

1 Introduction
From the beginning of the industrial age, mankind uses the combustion of fossil fuels as the main source of energy, powering developments in agriculture, industry, transportation, communication, and medicine [1]. However, the supplies of fossil fuels are not limitless [1]. In fact, the average worldwide energy consumption is 16.2 TW [2,3], and this value is doomed to rise. Besides the foreseen shortage of fossil fuels expected in the near future, a probably even worse problem is represented by the huge amount of CO2 released in the atmosphere by the combustion of fossil fuels: this CO2 release is responsible for global important issues, like Earth's warming and climate change [4]. Moreover, it cannot be disregarded that fossil fuels are mainly concentrated in limited regions of Earth, that are also politically unstable (and these facts are sometimes connected), and climatic change is probably also one of the reasons for the massive emigration from the South of the world. Considering these global problems, one of the major challenges for science is to find a renewable source of energy which is environmentally clean, inexpensive, distributed, and abundant [5,6]. Most of the energy sources that can satisfy these requirements are connected, directly or indirectly, with solar energy; among these, direct sunlight is the most attractive. Compared with the other sources, sunlight energy is available everywhere and completely sustainable. Actually, we are lucky that Spaceship Earth [6], which is otherwise a closed system, receives an inexhaustible power flow from the Sun: 120,000 TW of electromagnetic radiation of various wavelengths. So, the quantity of energy that continuously arrives from the Sun largely exceeds human needs [6–8]. However, light energy is intermittent and too diffuse to be profitably and immediately used by our civilization at its maximum potential, so it must be converted to some other type of energy for storage, and the most convenient way is to store it within chemical bonds. Indeed, this is what Nature did from the very beginning and allowed the birth of Life on Earth.
By mimicking natural photosynthesis, it is possible, in principle, to use sunlight to synthesize high-energy content chemicals (fuels) from low-energy content materials. A possible example is the preparation of molecular hydrogen and oxygen by a water splitting process [9]. Indeed, molecular hydrogen is considered one of the possible candidates to replace fossil fuels in the near future; it has several advantages, for example a high specific enthalpy value [10–12]. Moreover, technologies to store and transport hydrogen are available. The combustion of hydrogen regenerates water, releasing energy. The result is a sustainable cycle that is environmentally benign and generates no by-products [9,12]. Obviously, alternative chemical fuels from sunlight other than hydrogen and oxygen, like ammonia, CO, etc, can also be the object of artificial photosynthesis. In particular, a quite attractive field is photoreduction of CO2.
Whatever the final desired product is, an artificial, molecular-based photosynthetic system inspired by natural, molecular photosynthesis can be schematized as in Fig. 1 [13]. It basically includes (i) an antenna system, (ii) a reaction center, and (iii) multielectron transfer catalysts.
- (i) The light-harvesting antenna system. The antenna system is an assembly of a large number of chromophores. It has the role of collecting light energy and funneling it to a specific subunit (the energy trap) by a series of energy transfer processes. In essence, it converts light energy into electronic energy.
- (ii) The reaction center. The reaction center contains the energy trap and some electron donor and acceptor subunits coupled to the energy trap so that a sequence of photoinduced electron transfer processes can take place starting from the excited state of the energy trap, with the ultimate goal of producing a charge-separated state. Basically, the reaction center has the role of converting electronic energy into redox energy.
- (iii) The multielectron transfer catalysts. A single electron (or hole) is suitable to produce current, that is electricity, but to perform chemical reactions from low-energy content raw materials like water and carbon dioxide leading to high-energy content species like molecular hydrogen or reduced forms of CO2, multielectron processes are required. In fact, sequences of single electron steps cannot be used, since the intermediate states would have a too high energy potential. The consequence is that electrons (or holes) must be collected by some charge pool devices and used to drive the multielectron transfer processes. Such charge pools are catalysts for multielectron transfer reactions. Both catalysts for reduction and for oxidation processes are needed. Basically, the multielectron catalysts use redox energy to produce chemical energy.

Schematization of a possible artificial photosynthetic system. The colored triangles indicate the chromophores constituting the antenna systems; the D–P–A assembly corresponds to the reaction center; on the left and right extremes of the sketch, the multielectron transfer catalysts are represented.
Here we will concentrate on light-harvesting antenna systems. In particular, after a chapter in which some main requisites for designing an artificial light-harvesting antenna are summarized, we shall briefly review the work we have done in this field during a period of 25 years, starting from our research on the design and the photophysical studies of light-harvesting antenna dendrimers made of multinuclear Ru(II) and Os(II) polypyridine subunits to the energy transfer processes occurring in various multibodipy systems arranged on different scaffolds. The mechanisms of the energy migration processes occurring in the studied systems will also be discussed. All the reported experiments and data refer to acetonitrile fluid solutions at room temperature, unless otherwise stated.
2 Light-harvesting antennae. Basic requirements
To design an artificial antenna system, attention should be devoted to [14,15]: (i) the nature of chromophores/building blocks; (ii) connections between the chromophores; (iii) synthetic procedures.
2.1 Building blocks/chromophores
The first property an artificial antenna has to exhibit is, rather obviously, strong visible light absorption: as a consequence it has to contain a large number of chromophores having an absorption spectrum as much intense and broad as possible to span the whole visible spectrum. The chromophores must be stable in their ground and excited states: this requisite is more important for synthetic, artificial systems than for natural systems, since the latter can rely on self-repairing processes that are still largely unknown to the former ones. Because they should cover as much visible energy as possible, the chromophores should be easily tunable, so with little synthetic efforts it can be possible to prepare a variety of basic components with different absorption spectra from one another, and their combination in a supramolecular structure can therefore catch a large portion of light energy. It is not strictly needed that the chromophores are intrinsically luminescent, but is convenient as it can allow us to study the systems more easily, by taking advantage of luminescence spectroscopy, and also because their luminescence indicates that the nonradiative decay of the individual chromophores is not very fast: this is instrumental – though not strictly necessary – to obtain efficient energy transfer processes in the supramolecular antenna arrays.
2.2 Molecular connectors
In nature, the individual chromophores are arranged in supramolecular architectures by means of non-covalent interactions. Whereas search for similar non-covalent interactions to be used in synthetic antennae is increasing, the covalent interaction has dominated the field until now. Connectors are quite important: they must guarantee electronic coupling between the various chromophores they connect that is strong enough to allow fast and efficient energy transfer processes, but weak enough to let each chromophore/building block maintain its individuality. In other words, the system has to be supramolecular, according to the functional definition by Balzani and Scandola [16], and not delocalized; this anyway does not forbid that the properties of the individual building blocks can be largely perturbed in the assembly, in comparison with the isolated subunits.
