

benzene
C15C18alkanes with 15–18 carbon atoms
DEBdiethylbenzene
EBethylbenzene
Green LPGgreen liquid petroleum gas
HDOhydrodeoxygenation
LHSVliquid hourly space velocity
MeOmethyl oleate
mXmeta-xylene
pXpara-xylene
SEMscanning electron microscopy
Ttoluene
TCDthermal conductivity detector
1 Introduction
The increase in the number of vehicles and stringent regulations regarding the pollutant emissions abatement enforce the finding that alternative fuels and renewables have environmental benefits. Vegetable oils, for example, palm oil, canola, rapeseed oil, and cooking oils, are currently used for producing biodiesel. Resulting from the hydrotreatment of renewable feedstock, green diesel is considered a promising alternative to conventional diesel and stands out for the high cetane number value, the absence of sulfur and aromatic hydrocarbons, low cold filter plugging point, as well as lower emissions of greenhouse gases. During hydrotreatment, the side chains of triglycerides are fragmented and paraffinic hydrocarbons in the diesel range (C14–C18) are obtained (green diesel) [1].
The bio-oil conversion occurs at relatively high temperatures (above 300 °C) under high pressures over Pd-supported on mesoporous carbon, NiMo/CoMo on different supports, or Pd-amorphous SiO2Al2O3. The triglycerides were subjected to a complex reaction process comprising hydrocracking, hydrogenation, hydrodeoxygenation (HDO), and decarboxylation/decarbonylation [2–4].
Furthermore, hydrocracking involves destructive hydrogenation and is based on the conversion of compounds with high molecular weight into products with lower molecules. Isomerization reactions simultaneously occur, resulting in hydrocarbons with boiling points in gasoline or diesel distillation ranges. To diminish the possibility of either polymerization or coke formation reactions to occur, high reaction temperatures and hydrogen pressures are required. To accomplish this process, catalysts with acid function, provided by acid supports (e.g., amorphous aluminosilicates, silico-aluminous phosphates, or crystalline zeolites) are used [5–7].
Hydrotreating or hydrorefining is considered as a nondestructive hydrogenation reaction, enhancing the properties of petroleum fractions, without distillation range modifications. The process occurs at low temperatures and pressures (300–400 °C, 5–11 MPa), and unstable compounds, which may lead to gums or insoluble materials, are transformed into stable compounds [7–9]. Hydrotreating is favored by the metallic function of catalysts. Noble metals or transitional metals (e.g., Ni, Mo, and Co) supported on alumina were widely investigated [7,10,11]. Contaminants such as nitrogen or metals were removed during the process [7].
The oxygen removal from the triglycerides can be performed by HDO, decarboxylation, or decarbonylation. The n-paraffins with long chains are the main reaction products and oxygen is removed either in the form of H2O (via HDO) or H2O and CO (via decarbonylation) and CO2 (via decarboxylation) [12].
The operating conditions (e.g., temperature, pressure, and catalyst type) influence what reaction takes place first during the process, in addition to the yields and compositions of liquid products. Green gasoline (C5C10), green kerosene (C11C13), green diesel (C14C20), or green liquid petroleum gas can be obtained through hydrotreating vegetable oils. To convert vegetable oils into green diesel with higher yield, the catalysts should not induce sever hydrocracking and the temperature range should not be excessive (usually 450–500 °C) [13].
Because the n-paraffins with C17 and C18 are the main components of green diesel, the freezing point of green diesel can be relatively high [14]. For this reason, a selective catalyst (e.g., Ni promoted with transition metals such as Cu and Fe) may favor the hydroisomerization to obtain methyl isoparaffins with better cold properties. Several catalysts were studied for hydrotreating vegetable oils and for various hydroconversion technologies of oils and lubricants [15].
The study performed by Herskowitz et al. [16] shows that soybean oil is converted over Pt/SAPO-11–Al2O3 catalyst in a single-step process, at 375–380 °C and 30 atm. The yield in organic liquid product was approximately 80%, and the gas-chromatography coupled with mass spectrometry (GC–MS) proves the presence of straight and branched paraffins, naphthenes, and monoaromatics, producing a high quality diesel component [16].
Hanafi et al. [17] studied the conversion of waste cooking oil into renewable fuel by hydrocracking over NiW/SiO2Al2O3, in the temperature range of 375–450 °C under pressure below 6 MPa. Liquid products with a chemical composition similar to gasoline, kerosene, and diesel were obtained, and the yield of each fraction depends on the reaction conditions.
Ni–Mo/γAl2O3 is one of the most widely used catalysts for hydrotreating medium and heavy fractions because of its high activity and low acidity, which are convenient for the hydroconversion of triglycerides into green diesel [7,18,19]. The effect of a different atomic ratio of the NiMo/γAl2O3 reduced catalyst was recently investigated for hydrotreating triglycerides, indicating that changes in the atomic ratio influence the yield to hydrocarbons in the diesel range [12].
