait à la quantité minimum de charbon actif permettant d’obtenir un filtrat de l’extrait dichlorométhanique de couleur jaune à jaune orangé (élimination des pigments chlorophylliens verdâtres).
L’observation des chromatogrammes d’un extrait brut et traité au charbon actif de M. scaber (Fig. 7) indique la présence des mêmes pics chromatographiques. Cependant, on note des différences dans l'intensité des pics chromatographiques.
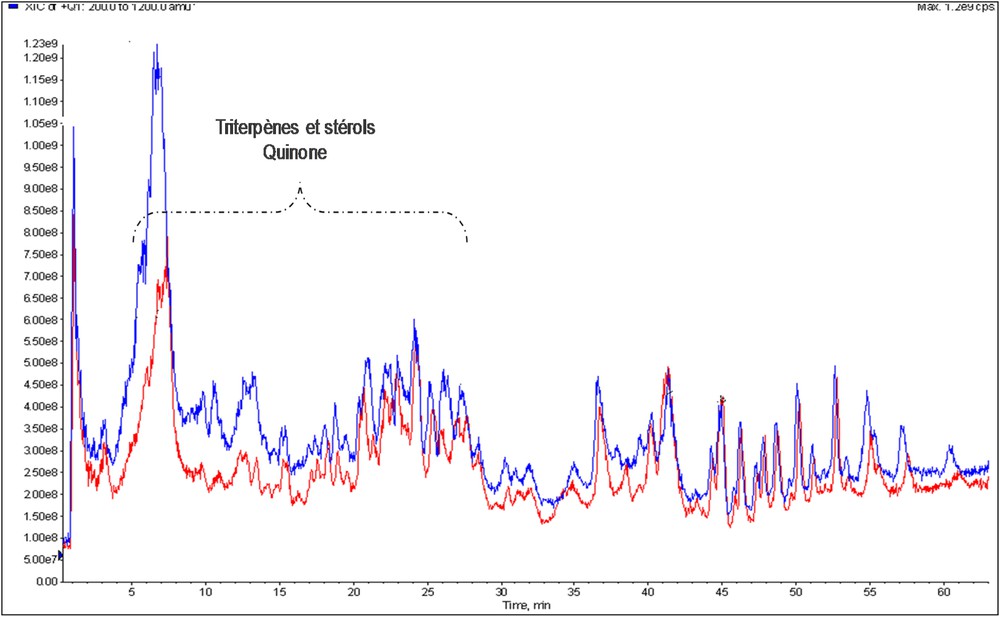
Comparaison des chromatogrammes TIC LC/MS d’un extrait de M. scaber (MS1) brut (bleu) et traité au charbon actif (rouge).
L’exploitation des S-Plot de l’analyse multivariée supervisée (PLS-DA) après correction orthogonale du signal (OSC) des profils chromatographiques des extraits de C. micranthum et M. scaber bruts et traités au charbon nous permet de dire que les composés phytochimiques défavorablement influencés par le traitement au charbon actif sont les triterpènes (temps de rétention (Rt) de 6 à 13 min), les tocophérols (tocophérol 13.04) et les phytostérols (Rt 15 à 25 min) (Fig. 7).
Cependant, les triterpènes, phytostérols et dérivés quinoniques ne sont que partiellement adsorbées par le charbon actif, ce qui se traduit par une diminution des intensités des pics chromatographiques. Une concentration du filtrat traité au charbon avant analyse LC/MS pourrait améliorer l'intensité des pics chromatographiques de ces classes de composés phytochimiques.
3.4.2 Différents lots de drogues végétales
L’analyse en composante principale (PCA) des profils chromatographiques des extraits de C. micranthum et M. scaber (Fig. 8) a indiqué une faible différence entre les lots de chaque drogue végétale :
- - le Score-Plot (Fig. 8A) des profils chromatographiques des extraits des 5 lots de C. micranthum montre : par rapport à la première composante (axe 1 : 22% de la variance totale), une variabilité liée à la préparation des échantillons. La deuxième composante (axe 2 : 17% de la variance totale), montre un rapprochement des lots en fonction du lieu d’obtention des échantillons : CM5 (Sénégal), CM1 et 4 (marché aux plantes) et CM2 (industrie de phtytomédicament). On note également un rapprochement selon l’année d’obtention : CM4 proche de CM5 et CM1 proche de CM2.