2.3 Synthetic procedures
Suitable procedures aimed to prepare supramolecular arrays containing a large number of chromophores with (relatively) few synthetic steps have to be designed. Besides the obvious reason of limiting the synthetic efforts as much as possible, the synthetic procedures have to be designed to build up the chromophores in desired, organized fashions. The assemblies must be structurally organized and functionally integrated in the domains of space, energy and time: in other words, a correct gradient for energy transfer must be present within the assemblies, and the rate constants for energy transfer have to largely exceed the intrinsic decay rate constants of the excited states localized in the individual chromophoric subunits. This allows us to maximize the efficiency of the energy transfer processes toward the population of the so-called energy trap, that is the subunit(s), where the lowest-energy excited state of the whole antenna is localized. This latter subunit will finally be interfaced with a component suitable to use the collected energy (a reaction center – that can be of molecular nature or also an electrode or any other bulk device – or directly a catalyst).
Clearly, the choice of a specific class of chromophores sometimes also drives the choices of the molecular connectors and of the synthetic procedures. Actually, chromophores, connectors, and synthetic procedures can be considered as sets of possible solutions, with one element of the set in some way dictating in large part the nature of the other elements of the set.
3 Multimetallic dendrimers as light-harvesting antennae
3.1 The building blocks, the connectors and the properties of the assembled dendrimers
Starting from a long-term collaboration involving the Universities of Bologna, Messina, and Pisa, we prepared and studied a large number of multinuclear compounds based on Ru(II) and Os(II) polypyridine subunits as building blocks. The choice of Ru(II) and Os(II) polypyridine compounds as chromophores for light-harvesting antenna (LHA) systems was justified by the properties of these compounds [17–21]. In fact, they absorb in the visible region of the spectrum, thanks to spin-allowed metal-to-ligand charge-transfer (MLCT) transitions (for Os(II) compounds, also formally spin-forbidden MLCT transitions significantly contribute to the visible absorption process); their lowest-energy excited state, typically a triplet MLCT level, is emissive and relatively long-lived; they are fairly stable, both in the ground and in their excited states (although photo-substitution is also possible, in specific cases, and has also been profitably used for various practical purposes [18–20,22,23]). Moreover, these absorption and luminescence properties – as well as the quite interesting redox properties of such compounds, which will not be discussed here – are tunable by a judicious choice and combination of ligands. As a matter of fact, all the above mentioned properties make Ru(II) and Os(II) polypyridine compounds quite interesting for several reasons, besides as chromophores for LHA purposes, so probably [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) and its derivatives are the class of transition metal complexes more investigated in the last 30 years [21].
As connectors, we used 2,3-dpp and 2,5-dpp bis-chelating ligands (dpp = bis(2′-pyridyl)pyrazine; see Fig. 2). These ligands can indeed coordinate two metal centers, behaving as bridging ligands. The dpp ligands allow a quite substantial electronic coupling, as demonstrated by the oxidation splitting of the metal-centered oxidation waves exhibited by the dinuclear compounds [(bpy)2Ru(μ-2,3-dpp)Ru(bpy)2]4+ (Ru2) [24] and [(bpy)2Os(μ-2,3-dpp)Os(bpy)2]4+ (Os2) [25]. For such compounds, in fact, the oxidation pattern shows two, well separated one-electron processes, assigned to successive M2+/M3+ processes. However, in spite of the strong electronic coupling offered by the dpp bridging ligands, oxidation remains localized on specific metal centers, as demonstrated by spectroelectrochemistry experiments and other techniques, indicating that Ru2 and Os2, as well as the other multimetallic compounds of the family, are Class II compounds according to the Robin and Day approximation [26–30]. The different potentials of these one-electron oxidation processes (+1.38 and +1.55 V vs SCE for Ru2 and +0.90 and +1.20 V vs SCE for Os2 [24,25]; similar values were obtained for the compounds containing 2,5-dpp as the bridge), yielded comproportionation constants KC of 7.5 × 102 and 1.2 × 105 for Ru2 and Os2, respectively, and also clarified that the electronic coupling via the bridging ligands 2,3- and 2,5-dpp was promoted by the superexchange mechanism mainly involving the LUMO of the bridge [31]. Incidently, the first investigations on the dinuclear compounds of these bridging ligands also contribute to rationalize the general redox behavior of dinuclear Ru(II) polypyridine compounds, that was attracting increasing interest in those years.
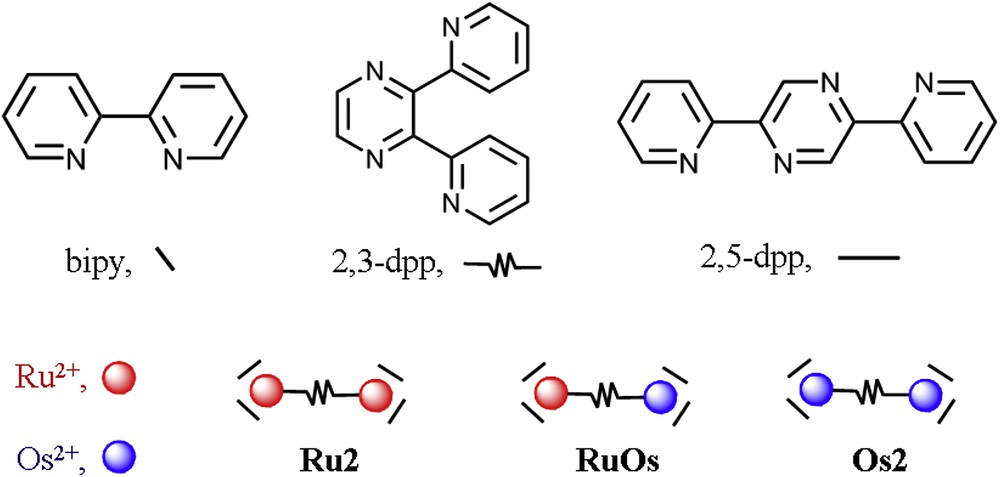
Structural formulas of the polypyridine ligands most mentioned in this work and their representations. Schematic representation of dinuclear Ru2, Os2, and RuOs compounds is also shown.
Finally, the synthetic strategy used was based on a dendritic approach, considering that dendrimers, prepared via divergent or convergent approaches, allowed us to assemble a large number of subunits (in this case, chromophores) with a relatively low number of synthetic steps [32]. More details on the dendritic approach will be discussed later. For the moment, it can be mentioned that the multinuclear species Ru4 and OsRu3, shown in Fig. 3, are examples of the first generation dendrimers of this class of compounds, Ru10 and OsRu9 are the second generation dendrimers and Ru22 and OsRu21 are the third generation dendrimers.