On the other hand, Sebos et al. [20] found that the CoMo/Al2O3 catalyst exhibits superior performance for the conversion of cottonseed oil–desulfurized diesel mixture into green diesel at 305–345 °C and 30 bar. The conversion of triglycerides was about 100% under the conditions examined [20].
A recent study regarding the bio-oil hydrotreating of diesel-like products over alumina-supported sulfided NiMo and CoMo catalysts was reported [21], suggesting the role of reaction conditions and additives.
Nowadays NiMo catalysts are widely used in the hydrotreating of petroleum products, focusing on hydrogenolysis and HDO of the heterocompounds [22]. This is the reason for choosing the NiMo/γAl2O3 catalyst for our study. To our knowledge, there are no studies available on the investigation of nonsulfided NiMo/γAl2O3 activity for the hydrotreatment of methyl oleate (MeO).
Several studies have been focusing on the hydrotreatment of vegetable oils mixed with petroleum products. An interesting finding was reported by several investigators [23–26], suggesting that the combination of various catalysts may contribute to the oxygen removal and CC double bonds, without hydrogenation of aromatics even if this could lead to a lower octane number of gasoline and increased hydrogen consumption [26–29]. The catalysts used to convert triglycerides, methyl esters, or ethyl esters in diesel-like components need to be selective toward deoxygenation, hydrogenation, and hydroisomerization, without coke formation.
The Zn-modified ZSM-5 support decreases the Brönsted acid sites and increases Lewis acid sites [30] while enhancing the production of aromatics by rapeseed oil conversion [31]. Different catalysts, such as La-modified Ni/γAl2O3 investigated for hydrotreating crude 2-ethylhexanol [32] and La/Zn-ZSM-5 tested for fluid catalytic cracking (FCC) gasoline upgrading [30] were reported. There are no reported studies on NiLa/Zn-ZSM-5 or NiLa/γAl2O3.
Ni, Mo, and W deposited on γAl2O3–ZSM-5 support was investigated for palm oil hydrotreating [33], suggesting that support acidity has a strong influence on the activity, selectivity, and stability of the catalyst. ZSM-5 support enhanced with Zn was reported for various applications including propane dehydrogenation [34] and methane activation [35]. Danuthai et al. [36] studied the performance of the Zn-modified H-ZSM-5 catalyst for the conversion of methyl esters to hydrocarbons, emphasizing that the addition of Zn has a promoting effect on the aromatization of light alkanes. The metal–support interactions on Ni/(H-ZSM-5 + Al2O3) for n-hexane conversion were reported [37].
In our research, we consider the Ni–La/Zn-ZSM-5–γAl2O3 catalyst to possess a higher acidity than Ni–Mo/γAl2O3 because of its support combination (Zn-ZSM-5 + γAl2O3). The hydroisomerization and hydrocyclization reactions require the catalysts to have high acidity [22], as proposed in our study. We propose that the combination of Ni–La/Zn-ZSM-5–γAl2O3 and Ni–Mo/γAl2O3 catalysts used in the reactor may lead, through the aforementioned reactions, to C15C18 hydrocarbons, components for clean diesel fuel.
In this study, novel tests of MeO hydrotreating were applied on the NiMo/γAl2O3 catalyst. Because acidity is a key point in hydrotreating, a combination of Zn-ZSM-5 and γAl2O3 supports was used. Thus, a new Ni–La/Zn-ZSM-5–γAl2O3 material was developed and first performed for the hydrotreatment of triglycerides in this study. In this article, we investigate the way in which the different nickel loadings can lead reactions toward the desired products.
On the other hand, our study proposes various sequences of reactions, which occur during the conversion of triglycerides into clean biofuel, according to the disposal of the Ni–Mo/γAl2O3 and Ni–La/Zn-ZSM-5–γAl2O3 catalytic layers in the reactor system. To emphasize the type and sequence of reactions that take place during the process, different disposals of catalytic layers were performed.
The influence of various operating conditions (e.g., temperature, pressure, and liquid hourly space velocity [LHSV]) on the distribution of reaction products over two catalysts represents another direction of this study.
2 Experimental section
2.1 Catalyst synthesis
Two catalysts supported on different acidic media (e.g., Ni–Mo/γAl2O3 and Ni–La/Zn-ZSM-5–γAl2O3) were used in this study. The catalytic materials are promoted with molybdenum or lanthanum.
The starting materials for the supports were the precursors of Zn-ZSM-5 (provided by Zecasin S.A.) and alumina (S.C.Vega S.A. Ploiesti). To prepare the Zn-ZSM-5–γAl2O3 support, the precursors were mixed in an aqueous solution of 10% HNO3 (from HNO3 67%, Merck). The mixture obtained was extruded, dried at 120 °C for 6 h, and calcined at 550 °C for 6 h.
In the next step, the metal components (e.g., Ni, Mo, and La) were loaded by the pore-filling impregnation method. The aqueous solutions of Ni(NO3)2, (NH4)6Mo7O24, and LaCl3 were used for impregnation. Appropriate amounts of solutions containing the Ni, Mo, and La precursors were used corresponding to the loadings of 4% Ni and 15% Mo for Ni–Mo/γAl2O3 and 10% Ni and 2% La for Ni–La/Zn-ZSM-5–γAl2O3 catalyst.
