

1 Introduction
The synthesis and transformation of α-diazocarbonyl scaffolds have aroused a steady interest in organic synthesis for many years [1]. These scaffolds are versatile intermediates that can undergo a large array of transformations, especially on the reactive CN2 site, including oxidations, reductions, CC, CH, heteroatom-H insertions and cyclopropanations. Numerous strategies have been designed towards their formation [1,2]. Among them, oxidation of α-keto hydrazones has been successfully developed (Scheme 1, route a), although limited by the availability of the substrates. Acylation of diazomethane (route b) is classically used to access terminal diazoketones but suffers from safety hazards. Diazo-transfer, typically from sulfonyl azides (route c), has been applied to a large range of active methylene compounds to elaborate diverse diazodicarbonyl scaffolds, which upon deacylation can provide terminal diazoketones. Substitution at the diazo carbon of terminal diazoketones, commonly prepared via route b or route c, while preserving the diazo moiety, constitutes a convergent strategy to access diversely functionalised diazocarbonyl scaffolds (route d).

Strategies towards the formation of diazoketone scaffolds.
An important reaction in this field (route d) is the aldol-type addition between terminal diazoketones 1 and various aldehydes, in basic media, providing a convergent route to β-hydroxy-α-diazoketones 2 (Scheme 2a). Strong bases such as lithium diisopropylamide (LDA) are commonly used in stoichiometric amount, at low temperature, to perform this reaction [3,4]. We recently reported an alternative procedure using nucleophilic and weakly basic conditions, consisting of the tetrabutylammonium fluoride (TBAF)-triggered aldol-type addition between α-triethylsilyl-α-diazoacetone (TES-diazoacetone, 3) and various aldehydes (Scheme 2b) [5]. A large range of β-hydroxy-α-diazoketones 4 could thus be synthesised under convenient experimental conditions.
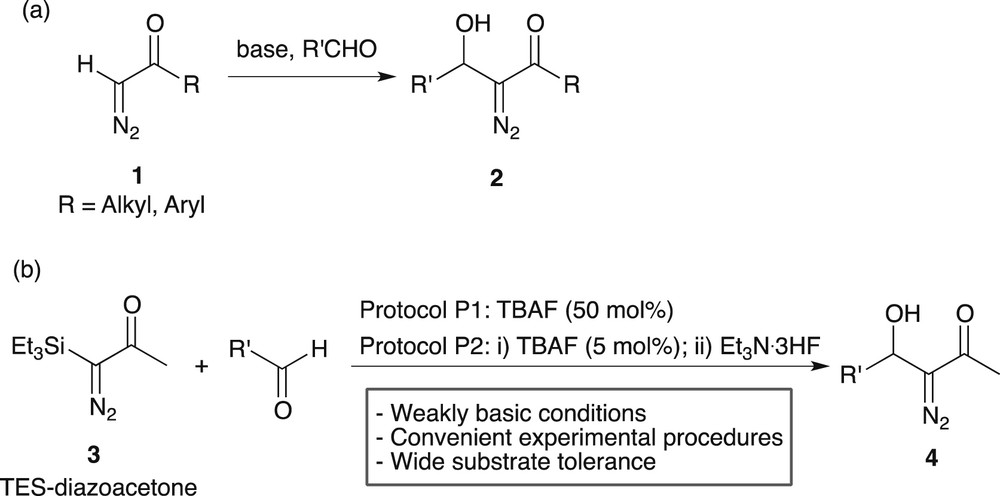
Base and fluoride-induced aldol-type additions on diazoketone scaffolds.
Beside, TES-diazoacetone 3 revealed itself to be a useful three-carbon building block, which could be introduced into functionalised carbon chains via a LDA-induced double cross-aldol addition sequence (Scheme 3) [4]. The presence of the TES protecting group was crucial to carry out first the LDA-induced “methyl-side” aldolisation, without any competitive aldol-type addition on the more reactive “diazo-side”. Methyl-side aldolisation resulted in a mixture of the C-silylated and C-desilylated aldols 5. Methanolysis on the crude mixture achieved complete desilylation before the diazo-side aldol-type addition could be performed classically with LDA, at low temperature, to produce diazodiols 6 (Scheme 3). This sequence allowed the fragile β-hydroxy-α-diazoketone moiety of compounds 6 to be successfully generated in the last step.