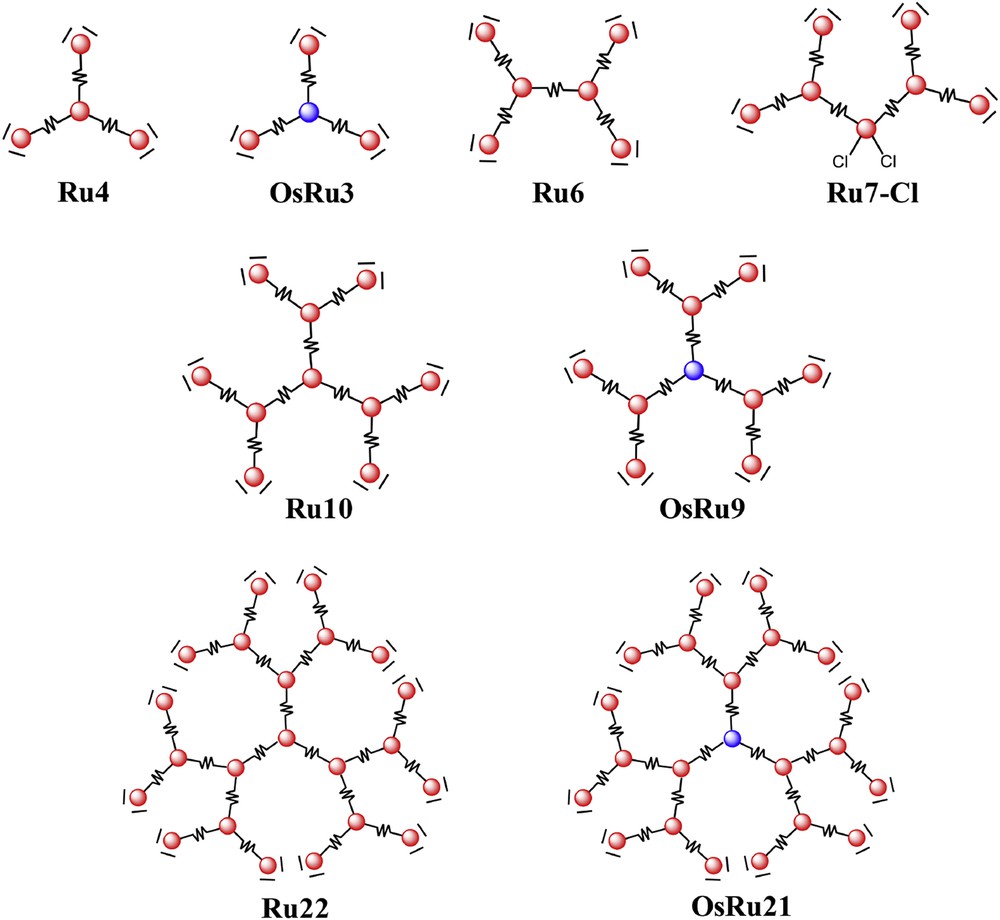
Schematic representation of some dendrimers discussed in this work. For symbols codes, see Fig. 2.
It must be considered that the core of the 2,3-dpp-based dendrimers has the general formula {M(μ-2,3-dpp)3}2+, and the same formula is that of the chromophoric units in the intermediate, “propagating” layers (see Fig. 3), the formula of a peripheral unit is {(μ-2,3-dpp)M(bpy)2}2+ (here, only species having bpy ligands in the peripheral layers are considered; dendrimers having different polypyridine ligands at the periphery, biquinolines, for example, have also been studied [24,25], but are not commented here). However, it should be noted that in all cases the dpp ligand is playing the role of the bridging ligand (as indicated by the greek letter preceeding it in the formula), except when it is protected (see later). This is important to consider, since the properties of a mononuclear species like [(2,3-dpp)Ru(bpy)2]2+ (here 2,3-dpp only chelates a single metal center) and [(bpy)2Ru(μ-2,3-dpp)Ru(bpy)2]4+ (in which the 2,3-dpp acts as a bridging ligand, chelating two metal centers) are quite different from one another: the most important difference is that the MLCT level, involving the dpp ligand, is strongly stabilized in the multinuclear species (for more details see the original references [14,15,24]). Indeed, when the additive properties of the absorption spectra of this class of LHA dendrimers are mentioned (see for example Fig. 4), indicating that each individual subunit brings its own properties in the dendritic assemblies, the individual subunits are referred to as containing dpp ligands in their “bridging” form and properties. In other words, the mononuclear [(2,3-dpp)Ru(bpy)2]2+ species, which does not contain bridging ligands, is not a good model as the building block for the peripheral units of the dendrimers, as far as its electronic properties are concerned.
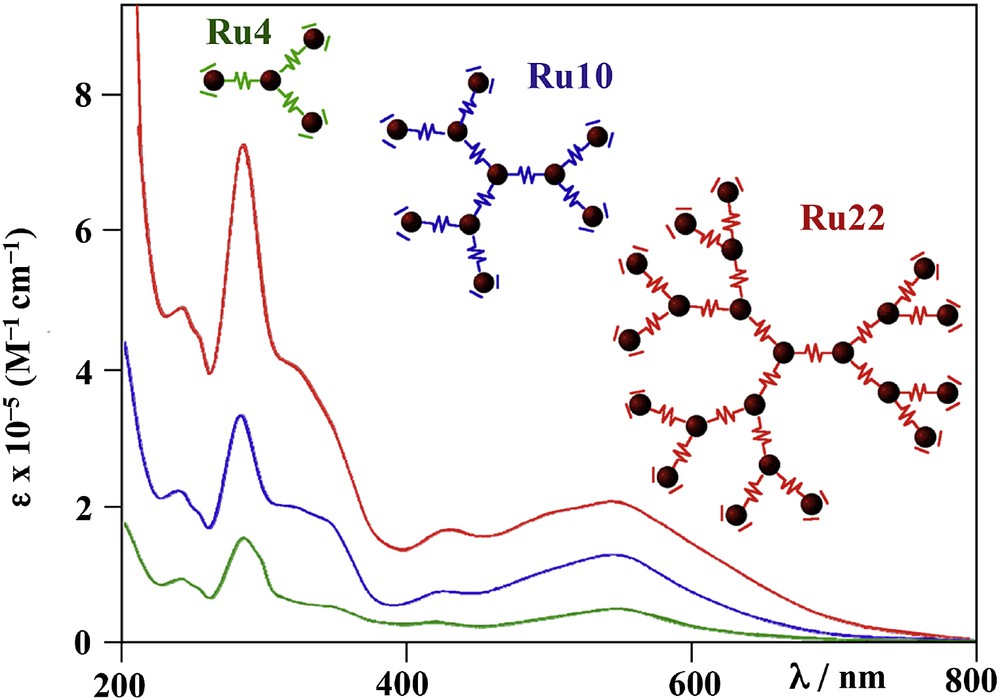
Absorption spectra of Ru4, Ru10 and Ru22 in acetonitrile at room temperature. For original data, see Refs. [24,36,48].