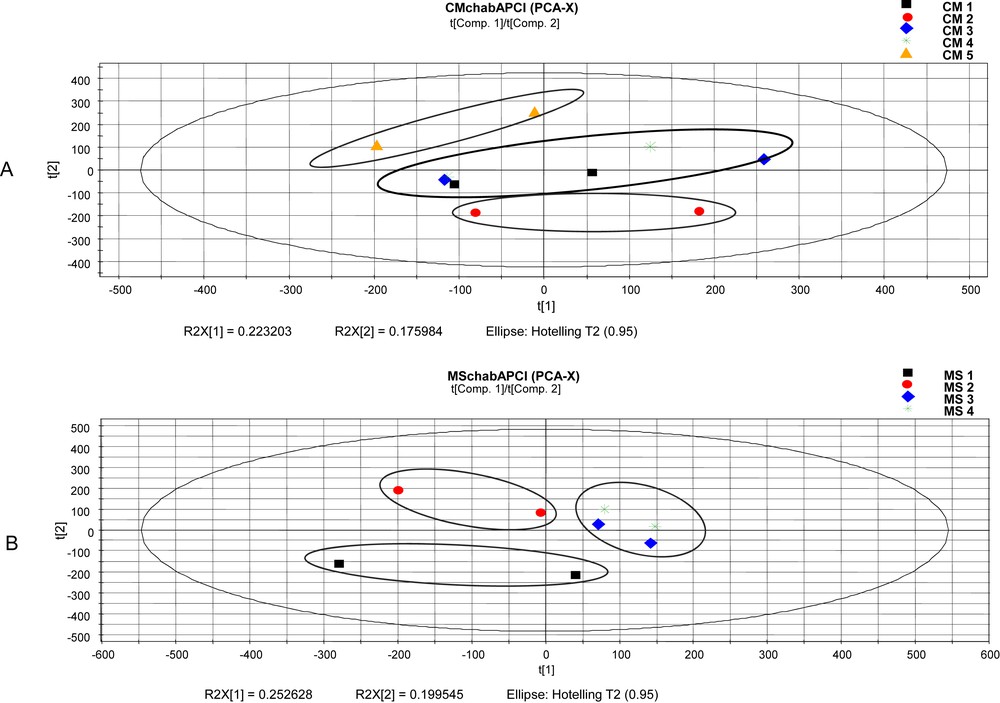
Score-plot des profils chromatographiques des différents lots de C. micranthum (A) et M. scaber (B).
Les échantillons des feuilles de C. micranthum proviennent de circuits commerciaux dont les lieux de récolte ne sont pas connus, mais à l’analyse de ces résultats, l’on pourrait supposer que le marché aux plantes et l’industrie de phytomédicament ont la même source d’approvisionnement, qui ne se trouve pas au Sénégal.
La même tendance peut être décrite dans le cas de M. scaber (Fig. 8B) : l’axe 1 (25% de la variance totale), une variabilité liée à la préparation des échantillons, et l’axe 2 (20% de la variance totale), une variabilité liée à la date de récolte (MS1 en 2008, MS2 en 2009 et MS3 et 4 en 2010 sur des lieux différents (jardin botanique et Bingerville).
En revanche, l’analyse croisée par projection des modèles C. micranthum et M. scaber l’un dans l’autre et la valeur calculée des distances entre les deux modèles supérieure à la limite de confiance fixée au risque de 5% indique que les modèles chimiométriques permettent de distinguer les deux drogues végétales.
4 Conclusion
La méthode que nous proposons dans cette étude utilisant la chromatographie liquide haute performance à polarité de phases inversées couplée à la spectrométrie de masse en milieux non-aqueux (NARP-LC/MS), après extraction au dichlorométhane sous reflux (1:10, m/v) et élimination des pigments chlorophylliens par adsorption sur charbon actif, permet d'établir des profils chromatographiques riches de métabolites apolaires, bioactifs ou non, de drogues végétales.
Cette méthode, associée à la chimiométrie, a été appliquée à différents lots de deux drogues végétales largement utilisées (feuilles de C. micranthum et M. scaber) : elle permet de distinguer les deux drogues végétales et éventuellement les produits à bases des feuilles de C. micranthum et M. scaber.
La méthode NARP-LC/MS permet de différencier les lieux d’approvisionnement des drogues végétales et également de mesurer les variations liées aux périodes d’obtention des échantillons.
En outre, elle permet de détecter la plupart des métabolites apolaires bioactifs ou non déjà identifiés dans des feuilles de C. micranthum et M. scaber.
La méthode NARP-LC/MS proposée dans cette étude, pour l’établissement de profil chromatographique des métabolites apolaires peut être utilisée pour l’évaluation de la qualité de divers médicaments africains à base de drogues végétales.
4.1 Remerciements
Les auteurs adressent leurs remerciements à l’Institut national de floristique de l’université Félix-Houphouët-Boigny d'Abidjan-Cocody pour l'authentification botanique.