In case of the Ni-Mobased catalyst, the metals were impregnated in successive steps. The Mo was first loaded onto the support. Ni was impregnated in the next step. For the Ni–La-based catalyst, a mixture of aqueous solutions of Ni and La precursors was prepared.
Both catalysts were dried at 120 °C for 3 h after the metals had been impregnated. The final samples were further calcined at 450 °C for 5 h.
Before the catalytic test, both catalysts were activated “in situ” in a hydrogen flow at 450 °C and at atmospheric pressure for 6 h.
2.2 Catalyst characterization
The supports and catalysts used were characterized in terms of texture with a Quantachrome NovaWin analyzer. The specific surface area was calculated by the Brunauer–Emmett–Teller (BET) method. The pore diameters and pore size distributions were determined by the Barret–Joyner–Halenda model. Substrate and acid strength of catalysts were determined by applying the Du Pont method using a Thermal Analyst 2000/2100 instrument coupled with a 951 Thermogravimetric Analyzer.
Metal loadings were determined by atomic absorption spectrometry method, using a spectrometer Varian AA240FS.
The morphology of the catalysts was determined by scanning electron microscopy (SEM). The measurements were performed on an FEI Quanta 200 (tungsten filament).
The behavior of the catalysts during thermal hydrogen reduction was investigated. The temperature-programmed reduction (TPR) tests were conducted on a ChemBET PULSAR TPR/temperature programmed desorption (TPD) instrument. Before reduction, catalysts were heated at 550 °C under 10% H2 flow diluted in Ar for 1 h. After the sample had been cooled down to 50 °C in the same atmosphere, TPR tests were performed using 10% H2 in Ar and the hydrogen consumption was recorded.
2.3 Catalytic testing
The feedstock used in the hydrotreatment process was MeO 99% (provided by Sigma–Aldrich). All experiments were carried out in a continuously operating reactor system, electrically heated. The temperature of the reactor was controlled by an SIMEX (Termodensirom) system. The feedstock was supplied using a Varian ProStar high performance liquid chromatography (HPLC) pump.
The aforementioned catalytic systems were arranged in different ways in the reactor, for example, Ni–Mo/γAl2O3 on the bottom part of the catalyst bed, followed by Ni–La/Zn-ZSM-5–γAl2O3 on the upper side. These aspects are explained in detail Section 3. Various proportions of the catalysts (e.g., 20 and 40 cm3) were used in this article.
The nonsulfided catalysts were loaded between two layers of glass beads into the middle of the reactor, ensuring an isothermal temperature profile along the length of the catalytic layer. The total volume of the catalyst loaded in the reactor was 80 cm3. The reaction mixture was cooled in a condenser and further separated; the liquid phase was evacuated through a valve at the bottom part of the separator.
vTen runs (numbered from 1 to 10) in the temperature range between 360 and 445 °C, at the pressures of 50 and 90 bar, were performed. The LHSV was varied from 0.38 to 1.5 h−1, respectively, corresponding to the total flows of 0.5, 1, and 2 cm3/min. The hydrogen flow rate was kept at 15 L/h [standard temperature and pressure (STP)] during all tests.
The effect of the paraffinic solvent on catalytic performances was investigated in this study and a mixture of MeO and n-octane at a ratio of 1:1 (v/v) was fed into the reactor. The n-octane (98% purity) was provided by Sigma–Aldrich.
The details regarding the experimental program are depicted in Table 1.
Experimental program for MeO hydrotreatment.
| Run | Catalytic system type | Feed | Temperature (°C) | Pressure (bar) | LHSV (h−1) | |
| 1 | Double-layered nonsulfided catalyst (40 cm3 + 40 cm3) | Ni–Mo/γAl2O3 Ni–La/Zn-ZSM-5–γAl2O3 | MeO | 360 | 90 | 0.75 |
| 2 | 1.5 | |||||
| 3 | 380 | 0.75 | ||||
| 4 | 400 | 0.75 | ||||
| 5 | MeO/nC8H18 = 1:1 | 360 | 0.75 | |||
| 6 | Ni–La/Zn-ZSM-5–γAl2O3 Ni–Mo/γAl2O3 | MeO | 400 | 0.75 | ||
| 7 | Triple-layered nonsulfided catalyst (20 cm3 + 40 cm3 + 20 cm3) | Ni–Mo/γAl2O3 Ni–La/Zn-ZSM-5–γAl2O3 Ni–Mo/γAl2O3 | MeO | 400 | 90 | 0.75 |
| 8 | 0.38 | |||||
| 9 | 420 | 50 | 0.38 | |||
| 10 | 445 |
The reaction products were obtained in two phases: the gas and liquid phases. Two layers of liquid were observed, the superior one was the organic phase, whereas the inferior layer was the aqueous phase. The liquid layers were left for decantation in a vertical separation funnel.