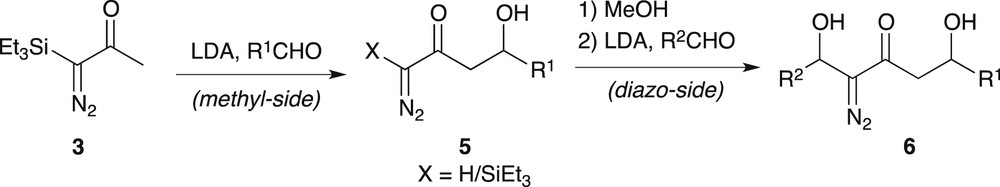
TES-diazoacetone chain-extension by double cross-aldol addition sequence.
To explore further the potential of the α-trialkylsilyl-α-diazoacetone scaffold as a three-carbon building block, it appeared necessary to prevent unintentional diazo-side desilylation, which readily occurs with the labile TES group. Thus, we thought of replacing the TES protecting group by the more robust triisopropylsilyl (TIPS) one [6]. To assess the relevance of this scaffold, we needed to investigate if the diazo-side aldol-type addition could still be conveniently performed under nucleophilic activation by a fluoride. To this aim, we report here the behaviour of α-triisopropylsilyl-α-diazoacetone (TIPS-diazoacetone, 7, Scheme 4) towards fluoride-induced aldol-type addition.

Preparation of TIPS-diazoacetone.
2 Results and discussion
TIPS-diazoacetone 7 [7] was prepared in a good yield by silylation of diazoacetone, using TIPS triflate/N,N-diisopropylethylamine as the silylating system, at 0 °C (Scheme 4) [5,8]. Diazoacetone, stable yellow oil that can be stored in freezer for months, was prepared beforehand on a multigram scale by diazo-transfer from tosyl azide to acetylacetone [5,9], followed by deacetylation [4] (Scheme 4). We were pleased to observe that TIPS-diazoacetone was stable, storable for months in the freezer, contrary to TES-diazoacetone, which rapidly underwent desilylation/degradation in a few weeks time, even in the freezer.
We investigated first the fluoride-induced aldol-type reaction between TIPS-diazoacetone and benzaldehyde (Table 1). TBAF, the optimal source of fluoride highlighted for TES-diazoacetone [5], was initially selected and the best catalytic conditions were applied: TBAF 5 mol %, 4 Å molecular sieves (MSs), anhydrous Et2O, −16 °C, 2 h (Table 1, entry 1). Unfortunately, very low conversion was achieved and aldol 4a, contaminated by traces of bis-aldol 8a (Scheme 5), was isolated in only 7% yield after column chromatography [10]. No O-TIPS–protected aldol was detected. This first result seems to exclude that a fluoride-triggered autocatalytic mechanism could proceed here. This hypothesis was supported by the fact that no reactivity was observed when TBAF was replaced by an equimolar amount of the tetrabutylammonium alkoxide of benzylic alcohol [11].
Fluoride-induced aldol-type addition between TIPS-diazoacetone and benzaldehyde.