The electronic coupling in mixed-metal dpp-bridged compounds is strong enough to allow a quantitative energy transfer from the higher-energy Ru → dpp CT state (in fact, the lowest MLCT state in Ru2 and Os2, and in the larger systems studied later on, involved the bridging ligand(s) as the acceptor) to the lower-energy Os → dpp CT level, the simplest case being [(bpy)2Ru(μ-2,3-dpp)Os(bpy)2]4+ (RuOs) [33], both in the 77 K rigid matrix and in fluid solution at room temperature (RT). The mechanism of the process was proposed to be Dexter electron-exchange energy transfer; more recently, it was demonstrated, investigating the photophysical properties of a series of mixed-metal compounds having various nuclearities, that the energy transfer partly occurred from non-equilibrated excited states [34,35], so that even singlet MLCT states are involved. This latter was a quite surprising result, since it was believed that intersystem crossing in Ru(II) and Os(II) complexes in general and in Ru(II)- and Os(II)-based subunits of multicomponent systems was so fast that any reaction (including inter-component energy or electron transfer) could not compete with it; intersystem crossing in fact takes place in the sub-picosecond regime and intercomponent energy transfer, expected to occur for equilibrated states on the ps regime, therefore was considered to involve exclusively triplet states. Indeed, in the tetranuclear [Os{(μ-2,3-dpp)Ru(bpy)2}3]8+ (OsRu3; see Fig. 3) energy transfer from the Ru → dpp CT state to the cored Os → dpp CT state takes place with a biphasic behavior, with both components in the sub-picosecond timescale: the fast component takes place with a time constant <60 fs (that is, it is faster or comparable to that of intersystem crossing, so it also involves the singlet MLCT Ru-centered state) and the slower component occurs with a time constant of about 600 fs [35], corresponding to triplet–triplet energy transfer. Differences between the singlet–singlet and triplet–triplet energy transfer processes were revealed on exciting OsRu3 at different wavelengths [35].
Other studies demonstrated that in larger dendrimers, where a metal-based subunit with a lower excited state compared to the other subunits is present, and the other subunits have roughly the same energy, the light absorbed by all the metal-based components is quantitatively conveyed to the lowest-energy site, on the picosecond timescale. This is the case of the decanuclear, all-Ru(II) second generation dendrimers [Ru{(μ-2,3-dpp)Ru[(μ-2,3-dpp)Ru(bpy)2]2}3]20+ (Ru10; see the structure in Fig. 3). In this latter species, although only Ru(II)-based components are present, the metal-based subunits have different properties, depending on their location in the supramolecular array: to make the story short, the MLCT state (in any case, a Ru → dpp CT state) of a peripheral {(μ-2,3-dpp)Ru(bpy)2}2+ subunit is lower in energy than the MLCT state of the core or the intermediate layer components, all of them of {Ru(μ-2,3-dpp)3}2+ type. The quantitative energy transfer occurring from the center to the periphery of Ru10 indicates that energy migration is very fast between isoenergetic chromophores when a driving force leading the excitation to a low energy site is present [36]. This result has also an intriguing consequence: assuming the intrinsic decay of a single subunit in fluid solution is of the order of 100 ns (for example, this is roughly the emission lifetime order of Ru2, Ru4 and Ru10 [24,36]; the estimated lifetime of a {Ru(μ-2,3-dpp)3}2+-type chromophoric unit should also be longer, according to the energy gap law [18]), and considering that down-hill energy transfer between nearby subunits is faster than 1 ps, and that isoergonic energy transfer between nearby subunits can be roughly estimated to be about 10 ps, this means that energy transfer is at least five orders of magnitude faster than the intrinsic decay of a single subunit. As a consequence, very large systems could be prepared, containing several chromophoric components, still exhibiting fast and efficient long-range vectorial energy transfer, at least until the correct energy gradient (iso-ergonic or down-hill steps) is present.
Whereas efficient energy transfer is obtained in metal dendrimers with down-hill or even iso-energetic units, the situation totally changed when up-hill energy transfer steps are present, that is when metal-based units having higher-energy MLCT states (“high-energy subunits”) are interposed between donor and acceptor energy transfer subunits. This was the case for the decanuclear mixed-metal compound [Os{(μ-2,3-dpp)Ru[(μ-2,3-dpp)Ru(bpy)2]2}3]20+ (OsRu9, see Fig. 3). Due to the different oxidation potentials of Os(II) compared to Ru(II) units and of the inner Ru(II) centers compared to the outer Ru(II) centers, the energy order of the three types of subunits of OsRu9 is {Os(μ-2,3-dpp)3}2+ (the core chromophore, Osc) < {(μ-2,3-dpp)Ru(bpy)2}2+ (the outer chromophore(s), Rup) < {Ru(μ-2,3-dpp)3}2+ (the intermediate chromophores, Ruint) [36]. So, in principle all the absorbed light should be conveyed into the Osc subunit. However, the emission spectrum is dominated by the emission of the peripheral Rup-type chromophores, with only a very small contribution from the core Osc chromophore [13a,15,36]. Evidently, energy transfer from Rup to Osc across the higher-energy Ruint interposed subunit is not effective [36]. This is even more evident in the docosanuclear species OsRu21 (see Fig. 3), where the Os(II) contribution is negligible [37]. In both cases, as well as in a similar mixed-metal decanuclear species in which 2,2′-biquinoline (biq) replaces bpy as the terminal ligands [36], the emission lifetimes recorded at the Rup dominant emission are also unchanged with respect to the emission lifetimes of the corresponding all-Ru dendrimers, confirming that Rup-to-Osc energy transfer is not a useful decay route in the presence of high-energy interposed chromophores [13a].