The quantitative analysis of the organic liquid layer was carried out by gas chromatography on a 5890 Network GC (Agilent Technologies) instrument, equipped with [flame ionization detector (FID)], a polar capillary column DB–WAX, 30 m × 0.32 mm inner diameter, operated at temperatures between 20 and 220 °C.
The gas phase coming from the separator was analyzed online with a VARIAN CP 3800 gas chromatograph, equipped with a valve system. A simultaneous analysis of hydrogen and carbon oxides on thermal conductivity detector (TCD) detector and the measurements of methane and other organic compounds by FID detector were performed. Separation of the components was achieved on capillary columns PoraPLOT 25 m × 0.53 + Mol sieve 5A 25 m × 0.53 (for H2 and COx) and CPWAX 57 CB 30 m × 0.32 (for organic compounds, respectively).
3 Results and discussion
The selection of target catalysts was focused on the optimization of their acidity, based on the nature of the catalytic processes are favored by these materials. Thus, a catalyst with low acidity generated by MoO3 species was designed for the hydrogenolysis of the oxygenated compounds. The acidity of molybdenum oxide mainly favors hydrotreating reactions (e.g., hydrodesulfurization, HDO, and hydrodenitrification). Another material with approximately 50% higher acidity than the Ni-Mo–based catalyst was chosen for the isomerization–dehydrocyclization reactions. The higher acidity is required to favor the isomerization of secondary carbocations. They are obtained by adding the proton given by the acid catalyst with the subsequent formation of tertiary carbocations. These final species are responsible for branched and cyclic hydrocarbon formation.
To design the new proposed catalytic materials (Ni–Mo/γAl2O3 and Ni–La/Zn-ZSM-5–γAl2O3), we have taken into account the role of each component that should induce the hydrotreating of triglycerides. The ZSM-5 zeolite support favors isomerization and dehydrocyclization reactions [38,39]. Zn was introduced into ZSM-5 to enhance zeolite acidity [40–42].
A low Ni content was used for synthesizing the Ni–Mo/γAl2O3 catalyst, similar to those usually loaded for industrial applications (e.g., 4%–6%). A catalyst with a low amount of Ni (e.g., 4% Ni–14% Mo/γAl2O3) only converts the heterocompounds through its dual hydrogenation–dehydrogenation function [43,44]. A high Ni loading is needed (e.g., 10% Ni–2% La/Zn-ZSM-5–γAl2O3) to dehydrogenate all compounds, with the formation of carbocations that subsequently contribute to inducing isomerization–dehydrocyclization reactions.
The addition of molybdenum provides the acidic function of the Ni–Mo/γAl2O3 catalyst. This material was intentionally calcined below 500 °C to get molybdenum oxides. These oxides enhance the catalytic acidity, favoring the hydrogenolysis reaction.
Regarding the role of lanthanum in the hydrotreating process, its presence lowers the proportion of strong acid sites. This behavior is connected with decreasing the undesired hydrocracking reactions during the isomerization–dehydrocyclization stage.
3.1 Characterization of catalysts
Table 2 shows the textural properties of the synthesized materials. As shown in Table 2, different BET surface area values of the catalysts were found, dependent on the support type and promoter addition. Furthermore, the materials differ in terms of their acidity and pore volume, as shown in Table 2. Comparing the surface area of alumina support alone (193 m2/g) with the Ni–Mo/γAl2O3 surface area, the increase in BET values indicates that metals were not incorporated into the substrate. The surface area of the Ni–La/Zn-ZSM-5–γAl2O3 catalyst is considerably lower (Table 2), likely because of the blockage of some pores by metal species.
Textural characteristics of the fresh nonsulfided catalysts.
| Fresh catalyst | Surface area (m2/g) | Average pore volumea (cm3/g) | Average pore diameterb (nm) | Acidity (mequiv/g) |
| Ni–Mo/γAl2O3 | 291.03 | 0.244 | 14.859 | 1.923 |
| Ni–La/Zn-ZSM-5–γAl2O3 | 150.45 | 0.120 | 14.402 | 2.854 |
Knowing that acid supports make a considerable contribution to the hydrogenation and dehydration, which are involved in the process studied, our results show significant changes in acidities for the materials analyzed. Ni–La/Zn-ZSM-5–γAl2O3 features higher acidity due to the combination of two strong acid supports that facilitate the hydroisomerization and dehydrocyclization processes. Furthermore, catalyst stability increases with the support of γAl2O3 [45,46].
The average pore diameter of Ni–Mo/γAl2O3 is slightly greater than that of Ni–La/Zn-ZSM-5–γAl2O3 (see Table 2), and from the BET analysis, it can be concluded that both synthesized catalysts have (a fully or partially ordered) mesoporous structure [47]. The lower values of the BET surface area, pore volume, and pore diameter for the fresh Ni–La/Zn-ZSM-5–γAl2O3 are because of the deposition of active metals inside the pores. A large drop in pore volume was observed in the case of the Ni–La/Zn-ZSM-5–γAl2O3 catalyst, probably because of structural changes in Zn-ZSM-5–γAl2O3 after metal loading.
