

1 Introduction
Syngas production from natural gas is an attractive process in the chemical industry because the carbon monoxide–hydrogen mixture produced can be converted in liquid fuels or valuable chemicals such as Fischer–Tropsch oil, methanol, or dimethyl ether [1]. Moreover, hydrogen has received considerable attention in the recent years considering the development of fuel cell technology [2]. Hydrogen is practically produced from natural gas and it is for the moment mainly used for the ammonia synthesis [3] and oil refining [4]. Nowadays, there are three common methods for producing syngas from methane: (1) the steam methane reforming (SMR) [5], (2) the dry methane reforming (DMR) [6], and (3) the partial oxidation of methane (POM) [7]. The POM possesses several advantages compared with the other two routes. In fact, POM reaction is a mild exothermic reaction (ΔrH°25 °C = −36 kJ mol−1), whereas SMR and DMR exhibit positive and much higher absolute values (205 and 247 kJ mol−1, respectively) indicating that the industrial process based on POM is energy saving [8] without critical thermal management problems inside the reactor. The POM process can be carried out thermally or with the use of a catalyst. Thermal process requires temperature greater than 1200 °C and leads to the formation of soot explaining why catalytic processes are more common and have the advantage to require a low amount of the catalyst meaning high gas hourly space velocity (GHSV) [9]. All these features have a consequence that POM process requires less sophisticated reactors with a low amount of the catalyst resulting in a lower investment compared with the SMR or DMR process. The H2/CO ratio obtained from POM is generally close to 2, which is of great interest for downstream processes, such as Fischer–Tropsch reaction, methanol, or dimethylether synthesis [10], whereas SMR and DMR processes lead to a ratio close to 3 and 1, respectively. All these considerations make the POM process very attractive and are expected to spread in the future [11].
The catalysts tested in CH4 partial oxidation can be classified into two types. The first one consists in supported noble metals, most commonly Rh, Ru, Pd, Pt, and Ir [7,12,13], and the second type corresponds to supported nonprecious metal catalysts. Because of their considerable activity, good stability, and lower cost, Fe, Co, and especially Ni catalysts were widely studied [7,14–17]. Among this series of metals, Ni exhibits the best catalytic activity, which is close to the one of Pt. To avoid deactivation, nickel-based catalysts are generally designed to exhibit low nickel sintering even at a reaction temperature close to 750 °C and low coke formation. Solid solution of NiO and MgO is well known to allow high nickel dispersion [18] resulting in the study of different materials containing Ni and Mg [19]. It is known that a good dispersion of the nickel is believed to reduce the coke formation according to the literature [20] but the addition of promoters such as Co [14], B [21], Cu [22], or Cr [23] is also used in this purpose. As the catalytic POM occurs at a high rate, it can be operated at high GHSV. To design a POM process with an optimal amount of the nickel-based catalyst, kinetic data relative to the activity of the catalyst are required. Many studies have been carried out to provide such data [17,24–26]. However, only a few of them are based on a detailed reaction mechanism [17,24], which is still under debate and controversy discussions.
In this article, we report a kinetic study of NiMgAl–based catalysts type Ni0.1Mg0.9Al2O4 and 10 wt % Ni/MgAl2O4 prepared by the coprecipitation (CP) and sol–gel (SG) methods in the POM reaction. All catalysts were characterized by several physicochemical methods of analysis. The study of the catalytic reactivity of these catalysts in the POM reaction was carried out as a function of the reaction time and temperature to investigate the role of the incorporation of nickel in the MgAl2O4 structure or its impregnation on the MgAl2O4 support; otherwise a comparative study was carried out between the physicochemical and catalytic properties of these two ways of preparation of NiMgAl catalysts. A kinetic study performed on the most efficient catalyst is also detailed in this work by varying the reaction temperature, the catalyst mass, and the total flow rate. The methodology of this kinetic study may be applied to other catalytic systems for POM.
2 Experimental section
2.1 Catalyst preparation
MgAl2O4 and Ni0.1Mg0.9Al2O4 samples were prepared using CP and SG methods. The catalyst 10 wt % Ni/MgAl2O4 was prepared by nickel wet impregnation of each support of CP and SG MgAl2O4.
2.1.1 Coprecipitation method
An aqueous solution of nitrates was prepared with an Al/M molar ratio of 2 (M = Ni + Mg) [27]. The precursor was prepared by adding the ammonia solution to the previous mixed nitrates solution at 40 °C under stirring, until reaching a pH value close to 10. The suspension was aged for 1 h at room temperature and then filtrated. The obtained precipitate was dried for 24 h at 80 °C.