| Entry | F− source (mol %) | Additivea | T (°C) | Isolated yield of 4a (%) |
| 1 | TBAFb (0.05 equiv) | 4 Å MSs | −16 | 7 |
| 2 | TBAF (1 equiv) | 4 Å MSs | −16 | 67 |
| 3 | TBAF (1 equiv) | 4 Å MSs | −78 | 66 |
| 4 | TBAF/AcOH: 1:1 (1 equiv) | −16 | –c | |
| 5 | TBAF/AcOH: 1:1 (1 equiv) | rt | 65 | |
| 6 | TBAF/AcOH: 1:1 (1 equiv) | 4 Å MSs | rt | 62 |
| 7 | TBAF/AcOH: 1:2 (1 equiv) | rt | –c | |
| 8 | TBAF (1 equiv) | −16 | 95 | |
| 9 | TBAF·3H2O (1 equiv) | −16 | 42d | |
| 10 | TBAT (1 equiv)e | −16 | 53 | |
| 11 | KF·18-crown-6 ether (1 equiv)e | −16 | 58 | |
| 12 | CsF (5 equiv)/n-Bu4NCl (0.1 equiv)f | −16 | 67 |
a Activated powdered 4 Å MSs (250 mg) were added per 0.5 mmol of substrate.
b 1 M/THF, commercial source.
c No conversion.
d Conversion estimated from 1H NMR of the crude product.
e For solubility reason, THF was chosen as the solvent.
f Reaction time: 24 h.

Formation of bis-aldols 8a and 8b in the course of the aldolisation process. a4a/8a = 9:1 from the 1H NMR spectrum of the crude product. b4b/8b = 7:3 from the 1H NMR spectrum of the crude product.
The use of an equimolar amount of TBAF (Table 1, entry 2) led to the formation of aldol 4a and bis-aldol 8a in a 9:1 ratio, from the 1H NMR spectrum of the crude product (Scheme 5). Pure aldol 4a was isolated in 67% yield after column chromatography. Bis-aldol 8a, slightly more polar than aldol 4a, could not be isolated and was recovered as a mixture with aldol 4a. When benzaldehyde was replaced by p-chlorobenzaldehyde, a NMR-based 70:30 ratio of the mixture aldol 4b/bis-aldol 8b was obtained (Scheme 5). Aldol 4b was isolated in 50% yield, and the corresponding bis-aldol 8b could be isolated as a single diastereoisomer in 20% yield [12] and fully characterised. Bis-aldols 8 are expected to result from methyl-side aldolisation, induced by the basicity of the medium. No improvement of the yield in aldol 4a was achieved when the reaction was conducted at −78 °C (entry 3), although bis-aldol 8a was no longer detected. To prevent the methyl-side aldolisation to occur, basicity of the TBAF was buffered by the use of a TBAF/AcOH system as a ratio of 1:1 mixture [13] (Table 1, entries 4–7). Although the reaction did not proceed at −16 °C (entry 4), increasing the temperature to room temperature resulted in the convenient formation of the expected aldol 4a in 65% yield (entry 5), which was not improved by the addition of MSs (entry 6). Traces of the bis-aldol were still recovered after chromatography in these two assays conducted at room temperature (entries 5 and 6). When a TBAF/AcOH ratio was changed from 1:1 to 1:2, reactivity was no longer observed (entry 7). At that time, we thought about the influence of the MSs on the reaction. Its main role is to trap some of the water present in various amounts in the commercial tetrahydrofuran (THF) solutions of TBAF, thus providing homogeneous quality of the reagent [5]. However, TBAF becomes at the same time more reactive and above all more basic [14]. We can suppose that the rate of desilylation of TIPS-diazoacetone by nucleophilic attack from TBAF is low, thus allowing degradation and side-reaction to proceed in the basic medium generated, like retro-aldolisation process and methyl-side aldolisation. To support our hypothesis, the reaction was conducted in the presence of an equimolar amount of TBAF without the addition of MSs (entry 8). Pleasingly, a drastic increase in the yield was achieved, from 67% (entry 2) to 95%, confirming that the addition of MSs was detrimental to the reaction. On the contrary, when the reaction was carried out using solid TBAF·3H2O instead of the commercial THF solution of TBAF, a moderate conversion of 42% was obtained (entry 9). The less basic and non-hygroscopic tetrabutylammonium triphenyldifluorosilicate [15] was also tested but led to the expected aldol 4a with a disappointing 53% yield (entry 10). The use of KF·18-crown-6 ether complex as an alternative fluoride source, which had proven suitable for TES-diazoacetone [5], resulted in a moderate 58% yield (entry 11). Finally, a solid–liquid phase-transfer system was assessed, consisting of anhydrous CsF (5 equiv)/n-Bu4NCl (0.1 equiv) in Et2O, at −16 °C (entry 12). The expected aldol was isolated in 67% yield after an extended reaction time of 24 h.