From the results of textural characteristics, it is evident that there is an increase in the pore diameters for both the NiMo/Al2O3 and the NiLa/Zn-ZSM-5–γAl2O3 catalysts after tests have been performed (Fig. 1a and b). The pore volume values are approximately 5%–8% greater than those of catalysts before experiments. Because a high amount of carbonaceous residues is deposited on acid sites after reactions, this results in a blockage of pores by the coke precursors, which are involved in the coke production during the process.
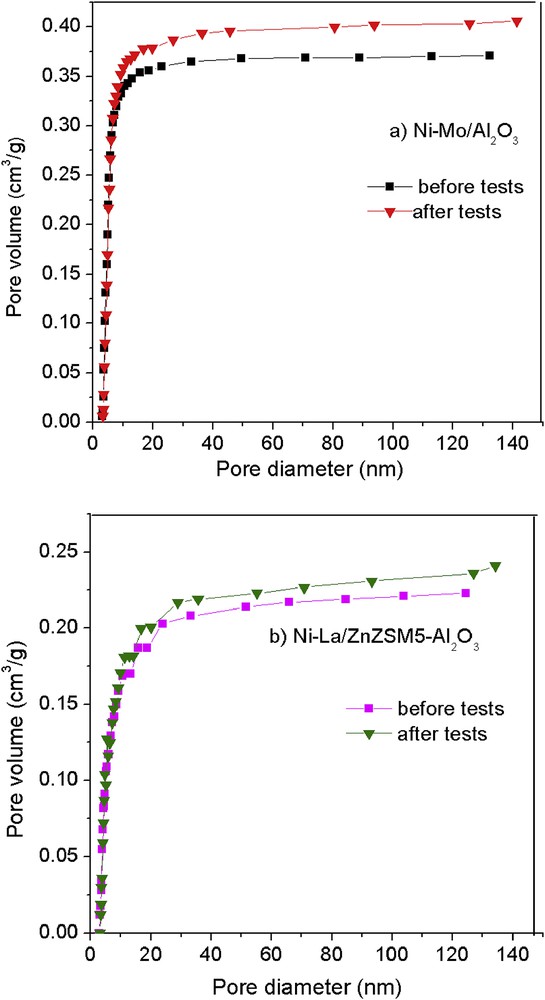
The profiles of pore diameter of (a) Ni–Mo/γAl2O3 and (b) Ni–La/Zn-ZSM-5–γAl2O3 catalysts before and after tests.
The isotherms of both catalysts during the nitrogen adsorption and desorption are depicted in Fig. 2. The isotherms are very similar for both catalysts and are attributed to type IV [48], whereas the hysteresis loops look relatively different.
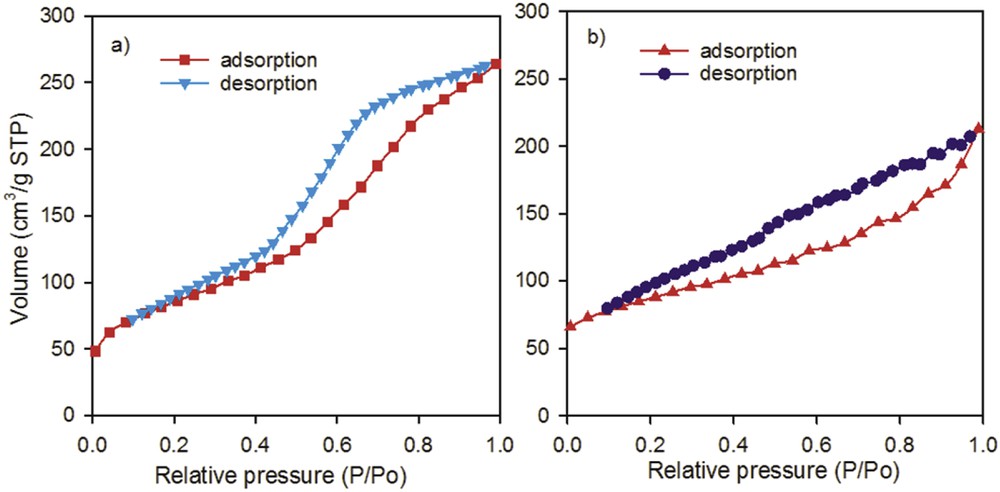
N2 adsorption–desorption isotherms of (a) Ni–Mo/γAl2O3 and (b) Ni–La/ZSM-5-Zn–γAl2O3 catalysts.
The metal loadings of Ni and Mo for Ni–Mo/γAl2O3 and the content of Ni and La for Ni–La/Zn-ZSM-5–γAl2O3 catalysts were determined by atomic absorption spectrometry (the targeted metallic contents discussed in Section 2). Our results are in agreement with the target metal contents of 4% Ni, 14.4% Mo, and 9.9% Ni with 1.9% La, respectively.
An important factor in the catalyst design consists of the mixed oxide reduction. TPR experiments over Ni–Mo/γAl2O3 and Ni–La/Zn-ZSM-5–γAl2O3 catalysts were performed in this study and the results are presented in Fig. 3.