The inefficiency of energy transfer across high-energy interposed subunits posed some doubts on the efficient employment of light-harvesting metal dendrimers of this family for practical use. In fact, it was clear that metal dendrimers could be connected with other systems (electron donor or acceptor subunits, for example) by a peripheral ligand. So, the light energy collected by all the chromophores should be channeled to that site to be used. In dendritic structures as most of those shown in Fig. 3, several identical energy traps are present (for example, in Ru10 or Ru6 the peripheral subunits – the energy traps – are respectively six and four) and if energy cannot travel from one trap to the other across the high-energy, the inner subunit(s), most of the absorbed light would have been lost. This consideration, together with the difficulty in characterizing larger dendrimers, led to a reduced activity on LHA metal dendrimers for some time. However, the photophysical study of a dendron-type, heptanuclear species like [Cl2Ru{(μ-2,3-dpp)Ru[(μ-2,3-dpp)Ru(bpy)2]2}2]12+ (Ru7-Cl) revealed interesting properties that inverted the trend [38]. The dendron Ru7-Cl, in fact, in principle could suffer from the same problem of OsRu9: The energy order of the MLCT states of its subunits was {Cl2Ru(μ-2,3-dpp)}2+ (the “core” excited state) < {(μ-2,3-dpp)Ru(bpy)2}2+ (the outer chromophore(s), i.e. Rup) < {Ru(μ-2,3-dpp)3}2+ (i.e., Ruint). As for OsRu9, it was therefore expected that energy transfer from the Rup chomophores to the core, lower-energy one, was inefficient. In contrast, luminescence spectroscopy demonstrated that all the absorbed light, independent of which the subunit is the initial absorber, leads to emission (that occurs in the near-IR region at about 900 nm) from the {Cl2Ru(μ-2,3-dpp)}2+ subunit, the energy trap of the whole dendritic system [38]. Detailed pump-probe transient absorption spectroscopy (TAS) revealed that the energy transfer process takes place in two successive electron transfer steps (see Fig. 5): first, photoinduced reductive electron transfer takes place from the metal center of the core {Cl2Ru(μ-2,3-dpp)}2+ to the MLCT excited state of the Rup subunit; this process occurred in less than 500 fs. The so-formed charge-separated state evolved in few ps to yield the MLCT state of the core [38]. This second step is indeed similar to electron hopping between identical ligands (the bridging ligand, in this case) in the excited state of a single metal complex (see Fig. 5); the difference, compared to reported cases for Ru(bpy)32+-type compounds [39] is that in Ru7-Cl the metal center is formally a Ru(II) center and not formally a Ru(III) center, as it occurs for MLCT states. However, the order of magnitude of the electron hopping between the ligands appears to be the same.
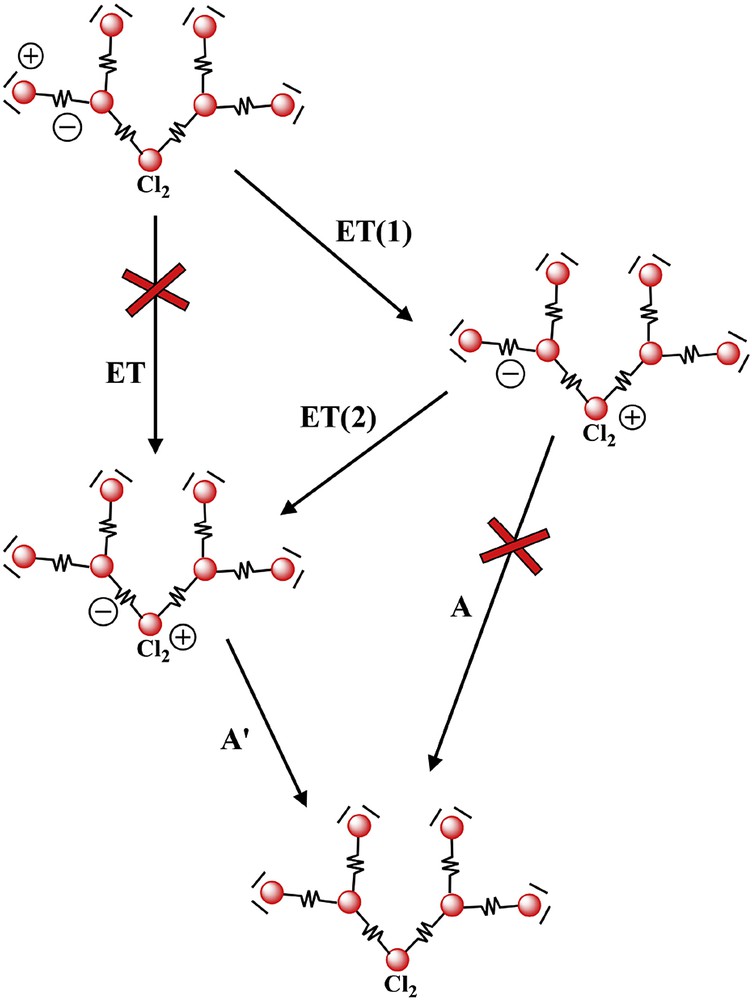
Representation of the two-step energy transfer process occurring in Ru7-Cl [38].
To summarize, Ru7-Cl demonstrates that the limit of the low efficiency of energy transfer across high-energy units can be overcome in LHA metal dendrimers by successive electron transfer steps (noteworthy, such a process is not possible in OsRu9 because of thermodynamic reasons [38]). Probably even more important is the information that long-range electron transfer is not limited by the presence of interposed high-energy subunits, since the first step of the overall process, photoinduced electron transfer from the metal center of the core unit to the MLCT state of the peripheral subunit, across the intermediate Ru-based subunit is ultrafast (<500 fs). This suggested that if a metal dendrimer is interfaced with an electron donor or acceptor subunit via one of its peripheral chromophores, electron transfer can be highly efficient, independently of which peripheral site the absorbed energy is channeled to. This information, further in agreement with the results obtained for a trinuclear dendron containing a tetrathiofulvalene electron donor subunit [40] and a tetranuclear mixed-metal dendrimer decorated with phenothiazine subunits (see Fig. 6 [41]), opened a way to use this family of dendrimers in several applicative studies, including photoinduced water oxidation [42,43].

Finally, the absorption properties of this class of LHA metal dendrimers have been implemented by adding other organic chromophores at the periphery. This is the case of OsRu3-pyr6 (Fig. 6), in which six pyrene derivatives are connected to the peripheral bpy ligands [44]. In this species, energy transfer from pyrene to the Os-based core (energy transfer to the Ru-based states are not considered, since once the light energy is transferred to the Ru-based subunit(s) it is channeled to the central Os core in the fs timescale, so the MLCT state centered in the Ru-based chromophores cannot accumulate) is biphasic and occurs from non-equilibrated and equilibrated excited states with time constants of about 6 ps and 45 ps, respectively [44].