The MgAl2O4 and Ni0.1Mg0.9Al2O4 prepared by CP are designated as CP-MA and CP-NMA, respectively.
2.1.2 Sol–gel method
M(NO3)2·6H2O (with M = Mg or Ni), Al(NO3)3·9H2O, citric acid, and ammonia solution (25 wt %) were used as starting materials in the synthesis of precursors. Nitrate salts were dissolved in 100 ml of distilled water. Then citric acid was added to the solution with a molar ratio of citrate to metallic ions equal to 0.5 [28]. The solution was agitated using a magnetic stirrer at room temperature for 1 h. The pH of the solution was adjusted to 6.0 by the addition of ammonia [29,30]. This solution was gelled by heating at 120 °C. Thus, the gel was heated at 180 °C in an electric oven.
MgAl2O4 and Ni0.1Mg0.9Al2O4 solids synthesized by the SG method are designated as SG-MA and SG-NMA, respectively.
2.1.3 Impregnation method
Samples of 10 wt % Ni/MgAl2O4 were prepared by impregnation of both CP-MA and SG-MA in an aqueous solution of nickel nitrates. The catalysts were dried overnight at 80 °C. The obtained catalysts are designated as 10Ni/CP-MA and 10Ni/SG-MA.
For all solids, the calcination was performed at 700 °C under air for 4 h with a heating rate of 5 °C min−1.
2.2 Catalyst characterization
X-ray diffraction (XRD) patterns of a fresh and used powdered sample were recorded using a Brucker AXS-D8 diffractometer with a Cu Kα irradiation source (λ = 1.5406 Å). The scan range was 10°–80° with a 0.020° step and an acquisition time of 0.80 s at each step. The diffraction patterns were identified by comparing with those in the Joint Committee on Powder Diffraction Standards (JCPDS) database. The crystallite sizes of metal Ni were estimated using the full widths at half maximum (FWHMs) of the Ni (200) peaks at 2θ = 51.80°, through the Scherrer equation: d = 0.89λ/(β cos θ), where λ is the wavelength of the radiation, β is FWHM (in radian), and θ is the half of the diffraction angle (25.90°). Average crystallite sizes of spinel oxides were estimated through the Scherrer equation assuming a same diffraction angle for both oxides (2θ = 44.99° for NiAl2O4 International Centre for Diffraction Data – Powder Diffraction File (ICDD-PDF) 10-0339 and 44.83° for MgAl2O4− ICDD-PDF 21-1152). The FWHMs measured were always much higher than the difference between these two theoretical diffraction angles, leading to an only small overestimation of the spinel crystallite sizes.
The thermogravimetric analysis (TGA) was recorded on a Q5000 apparatus. Analysis was carried out from room temperature to 900 °C with a heating rate of 10 °C min−1 under an airflow of 25 mlSATP min−1.
Temperature-programmed reduction (TPR) analysis was performed to determine the reducibility of the catalyst and the corresponding temperature. The total H2 consumption was calculated by the integration of the TPR curve, thanks to a calibration. TPR measurements were carried out using Micromeritics Auto ChemII 2920 equipment; a mass of 150 mg of the fresh calcined catalyst was heated under a flow of 10% H2 in argon (50 mlSATP min−1) from room temperature to 900 °C at a heating rate of 15 °C min−1.
N2 physisorption was conducted at −196 °C on a Micromeritics ASAP 2420 instrument. The sample was pretreated at 200 °C for 12 h in vacuum before N2 adsorption.
2.3 Catalytic tests
Three types of catalytic tests of POM were carried out at atmospheric pressure in a quartz tube with an inner diameter of 7 mm.
First series of tests (called “test-A”) were performed on the four samples with a mass of the catalyst of 200 mg. Before reaction, the catalyst was pretreated from room temperature to 500 °C under N2 with a 10 mlSATP min−1 flow rate (unless otherwise stated). The gas mixture CH4/O2/N2 = 1.9:1.0:4.0 was introduced into the reactor at the reaction temperature with a total flow of 50 mlSATP min−1, corresponding to a GHSV around 15,000 h−1. The catalytic activity was recorded during 90 min in isothermal conditions every 100 °C from 500 to 800 °C.
The second series of tests (called “test-B”) consisted in long-term stability of catalysts. Before reaction, 200 mg of the catalyst (unless otherwise stated) was pretreated from room temperature to 800 °C under N2 with a 10 mlSATP min−1 flow rate. Total inlet flow and its composition were the same than the ones used for test-A. The catalytic activity has been measured during 64 h in these conditions.