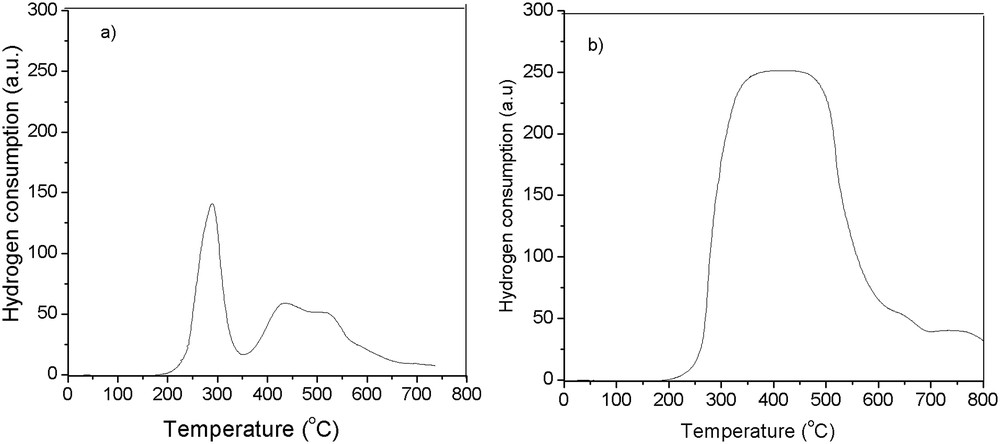
H2-TPR profiles over (a) Ni–Mo/γAl2O3 and (b) Ni–La/Zn-ZSM-5–γAl2O3 catalysts.
For the Ni–Mo/γAl2O3 catalyst, the H2-TPR profile exhibited three main reduction peaks (Fig. 3a). The reduction of nickel and molybdenum oxides usually occurs at high temperatures (700–1000 °C) [49–52]. When molybdenum oxide is supported on alumina, its reduction can be shifted to lower temperatures. The large peak corresponding to the maximum 298 °C can be assigned to the reduction of molybdenum from 6+ to 4+ for the octahedral Mo-based species. In addition, the broad peak that may include two small peaks attributed to the 440–525 °C temperature range indicates the presence of MoO3 on alumina support [53]. The absence of a peak at higher temperature (above 550 °C) is an indication of the lack of metallic molybdenum on the catalytic surface [51].
Fig. 3b shows the comparative behavior of the Ni–La/Zn-ZSM-5–γAl2O3 catalyst during the TPR run. The presence of La as promoter shifted the reduction peaks (of α-type NiO) to the lower temperature of 390–430 °C [54].
The higher reduction temperature for Ni–Mo/γAl2O3 suggests a strong interaction between alumina support and the metal oxides of the active phase [33,55], which can be attributed to a high dispersion of Ni–Mo loadings on the catalytic support. On the other hand, alumina–metal oxide interaction can improve catalyst stability [51].
SEM micrographs recorded at 10 nm are depicted in Fig. 4, which shows the presence of aggregated irregular particles of nickel and molybdenum in oxide form (dark area) distributed over the alumina support (Fig. 4a). These species could be a mix of mono- and/or bimetallic entities. It should be noted that the catalysts have not been reduced before the analysis, so the metallic oxides were present in the catalyst composition. Furthermore, isolated nanoparticles were detected during the analysis.
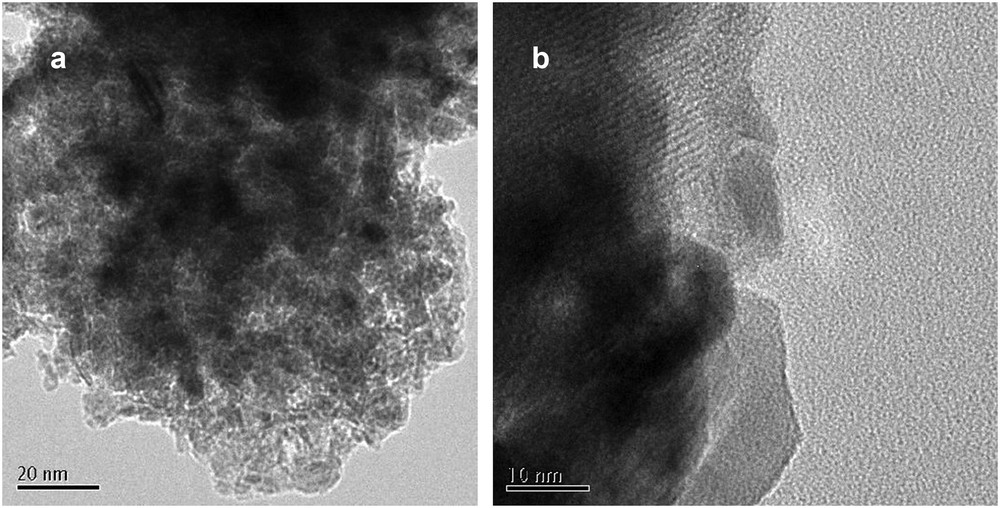
SEM micrographs of fresh (a) Ni–Mo/γAl2O3 and (b) Ni–La/Zn-ZSM-5–γAl2O3 catalysts.