With optimal conditions in hand (entry 8, Table 1), we investigated the scope of the TBAF-induced aldol-type reaction of TIPS-diazoacetone (Scheme 6). The results were systematically compared with those obtained from the TBAF-triggered aldol-type reaction of TES-diazoacetone, recently studied by our team [5]. Benzaldehyde and electron-poor p-Cl, o-Cl and p-CF3 benzaldehyde led to excellent yields of aldols (4a–c, 4e), very similar to those obtained from TES-diazoacetone. We were pleased to isolate diazoaldol 4d in a high 88% yield from m-Cl-benzaldehyde. Indeed, this original diazoaldol had not been isolated with the protocols previously developed from TES-diazoacetone [5]. The less reactive, electron-rich anisaldehyde provided aldol 4f with a moderate 58% yield, substantially lower than previously obtained from TES-substrate. Although some degradation was obvious, no side product could be identified. Moreover, mixing together TBAF and anisaldehyde under the reaction conditions (Et2O, −16 °C, and 2 h) resulted in clean recovery of the aldehyde. In addition, we were delighted to observe that sensitive heteroaromatic aldehydes like furfural and 2-thiophenecarboxaldehyde behaved as excellent substrates. Such access to diazoaldols 4g (90%) and 4h (80%) is more rapid, and thus more efficient, than the two-step procedure previously set up from TES-diazoacetone (Scheme 6, note c) [5]. Hindered and/or enolizable alkyl aldehydes behaved equally well, providing the expected diazoaldols (4i–k) in 91–95% yields. With sensitive and less reactive α,β-unsaturated aldehydes like octynal (aldol 4l, 39%) and cinnamaldehyde (aldol 4m, 26%), low yields of the expected aldols were obtained [16].
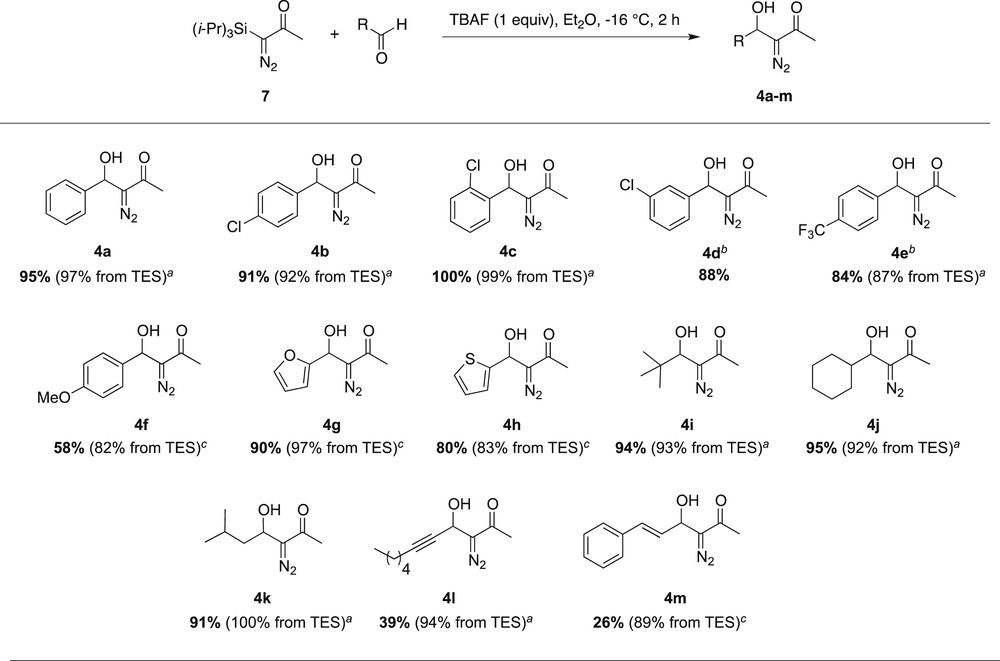
Scope of the TBAF-induced aldol-type addition of TIPS-diazoacetone. aYield resulting from TBAF-induced aldol-type addition to TES-diazoacetone according to protocol P1: TBAF 50 mol %, Et2O, 4 Å MSs, −16 °C, 2 h. See Ref. [5]. bTraces of the corresponding bis-aldol 8 were detected as a mixture with aldol 4 in a minor discarded fraction of chromatography. cYield resulting from the two-step TBAF-induced aldol-type addition to TES-diazoacetone according to protocol P2: (1) TBAF 5 mol %, Et2O, 4 Å MSs, −16 °C, 2 h; (2) Et3N·3HF (2 equiv), THF, rt, 16 h. See Ref. [5].