Last series of tests (called “test-C”) were performed with the aim to acquire data for kinetic modeling. Before reaction, 20–67 mg of the catalyst was pretreated from room temperature to 800 °C under N2 with a 10 mlSATP min−1 flow rate. Inlet CH4/O2/N2 gas mixture was the same than the one used for test-A with a total flow rate of 33 or 50 mlSATP min−1. The catalytic activity was recorded during 60 min in isothermal conditions every 50 °C from 800 to 700 °C.
Water was trapped at the outlet of the reactor and the dried gas composition was analyzed by a gas chromatography line Thermal Conductivity Detector (Agilent 6890N) equipped with a carbosphere column with Ar as carrier gas. N2 was used as an internal standard to calculate outlet partial flow rates as it is inert during catalytic tests. In addition to compounds present in the feed, CO, CO2, and H2 were detected, whereas no C2H6 or other hydrocarbons except methane were detected.
Methane conversion, selectivity, and yields were calculated according to the equations given below:
3 Results and discussion
The XRD patterns of fresh and spent (after H2-TPR and after test-B) samples are presented in Fig. 1. The fresh CP-MA and SG-MA solids showed the formation of a pure spinel phase MgAl2O4. For the bulk NMA catalyst containing nickel, the absence of the free nickel oxide NiO indicates the formation of a NiAl2O4 phase whose peaks may be overlapped by those of a MgAl2O4 phase. In fact, the spinel structure consists of A2+ and B3+ cations where they can easily displace each other. Therefore, Mg2+ ions in MgAl2O4 are partially substituted by Ni2+ on heat treatment in oxidizing atmosphere air and then creates a spinel phase NiAl2O4, which is in good agreement with the literature [31,32]. As for the bulk catalysts, the impregnated ones 10Ni/CP-MA and 10Ni/SG-MA revealed only the formation of spinel phases of MgAl2O4 and NiAl2O4, free NiO is not observed, which confirms the formation of a NiAl2O4 spinel structure.
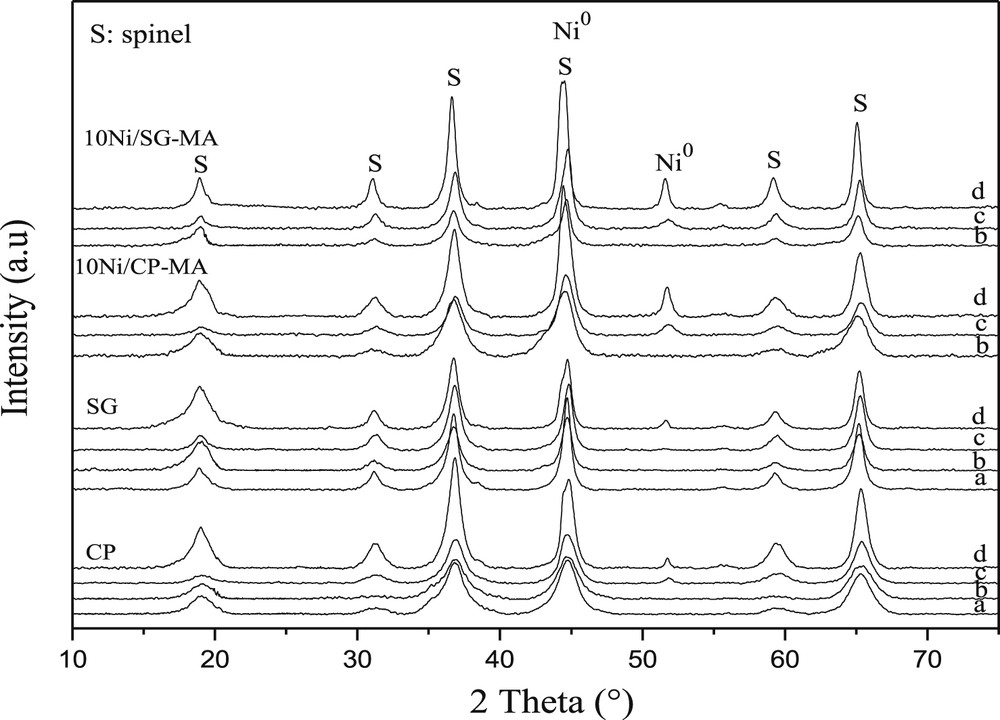
XRD patterns of the catalysts prepared by different methods and calcined at 700 °C: (a) MA fresh, (b) NMA fresh, (c) after H2-TPR, and (d) after test-B.