The electron microscopy image of Ni–La/Zn-ZSM-5–γAl2O3 described the highly concentrated zone of agglomerated Ni and La particles on the porous support as the particle aggregation mostly increase with the metals loading (Fig. 4b). This finding can be associated with the fact that the metallic clusters of oxide species are usually too large to be accommodated inside zeolite pores; thus, they can be distributed over the external surface of zeolite, which corresponds to the concentrated area.
The acidity of the catalysts plays an important role in hydrotreating processes, especially in the dehydration reaction, with the formation of unsaturated intermediary compounds. In other words, the catalysts make an important contribution in breaking the CO bond. Furthermore, the adsorption and activation of the OH group mainly occurs on the acid component of the catalyst, followed by water removal. The acidity of the materials is provided by the used support and details regarding their acidity are presented in Table 3.
Acid strength distribution of the synthesized catalysts.
| Catalyst | Weak strength acid centers (%) | Medium strength acid centers (%) | Strong strength acid centers (%) |
| Ni–Mo/γAl2O3 | 52.88 | 21.19 | 25.92 |
| Ni–La/Zn-ZSM-5–γAl2O3 | 44.19 | 25.83 | 29.98 |
The high concentration of weak strength acid centers can be explained by the existence of a mixed oxide phase on the catalytic surface, caused by the incomplete reduction of precursors and possible structural defects. The results of the experimental tests performed on the prepared catalysts support the hypothesis presented in the literature that an increase in acidity favors a strong interaction between metal and support, making it difficult to reduce metallic oxides on the catalytic surface, thereby affecting the CC bond breaking [56].
3.2 Catalytic activity tests
The experimental tests were carried out using a double and a triple layer catalytic disposal in the reactor system. It should be mentioned that a double layer represents 40 cm3 Ni–Mo/γAl2O3 as the first layer from the top of the reactor followed by 40 cm3 of Ni–La/Zn-ZSM-5–γAl2O3 as the second layer. The triple-layered catalyst has first 20 cm3 of Ni–Mo/γAl2O3, followed by 40 cm3 of Ni–La/Zn-ZSM-5–γAl2O3 and 20 cm3 of Ni–Mo/γAl2O3 as a third layer.
It should be pointed out that MeO, being an unsaturated compound has been chosen as feed owing to its several advantages including the easy emphasis of its structural modifications during the process when it is used as a single, pure component; straight or branched paraffins with 15–18 carbon atoms in the molecule, which are similar to those of classical diesel fuel. Good service properties can be obtained by MeO hydrotreatment.
Several compounds belonging to different hydrocarbon classes (e.g., n-paraffins, isoparaffins, aromatics, and small amounts of alkyl naphtenes) were identified in the organic liquid phase collected at the end of the experiments. It should be noted that unsaturated hydrocarbons were not identified in the tests performed. Benzene (B), toluene (T), ethylbenzene (EB), o-xylene, m-xylene (mX), p-xylene (pX), propylbenzene, isopropylbenzene, n-dodecane, n-nonadecane, dimethylcyclohexane, ethyl-methylcyclohexane, ethyl tetraline, dimethyl tetraline, and n-paraffinic hydrocarbons C15–C19 were identified as individual compounds. The chromatographic analysis of the gas phase reveals the presence of hydrogen as a major component, with concentrations higher than 99.2%, whereas the other identified gases were CO and CO2.
3.3 Double-layered catalytic systems
The double-layered catalytic system was used for the first six runs (see Table 1) at different temperatures (360, 380, and 400 °C) and a pressure of 90 bar. Two LHSVs were used and n-octane was added in the feed. The double-layered catalytic system was designed owing to its high deoxygenation activity and good selectivity in COH bond hydrogenolysis [57].
3.4 The influence of temperature
To study the temperature effect on the MeO hydrotreatment performance, the experiments were performed at 360, 380, and 400 °C (runs 1, 3, and 4) and at 90 bar with LHSV of 0.75 h−1. The experimental results are presented in Fig. 5.