3.2 Synthetic strategy. The “complexes as ligands/complexes as metals” approach and the iterative protection/deprotection procedure
We cannot conclude this section devoted to metal dendrimers without briefly mentioning the synthetic procedures that allowed their synthesis. The key point was to differentiate between metal complexes that contain free chelating sites, and therefore can behave as ligands towards other metals (complexes-as-ligands) and metal complexes that have some ligands that can be easily substituted, so that they can be considered as nude metals and can be complexed by ligands (complexes-as-metals) [45,46]. Typical complexes-as-ligands are metal complexes such as [Ru(bpy)2(2,3-dpp)]2+ or [Ru(2,3-dpp)3]2+, that can still react as ligands and coordinate one and three other metal centers (or complexes-as-metals, respectively. A typical complex-as-metal is Ru(bpy)2Cl2, that can easily lost the two chloride ligands (for example, by refluxing in alcoholic solvents) and promptly be coordinated by a ligand (or a complex-as-ligand). Besides simple mononuclear complexes-as-ligands and complexes-as-metals, more precious synthons are multinuclear species, like the trinuclear complex-metal [Cl2Ru{(μ-2,3-dpp)Ru(bpy)2}2]4+ that in combination with [Ru(2,3-dpp)3]2+ yields Ru10.
The use of the complexes-as-ligands/complexes-as-metals strategy was further implemented by the development of a protection/deprotection procedure of chelating sites. For example, 2,3-dpp was transformed into a methylated form, 2,3-dpp-Me+ (see Fig. 7 [47,48]), and used as a terminal ligand, similarly to bpy. Once a protected multinuclear species is prepared, it was deprotected and the chelating site so restored is used for building up larger species. This procedure was iterated to prepare species like docosanuclear dendrimers, even of mixed-metal nature [13a,15,37,48]. The process, exemplified in Fig. 7, shows the convenience of using such an iterative dendritic approach, that allows us to insert a large number of chromophores (in practice, a layer of chromophores) in a single reaction steps, for preparing multichromophoric LHA based on covalent linkages.
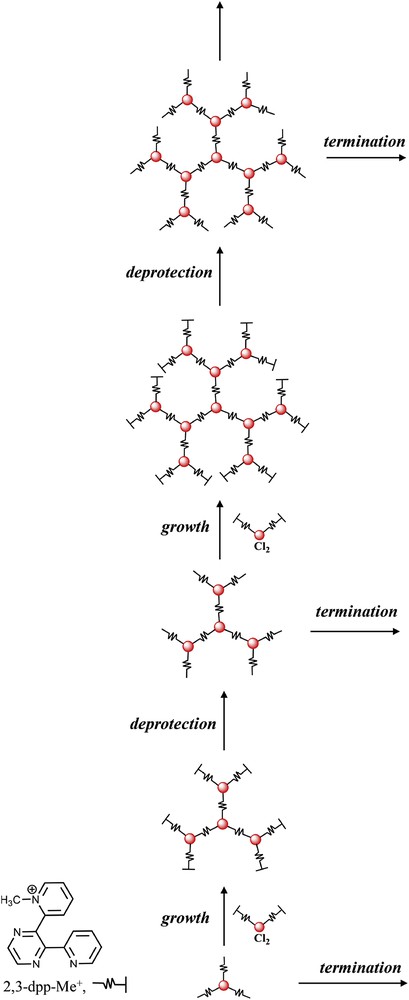
Divergent synthetic strategy for the preparation of polynuclear metal complexes of dendrimer shapes [48]. Each deprotected compound can be employed in termination steps, yielding sterile dendrimers. The structural formula of the protected 2,3-dpp-Me+ species is also shown on the bottom left corner.
4 Light-harvesting antennae containing multi-bodipy chromophores
Boron dipyrromethene dyes, abbreviated as bodipy dyes (see Fig. 8 for a representative example), are quite appealing compounds to be used as building blocks of light-harvesting antenna systems [49]. Indeed, bodipy species exhibit intense visible absorption, their lowest-energy, singlet excited state is usually strongly emissive (in some cases, emission quantum yields >0.95 have been reported), with lifetimes in the ns timescale. Moreover, the spectroscopic and photophysical properties of bodipy dyes are extremely tunable, by inserting appropriate substituents in various positions of their structure [49,50]. During the last 10 years, this stimulated an intense research activity on bodipy dyes, for several, also applicative reasons. For example, bodipy systems have been used as molecular components of fluorescent logic gates [51] and – more relevant to the topic of this work – as components of multichromophoric systems for investigating photoinduced energy transfer [49,50,52].
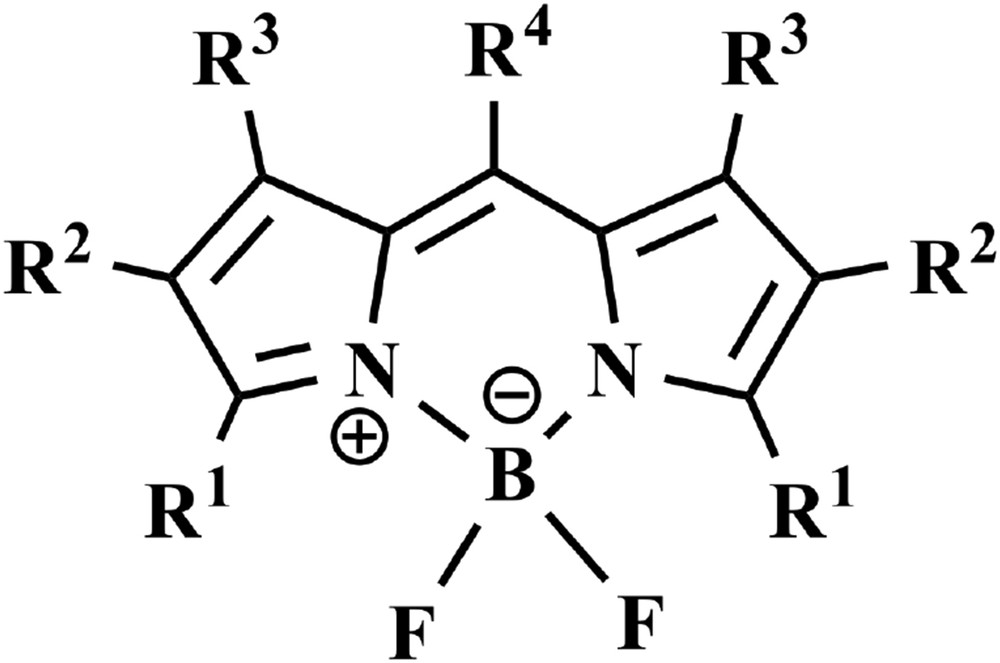
General formula of a bodipy core. The groups indicated with R can be various substituents. The fluoride ligands can also be substituted by alkyl groups [49,50].