To study the evolution of the size of the crystalline domains, the average crystallite size of the fresh, the reduced, and the used (after test-B) samples were calculated using the Scherrer equation (Table 1). For the fresh CP-MA and SG-MA samples, an average spinel crystallite size of about 14 nm was found, no influence of the preparation method on a spinel crystallite size was observed. For the NMA catalysts containing nickel, as can be seen, they have an average spinel crystallite size smaller (5–11 nm) with respect to that of MA samples, the smallest crystallite size being observed for CP-NMA. This difference can be attributed partially to the interactions between different species in NMA because of the presence of nickel. These interactions can limit particle agglomeration. After reduction under hydrogen (TPR analysis) and catalytic test-B (during 64 h), the patterns of all NMA and 10Ni/MA solids prepared by CP and SG methods revealed, besides the spinel structure as the main phases, the presence of metallic nickel Ni0 characterized by one of the peak clearly observed at around 2θ=51.8° and well isolated from those corresponding to the spinel structures. The crystallite size of the metallic Ni was calculated using this peak. For the reduced samples, we observe that the impregnated solids (10Ni/MA) compared to the bulk NMA samples (with nickel incorporated directly in the spinel structure NiAl2O4) lead to lower Ni0 crystallite sizes. For all the used samples (after test-B), a noticeable increase in the Ni0 crystallite size is observed, which could be because of the sintering of nickel particles during the catalytic reaction.
Structural, textural, and redox properties of catalysts.
| Catalysts | Crystallite size (nm) | Specific surface area (m2/g) | Temperature-programmed reduction | |||||
| Spinel(s) in fresh samples | Ni0 after reduction | Ni0 after catalytic test-B | Fresh catalysts | After test-B | H2 consumption (mmol gcat−1) | Tmax (°C) | Degree of reduction of nickel (%) | |
| CP-MA | 14 | – | – | 150 | – | – | – | – |
| SG-MA | 14 | – | – | 136 | – | – | – | – |
| CP-NMA | 5 | 15 | 29 | 132 | 55 | 0.22 | 853 | 32.2 |
| SG-NMA | 11 | 14 | 19 | 81 | 67 | 0.65 | 853 | 95.4 |
| 10Ni/CP-MA | 6 | 9 | 15 | 89 | 53 | 1.62 | 842 | 95.4 |
| 10Ni/SG-MA | 12 | 9 | 18 | 93 | 32 | 0.02 1.61 | 556 820 | 1.1 94.3 |
Specific surface areas of the fresh and used catalysts are listed in Table 1. Similar surface areas were found for both CP-MA and SG-MA fresh samples; these decreased after impregnation by the nickel. The loss of surface area after introducing nickel could be related to pores filling and/or blocking by nickel particles but may also be related to the presence of NiAl2O4 after introducing nickel. After the catalytic test-B, we note a loss of the specific surface area for all the used samples. According to some authors [33,34], this result can be assigned to the sintering of crystallites and migration of nickel out of the spinel structure. The migration from microcrystallites to large aggregates in the catalyst is attributed to the influence of the reaction conditions (time and temperature). These assumptions are confirmed by the XRD analysis results, which showed for samples after catalytic test-B (Fig. 1d) thin diffraction peaks associated with Ni° that correspond to large particles of metallic nickel after the POM reaction.
The H2-TPR profiles of catalysts containing nickel NMA and 10Ni/MA are shown in Fig. 2. The MA samples (without nickel) were found to be irreducible in the studied temperature range as reported by Djaidja et al. [35]. The Ni-based solids were gradually reduced from 550 to 850 °C with two distinct peaks clearly observed for 10Ni/MA supported catalysts. The TPR profiles of the NMA samples show that the reduction of the fresh samples occurred in the temperature range between 550 and 850 °C. Nickel in the supported catalysts (10Ni/CP-MA and 10Ni/SG-MA) and in the bulk catalyst prepared by the SG method (SG-NMA) is close to be totally reduced after TPR until 800 °C, whereas only a degree of reduction of nickel of 32% was recorded for a sample prepared by the CP method (CP-NMA) (Table 1). This may indicate a greater stability of the Ni2 + atoms in the spinel structure for this sample. The hydrogen consumption peaks observed at low temperature correspond to reduction of Ni2+ in the spinel structure NiAl2O4 in the octahedral sites, whereas the peaks at high temperature are attributed to the reduction of Ni2+ ions (Ni2+ → Ni0) in the tetrahedral sites. In fact, in this spinel structure Ni2+ exists in two forms, octahedral and tetrahedral, which indicate the presence of a partially inverse NiAl2O4 spinel. The tetrahedral sites are reported to be more difficult to reduce than the octahedral ones [36,37].

H2-TPR profiles of catalysts calcined at 700 °C.