3 Conclusions
This study highlighted that TIPS-diazoacetone constitutes a stable, storable and thus convenient surrogate for TES-diazoacetone in the fluoride-induced Mukaiyama-type aldol addition towards alkyl and aryl aldehydes. TBAF proved to be a particularly suitable fluoride source, required in stoichiometric amount, whereas MSs played a detrimental role in the reaction. This aspect of the reactivity of TIPS-diazoacetone, combined with the robustness of the TIPS protecting group, makes this building block valuable for further study towards the convergent elaboration of highly functionalised α-diazocarbonyl chains. This investigation is underway in our laboratory.
4 Experimental section
Caution: Although we never had any trouble with the handling of the diazo compounds described in this study, diazo compounds are potentially explosive and should be handled with care in a well-ventilated fume hood.
4.1 General information
All reactions were performed under an argon atmosphere. Et2O and THF were dried through activated alumina columns. DIPEA and CH3CN were distilled over CaH2. Commercial aldehydes were distilled or recrystallised before use. TBAF (1 M/THF, 50 mL) was used as received. MSs (4 Å, powder) were activated by hot gun heating under vacuum. Reactions at −16 °C were performed using an ice/NaCl bath or using a bath cooled by cryogenic flow. Melting points were measured with a Büchi B-540 apparatus and are uncorrected. Column chromatography was performed using 60 μm silica gel. Thin-layer chromatography was performed with silica gel 60F254 precoated thin-layer chromatography (TLC) sheets, and products were detected by UV light or vanillin ethanolic solution. 1H NMR (200 or 400 MHz) and 13C NMR (100.6 MHz) spectra were recorded in CDCl3. Chemical shifts are reported as parts per million (ppm) relative to Me4Si and coupling constants are expressed in hertz. The splitting patterns are designated as follows: s, singlet; br s, broad singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Proton and carbon assignments were established using correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC) and distortionless enhancement by polarization transfer with retention of quaternaries (DEPT-Q) experiments. IR spectra were recorded on a Fourier transform infrared (FTIR) spectrometer equipped with an attenuated total reflectance (ATR) unit. The wavenumbers of representative absorption peaks were given in cm−1. High-resolution mass spectra were recorded on an electrospray ionization-quadrupole-time-of-flight (ESI-QTOF) apparatus.
4.2 General procedure for the fluoride-induced aldol-type addition of TIPS-diazoacetone
To a stirred solution of TIPS-diazoacetone (0.42 mmol, 1 equiv) and aldehyde (0.46 mmol, 1.1 equiv) in anhydrous Et2O (4 mL) at −16 °C was slowly added TBAF (1 M/THF, 0.42 mmol, 1 equiv). After 120 min stirring at −16 °C under Ar, the reaction mixture was quenched by the addition of a saturated aqueous NH4Cl solution (8 mL). The aqueous layer was extracted with diethyl ether (3 × 10 mL) and the combined organic layer was dried over Na2SO4, filtered, and the filtrate was concentrated under reduced pressure. The oily residue was purified by column chromatography (silica gel, cyclohexane/ethyl acetate = 95:5 to 60:40) to isolate the expected aldol 4.