We have investigated several multibodipy systems, also made of different bodipy subunits, capable of collecting a large area of the visible spectrum. The first species we studied is 1T-ABC, in which three different bodipy units, named A, B, and C are arranged around a truxene core (Fig. 9) [53]. In this species, four different chromophores, all of them potentially fluorescent, are present, the three bodipy dyes and the truxene core. The absorption spectrum of 1T-ABC perfectly overlaps the arithmetic sum of the absorption spectra of its individual components, demonstrating that the electronic coupling between the components is very weak. Steady-state and time-resolved luminescence experiments showed that quantitative Dexter electron-exchange energy transfer takes place from the truxene core to the lower-lying bodipy subunits, and that Coulombic, Förster energy transfer occurs between the various bodipy subunits, finally leading to the population of the lowest-energy excited state of the whole structure, centered in the amine containing component C, with at least 95% of quantum efficiency. According to Förster theory, spectral overlap between donor emission and acceptor absorption spectra affects the rate constants of the processes, so that the electronic energy preferably travels from A to C via B, rather than using the direct A-to-C pathway. The rate constants of the bodipy-bodipy energy transfer processes are in the 108–1010 s−1 range [53].
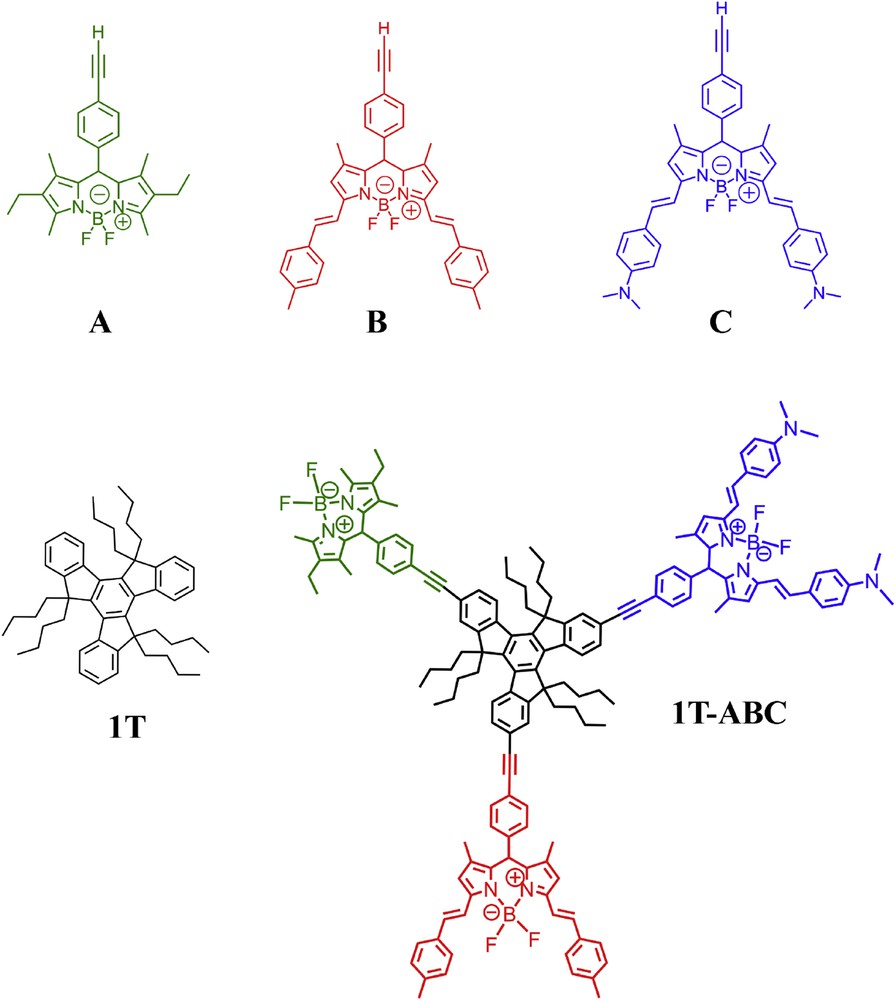
Structural formula of 1T-ABC and of its isolated components [53].
Another multibodipy species, containing three different bodipy dyes, is the trypticene-cored compound 2T-DEF shown in Fig. 10 [54]. Even in this case, the electronic coupling between the various components was very weak, as indicated by the absorption spectra of 2T-DEF and of its individual components. The photophysical properties of 2T-DEF and its parent compounds (including some bichromophoric simplest species) were investigated both by luminescence (steady-state and time-resolved) spectroscopy and by pump-probe femtosecond transient absorption spectroscopy [54]. The efficiency of the population of the lowest-energy excited state of the whole multibodipy species was very close to 100%, indicating that energy transfer between the various bodipy subunits was essentially quantitative. In fact, the rate constants for the various processes, also controlled by the Förster mechanism, were of the order of 1010–1011 s−1. In some cases, biphasic energy transfer processes are evidenced, probably due to the presence of different conformers [54]. The faster rate constants for inter-bodipy energy transfer in trypticene-cored species with respect to the similar truxene-based compounds is probably due to the shorter bodipy-bodipy distance in the former with respect to the latter assemblies.

Structural formula of 2T-DEF and of its components [54].
Finally, multibodipy systems have been made by using a carbohydrate unit as the scaffold. Compounds C1 and C2 (Fig. 11) have been prepared and studied [55]; C1 contains two different bodipy chromophores, while C2 contains four bodipy units, three of them identical and one having a lower-energy singlet state. In spite of the small dimension of the carbohydrate scaffold, even in this case the comparison between the experimental absorption spectra of C1 and C2 with the calculated spectra obtained by adding the absorption spectra of their individual components (see Fig. 12 for the case of C1) indicated that ground state electronic interactions were negligible. Excitation and emission spectroscopies showed that energy transfer was quantitative for both C1 and C2. By taking advantage of the different signatures of the transient absorption spectra of the two different types of chromophores that are present in C1 and C2 it was possible to measure the rate constants of the photoinduced energy transfer processes [55]. For C1, the energy transfer rate constant was estimated to be faster than 4 × 1012 s−1, since the donor transient signature, initially present, disappears with a time constant comparable with that of the laser pulse (120 fs). Compound C2 exhibits two energy transfer rate constants: a faster one, of the same order as the energy transfer rate constant of C1, and a slower one, corresponding to 8 × 1010 s−1 [55]. The faster rate constant was assigned to energy transfer involving the donor bodipy dye closest to the (single) amino-containing acceptor bodipy dye, and the slower component was assigned to energy transfer involving as donors the more distant high-energy bodipy subunits.