POM catalytic tests were performed by increasing the reactor temperature without H2-pretreatment of the catalysts. The results are shown in Table 2. For all catalysts, the methane conversion increases with increase in the reaction temperature. The 10Ni/NMA supported catalysts prepared by CP and SG methods exhibited the highest methane conversions at 700 °C, which is close to thermodynamic equilibrium beyond this temperature. The higher activity of supported catalysts was in accordance with their higher reducibility observed during TPR analysis, the lower Ni0 particle size, and the higher nickel content compared with the bulk catalysts. Similar activity to that of supported catalysts was reached over the SG-NMA bulk catalyst to higher temperature (toward 800 °C). Although having the same composition of SG-NMA over CP-NMA, the methane conversion remained low (less than 44%), irrespective of the reaction temperature. This catalyst, despite its higher surface area, presents the lower POM activity, which could be explained by a small proportion of the Ni0 metallic phase as observed in H2-TPR analysis.
CH4 conversion, H2 and CO selectivities, and H2/CO ratios at the outlet in the POM tests over bulk and supported catalysts as a function of reaction temperature.
| Catalysts | Treaction (°C) | SCO | H2/CO | ||
| CP-NMA | 600 | 33.6 | 18.0 | 19.0 | 1.8 |
| 700 | 36.0 | 20.0 | 18.2 | 2.2 | |
| 800 | 44.0 | 38.1 | 39.9 | 2.0 | |
| SG-NMA | 600 | 34.3 | 23.1 | 18.5 | 2.5 |
| 700 | 38.0 | 23.0 | 17.8 | 2.6 | |
| 800 | 95.0 | 94.5 | 85.7 | 2.2 | |
| 10Ni/CP-MA | 600 | 34.6 | 19.0 | 14.7 | 2.6 |
| 700 | 82.1 | 90.0 | 75.8 | 2.4 | |
| 800 | 95.0 | 95.2 | 85.6 | 2.2 | |
| 10Ni/SG-MA | 600 | 33.4 | 12.2 | 2.4 | 2.6 |
| 700 | 81.0 | 91.2 | 77.3 | 2.5 | |
| 800 | 94.3 | 96.0 | 86.0 | 2.2 |
The higher selectivity to H2 (95.2% and 96.0%) and CO (85.6% and 86.0%) was obtained at 800 °C over the 10Ni/CP-NMA and 10Ni/SG-NMA supported catalysts, respectively. The values of the H2/CO ratio vary as a function of the reaction temperature (Table 2), the values close to 2.2 were observed at 800 °C for all the samples except for the bulk CP-NMA sample for which the H2/CO ratio was about 2.0. The activity of the catalysts after catalytic test-B is shown in Fig. 3. All catalysts showed a stable methane conversion estimated at about 95% except for the catalyst CP-NMA, which exhibited poor catalytic performance because of the low nickel reduction and the formation of large nickel metal particles (Table 1). It is worth noting that the different activities observed in Table 2 and Fig. 3 are because of the use of a time much longer under flow of a reactant mixture at 800 °C, which enabled a better reducibility of Ni2+ ions into metallic Ni0.
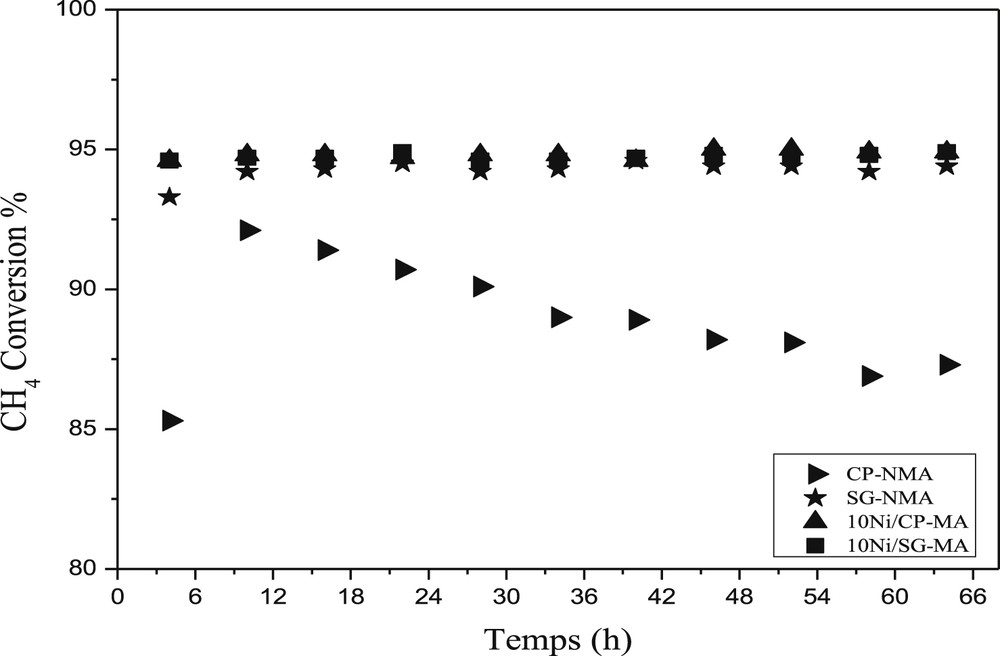
Long-term activity for the different catalysts at 800 °C.