4.2.1 4-(3-Chlorophenyl)-3-diazo-4-hydroxybutan-2-one (4d)
Prepared from TIPS-diazoacetone and m-chlorobenzaldehyde according to the general procedure. Aldol (4d) was obtained after column chromatography (cyclohexane/ethyl acetate = 95:5 to 60:40) as a yellow solid (83 mg, 88% yield): mp = 98 °C; Rf = 0.12 (petroleum ether/ethyl acetate = 80:20); IR (film) νmax (cm−1) 3363 (νOH), 2079 (νCN2), 1613 (νCO), 1336, 1025, 766, 736; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.43 (s, 1H), 7.27–7.34 (m, 3H), 5.98 (br s, 1H), 3.72 (br s, 1H, OH), 2.27 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 191.0, 140.9, 134.8, 130.1, 128.6, 126.0, 123.9, 73.9, 67.3 and 25.8; HRMS m/z calcd for C10H9ClN2NaO2 [M + Na]+ 247.0245; found, 247.0238.
The spectroscopic data for aldols (4a), (4b), (4c), (4e), (4f), (4g), (4h), (4i), (4j), (4k), (4l) and (4m) proved consistent with the literature [5].
4.3 Formation of the bis-aldols 8
4.3.1 1,5-Bis(4-chlorophenyl)-2-diazo-1,5-dihydroxypentan-3-one (8b)
To a stirred solution of TIPS-diazoacetone (100 mg, 0.42 mmol, 1 equiv), p-chlorobenzaldehyde (65 mg, 0.46 mmol, 1.1 equiv) and 4 Å MSs (210 mg, 500 mg/mmol) in anhydrous Et2O (4 mL) at −16 °C was slowly added TBAF (1 M/THF, 0.42 mL, 0.42 mmol, 1 equiv). After 120 min stirring at −16 °C under Ar, the reaction mixture was quenched by the addition of a saturated aqueous NH4Cl solution (8 mL). The aqueous layer was extracted with diethyl ether (3 × 10 mL) and the combined organic layer was dried over Na2SO4, filtered, and the filtrate was concentrated under reduced pressure. The oily residue, containing a 70:30 mixture of aldol 4b/bis-aldol 8b, evaluated from its 1H NMR spectrum, was purified by column chromatography (silica gel, cyclohexane/ethyl acetate = 95:5 to 60:40). Aldol 4b was isolated as a yellow solid (47 mg, 50% yield) [5], and bis-aldol 8b was isolated as an orange oil (16 mg, 20% yield based on the limiting reactant p-Cl-benzaldehyde): Rf = 0.12 (petroleum ether/ethyl acetate = 80:20); IR (film) νmax (cm−1) 3379 (νOH), 2924, 2088 (νCN2), 1606 (νCO), 1490, 1090, 1013, 780, 742; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.37–7.23 (m, 8H), 6.01 (s, 1H), 5.25 (dd, 1H, J = 9.0, 3.2 Hz), 2.87 (dd, 1H, J = 15.6, 9.0 Hz), 2.75 (m, 1H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 192.2, 141.2, 137.3, 134.5, 133.9, 129.2, 129.0, 127.3, 127.2, 75.6, 70.3, 67.3, 47.2. HRMS m/z calcd for C17H15Cl2N2O3 [M + H]+ 365.0460; found, 365.0469.
Acknowledgements
We gratefully acknowledge the “Office méditerranéen de la jeunesse” (OMJ) and Erasmus+ Program – “Mobilités internationales de crédits de l'enseignement supérieur” (call for proposals 2015) for I. Abid's Ph.D. funding. We are deeply grateful to Ligue contre le cancer”the “ for financial support (CSIRGO 2013). We thank Amélie Durand and Corentin Jacquemmoz for the NMR analyses and Patricia Gangnery and Emmanuelle Mebold for the HRMS analyses.