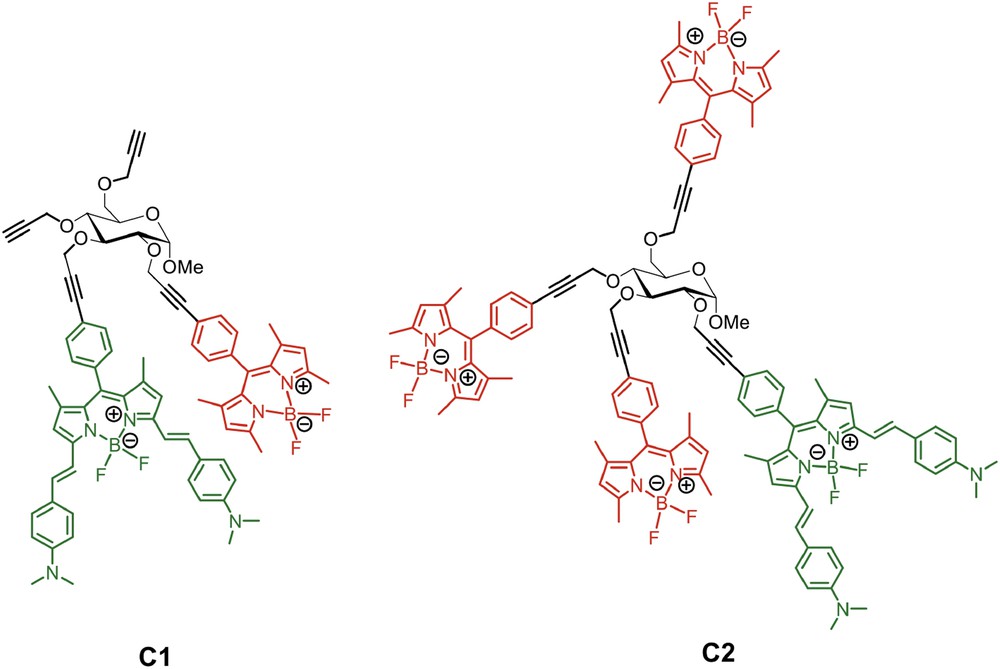
Structural formulas of the multibodipy assemblies C1 and C2 [55].
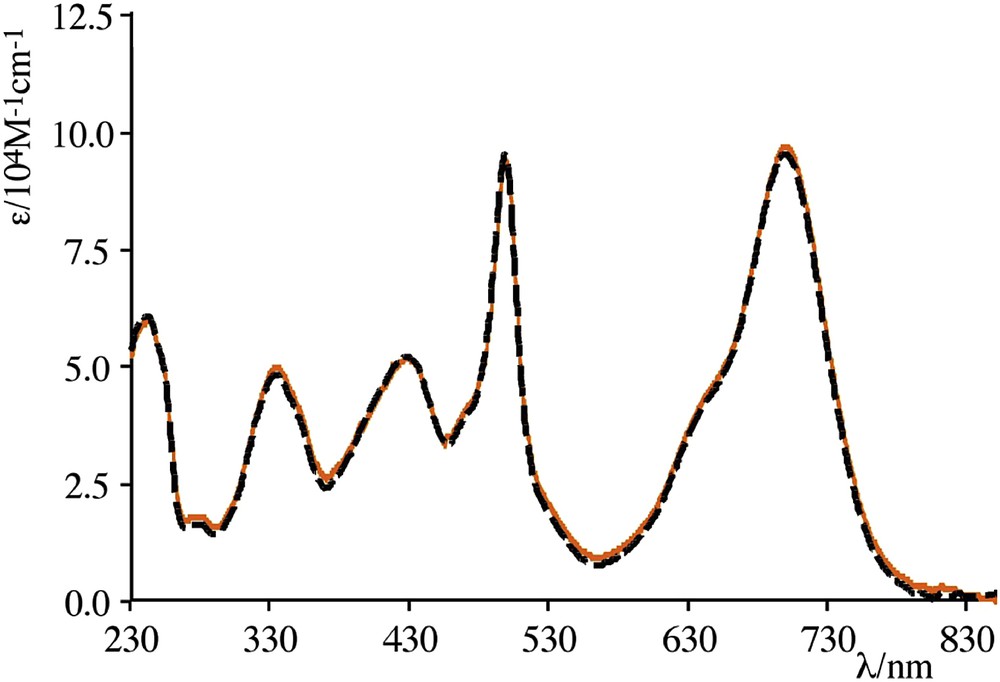
Experimental absorption spectrum of C1 (black dashed line) versus the calculated curve (orange line) obtained by adding the absorption spectrum of the isolated red bodipy species to the absorption spectrum of the isolated green bodipy species (color codes refer to the colors in Fig. 11). Details in Ref. [55].
Compounds C1 and C2 are particularly interesting examples of multichromophoric assemblies having negligible electronic coupling in the ground states but capable of exhibiting very fast energy transfer between subunits: in fact, energy transfer time constants faster than 250 fs, as for C1, are rarely reported for non-interacting subunits. Moreover, they are perfect examples of well-behaved Förster energy transfer: indeed, by using the simplified Förster equation [56] reported in Eq. (1),
| (1) |
| (2) |
5 Concluding remarks
Here we have reported a summary of some research studies we developed in the last few decades on artificial LHA systems. Two classes of systems have been taken into account, metal dendrimers and multibodipy species. The Dexter electron-exchange mechanism involving triplet and singlet states drove energy transfer processes in metal-based species, whereas Förster energy transfer involving singlet states occurs in multibodipy systems. In any case, it is clear that there are various options to design efficient LHA, and all of them can be used. More recently, research has been oriented to interface such LHA systems with reaction centers or multielectron catalysts [42,43], and this will probably be one of the next frontiers of research on artificial photosynthesis.
Acknowledgments
Financial support from MIUR (FIRB project RBAP11C58Y Nanosolar, PRIN 2010-2011 Hi-Phuture) and COST Action CM1201 Perspect-H2O is gratefully acknowledged.