The spent catalysts (after test-B) were characterized by TGA and XRD analysis as can be seen in Fig. 4. All the used catalysts present similar TGA curves with three thermal events. The first weight loss occurring around 150 °C is attributed to the desorption of physisorbed water molecules. The second thermal event is observed between 250 and 450 °C and is assigned to the oxidation of metallic Ni0 particle surface to Ni2+ to form NiO. This oxidation step is confirmed by the XRD analysis effectuated after the test-B (Fig. 1) and after the TGA (Fig. 4). In fact, the XRD profiles of the used samples after TGA revealed, in addition to the presence of a spinel phase, the formation of peaks attributed to NiO (PDF-ICDD 47-1049). Finally, the last thermal effect observed at high temperature (between 650 and 900 °C) is assigned to the oxidation of bulk nickel Ni0 species. Furthermore, on the basis of the nickel content and the increase in mass during TGA experiments, it was estimated that the amount of coke present in this sample was so small (a maximum of 1 wt % for the bulk samples and undetectable for the impregnated ones) that its oxidation process was hidden by the oxidation of metallic Ni0 particles in the bulk. This suggestion is in agreement with the absence of a clearly visible diffraction peak assigned to carbon (Fig. 1). This phenomenon was also obtained by López-Fonseca et al [7] in the study of POM (for more than 35 h) over NiAl2O4 catalysts.

TGA profiles of spent catalysts (a) and XRD of catalysts after TGA (b).
3.1 Kinetic modeling
To better understand the mechanism of the POM reaction, we have selected the 10Ni/SG-MA sample to study the kinetics of the reaction by varying several parameters such as reaction temperature, the solid mass, and the total flow.
According to a survey of the literature [16,17,38–40] elementary steps involved during POM in the presence of a nickel-based catalyst can be listed as following:
Dissociative adsorption of oxygen:
| (1) |
Dissociative adsorption of hydrogen:
| (2) |
Dissociative adsorption and decomposition of methane:
| (3) |
| (4) |
| (5) |
| (6) |
Formation of water:
| (7) |
| (8) |
Formation and desorption of CO:
| (9) |
| (10) |
| (11) |
Formation of CO2:
| (12) |
| (13) |
POM reaction is favored by thermodynamic in the considered range of temperature and at atmospheric pressure with equilibrium constant over 1011. As a consequence, only the forward reaction is taken into account. The CO formation by POM is reported to be limited by a reaction (9) on nickel catalysts [17], which enables to write the expression of the POM rate rPOM as follows:
Assuming reactions (1)–(6) and (8), (11), and (12) to be close to thermodynamic equilibrium in steady conditions and that surface species mostly consist of adsorbed O and C [17], one can express rPOM as a function of k9, of thermodynamic equilibrium constants of elementary reactions (Ki), and of the surface concentration of nickel active sites L (assumed to be constant over the range of temperature studied).
CO2 is supposed to be formed by CO oxidation, and its rate of formation was considered to be limited by a reaction (12) because of high NiO bonding strength [17,41], which allows to establish the following expression for the total oxidation of methane (TOM) rTOM (reverse reaction is not considered as the corresponding thermodynamic equilibrium constant is over 109 in the considered range of temperature):
The produced hydrogen can react with oxygen from the feed to form water (oxidation of hydrogen noted “+OH”) by steps (7) and (8), with the former considered as rate determining. The rate r+OH can thus be expressed as
Using the same methodology as before, global reaction rate for the WGS reaction, noted rWGS, can be expressed as follows (with K8 and K11 included in the apparent kinetic constant kWGS):
The influence of thermodynamic is taken into account with the rate of the reverse SMR reaction with rate r−SMR, which can be expressed as
Using the same methodology as before, global reaction rate for the SMR reaction, noted rSMR, can be expressed as follows (with K8 included in the apparent kinetic constant kSMR):
Kinetic modeling was performed using experimental data from catalytic test-C at three different temperatures and for each temperature at six or seven different GHSVs (19 experiments in total in steady state). External heat and mass transfer limitations were excluded as two experiments with the same GHSV but with a different amount of the catalyst led to the same results. Catalyst grains were diluted by a mass factor close to 10 in inert SiC to keep a similar catalytic bed for all experiments and to favor heat transfer phenomena. As a consequence, temperature was assumed to be constant along the catalytic bed even if this assumption is less justifiable in the first part of the reactor as discussed subsequently. Only mass balances in the steady state were taken into account. GHSV is expressed as the ratio of a volume flow rate in standard ambient temperature and pressure (SATP) conditions and apparent volume of a catalyst (without SiC).
Catalytic reactor was assumed to exhibit a plug flow behavior as criteria of the literature were respected [43,44]. Catalytic bed was decomposed in slices of 1.0 mg of the catalyst each with an equivalent total flow rate of 50 mlSATP min−1 at the inlet of the reactor. Each slice was considered as a continuous stirred tank reactor (CSTR) in a steady state and the following mass balance equations were applied:
Influence of the GHSV on the partial flow rate of each compound at the outlet of the reactor is illustrated in Fig. 5. It can be clearly seen that the presence of oxygen is a key factor for the catalytic activity as already reported by many authors [24,45,46]. In fact, it has been shown that the state of reduction of surface nickel is directly correlated to the presence of oxygen [24]. In other words, when oxygen is present in the gas flow, nickel surface is almost entirely covered by oxygen, which limits the catalytic activity as Ni0 is known to be much more active than NiO [16]. When all the oxygen is consumed by methane oxidation, surface nickel atoms are in reduced state and catalytic activity for syngas formation starts to really increase. As a consequence, the catalytic bed can be decomposed into two parts. A first small one at the inlet in which oxygen is consumed by steps (7), (9), and (12) to form CO2 (not shown but corresponds to the difference between CH4 conversion and CO yield) and water. The second part, in which oxygen is absent, involved metallic nickel surface as the active phase, which enables SMR and RWGS with high rates of a reaction. As a consequence, most of the CO and hydrogen are produced on a thin layer of the bed located just after the part in which oxygen is consumed. The remaining part of the catalytic bed allows to increase hydrogen and CO yield but in small proportion because of the thermodynamic limitation of RWGS and SMR reactions, which are almost reached at 800 °C for a GSHV of 15,000 h−1.
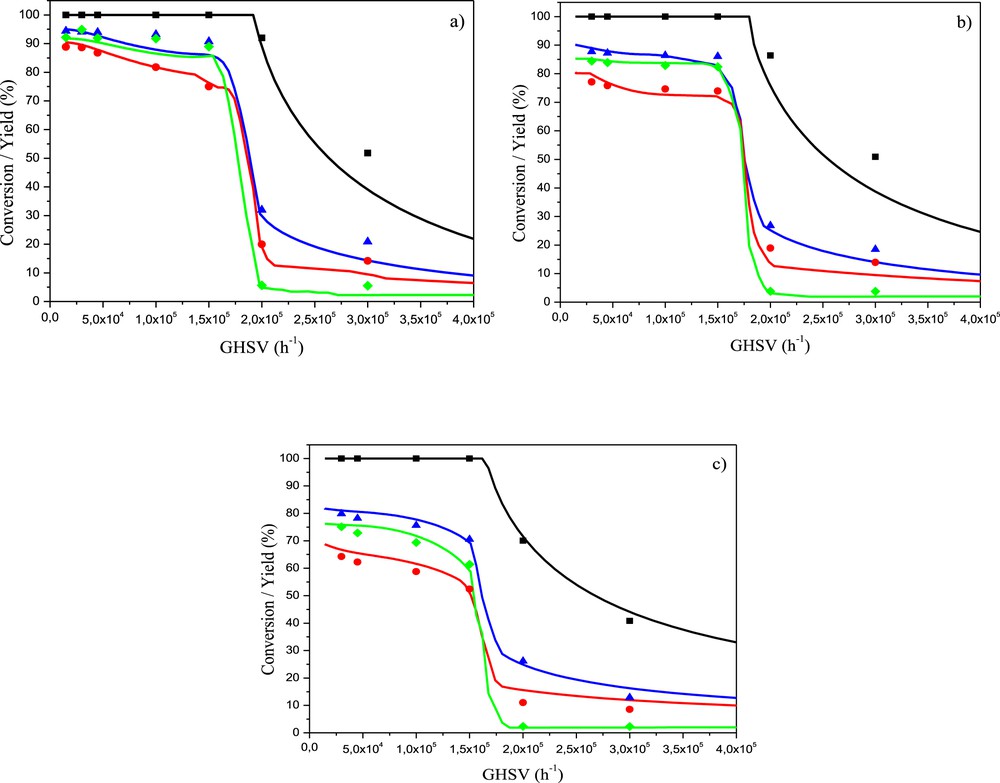
Experimental CH4 (blue triangles) and O2 (black squares) conversions and CO (red circles) and H2 (green diamonds) yields for the NMA-SG-Imp catalyst during catalytic tests C as a function of the GHSV at 800 °C (a) 750 °C (b), and 700 °C (c) and modeled CH4 (blue curves) and O2 (black curves) conversions and CO (red curves) and H2 (green curves) yields at the corresponding temperature.
The five global reactions used to model POM require five kinetic constants and two adsorption constants (for O2 and CH4), which lead to a total of seven parameters at each of the three temperatures investigated. Assuming an Arrhenius dependency of the kinetic constant with temperature and a constant heat of adsorption for the adsorption constants, the modeling was performed simultaneously on the experimental results obtained at the three temperatures with 14 parameters: kinetic and adsorption constants at 800 °C as well as activation energies and heats of adsorption. They were adjusted to minimize the global difference between measured and simulated partial flow rates for all the different GHSVs tested. Values of variable parameters were first roughly estimated by ‘‘curve fitting by hand’’ to reach a consistent range and then optimized by using the Levenberg–Marquardt algorithm. Pre-exponential factor was then calculated for all kinetic and adsorption constants. A quite good agreement was found (Fig. 5) with values reported in Table 3. It has to be mentioned that in the case of oxygen adsorption, no dependency of adsorption constant with temperature in the considered range was found. Differences between experimental and simulated values are the most significant in the first part of the catalytic bed (where oxygen is still present), which may be because of the high exothermicity of the reactions involved in this part (mostly TOM) that may lead to hot spots in the catalytic bed even with the use of SiC as dilutant.
Kinetic parameters obtained for modeling curves presented in Fig. 5.
| Kinetic constants | Adsorption constants | |||||||
| kPOM | kTOM | kOH | kWGS | kSMR | ||||
| Ea (kJ mol−1) | 87.3 | 101.0 | 33.9 | 59.7 | 232.4 | Qads kJ mol−1 | – | 25.6 |
| A unit | 1.5 × 103 mol s−1 g−1 | 8.9 × 107 mol bar−1 s−1 g−1 | 2.7 × 104 mol bar−0.5 s−1 g−1 | 1.7 mol bar−1.5 s−1 g−1 | 1.3 × 1011 mol bar−0.5 s−1 g−1 | A unit | 13.1 bar−0.5 | 3.2 × 10−3 bar |
To be precise, for POM and SMR reactions the activation energy reported in Table 3 is not the apparent activation energy of the reaction because of the presence of the term, which implies to subtract the heat of adsorption of methane leading to apparent activation energies of 61.7 and 206.8 kJ mol−1 for POM and SMR reactions, respectively.
The high activation energy values for SMR compared to other reactions for nickel-based catalysts were also reported by other authors [26] (240.1 and 106.9 kJ mol−1, respectively) who also reported similar values for WGS activation energy (67.1 and 54.5 kJ mol−1, respectively).
Apparent activation energy of the POM reaction estimated in this work was close to values reported in the literature with 65 kJ mol−1 for Theron et al. [47] and 77 kJ mol−1 for Enger et al. [16] in which similar heat of adsorption of methane is mentioned (between 19 and 25 kJ mol−1). This activation energy was found to be close to the one study of TOM by Nguyen et al. [48], which is also the case in this study with a value of 101 kJ mol−1 for TOM activation energy also close to the value of 86 kJ mol−1reported by Trimm and Lam [49].
4 Conclusion
Nickel-based materials with spinel structures MgAl2O4 and NiAl2O4 were prepared using CP, SG, and impregnation methods. The elaborated materials presented small crystallite sizes (5–14 nm) and appreciable specific surface areas. Catalysts with nickel that show a good reduction of the nickel could be attributed to the partial formation of the inverse spinel NiAl2O4 as a portion of Ni2+ cations occupied on the octahedral sites are more reducible because they are less stable. The study of the reactivity in the POM reaction showed that the supported catalysts (obtained via the impregnation method) exhibited better catalytic performances (high activity and selectivity to syngas and the better stability for more than 64 h under the catalytic test), which can be assigned to their high reducibility of Ni2+ to Ni0. The kinetic study of the POM reaction carried out over the 10% Ni/MgAl2O4 (support prepared via the SG method) catalyst showed that the production of syngas (CO + H2) occurred on a thin part of the catalytic bed, in which we have low participation of side reactions such as steam reforming and WGS.