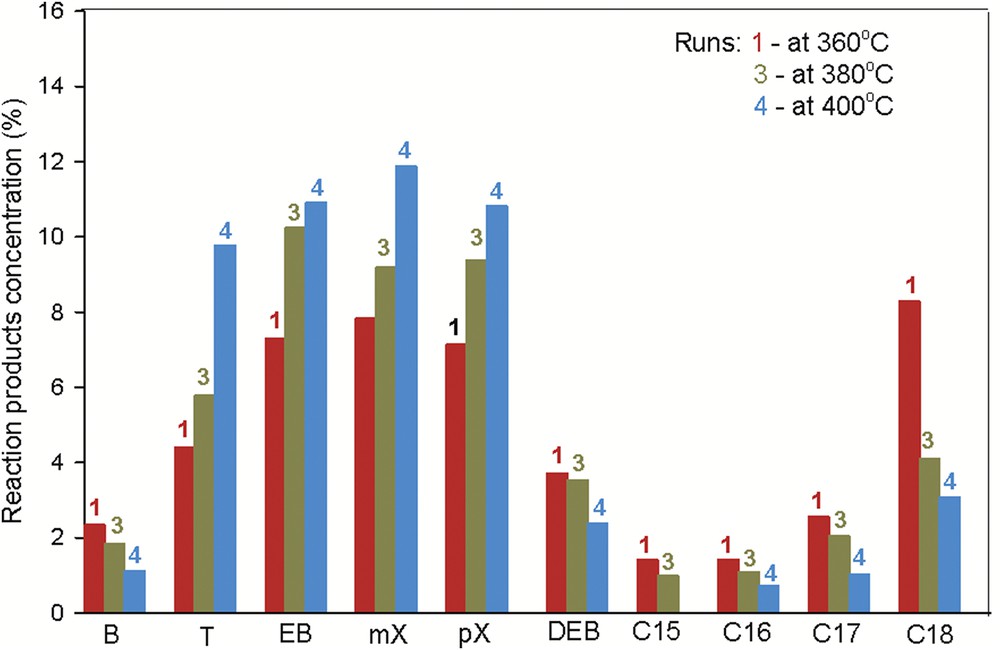
The distribution of the reaction products at various temperatures (360, 380, and 400 °C) over (Ni–Mo/γAl2O3 + Ni–La/Zn-ZSM-5–γAl2O3) double-layered catalysts. The experimental details are presented in Table 1.
It is known that the textural properties (e.g., pore diameter, pore volume, and surface area) play an important role regarding catalytic performance, especially for large molecule reactants, because they can affect the diffusion of these molecules. In comparison with the Ni–La/Zn-ZSM-5–γAl2O3 catalyst, the Ni–Mo/γAl2O3 catalyst possesses a slightly larger pore size, which is in favor of the diffusion of MeO, consequently it displays a higher efficiency toward C18 hydrocarbons at 360 °C (Fig. 5).
As seen in Fig. 6, the process leads mainly to aromatic hydrocarbon formation over double-layered catalytic systems. The aromatization of MeO is a multistage process. The hydrogenolysis–hydrocracking reactions occur on the first catalytic layer, NiMo, with the formation of paraffins. The hydrogenolysis is favored by the acidic support of the first catalytic layer (γAl2O3), but is not so acidic as to break the C–C bonds. Subsequently, dehydrocyclization reactions may occur by a carbocationic mechanism on strong acid centers over the second catalytic layer, Ni–La. It possible for hydrocracking reactions to takes place simultaneously due to the strong acidity of the second catalytic layer.
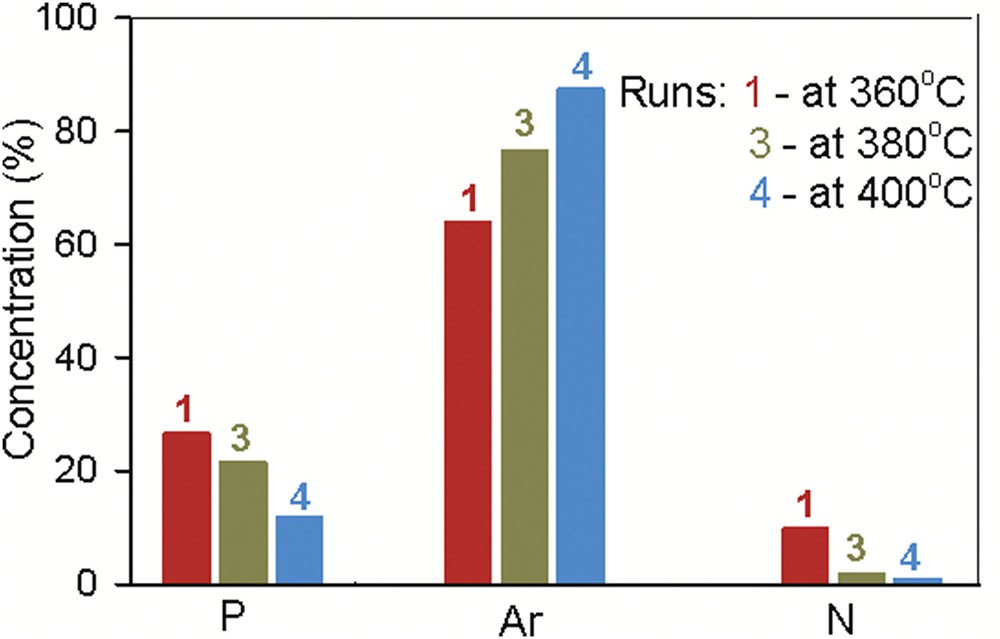
Hydrocarbon classes (P, Ar, and N) distribution at different temperatures (360, 380, and 400 °C) over (Ni–Mo/γAl2O3 + Ni–La/Zn-ZSM-5–γAl2O3) double-layered catalysts.
Advanced breaks and reforming reactions are favored with increasing temperature. The advanced breaks are reflected by a decrease in the octadecane proportion. The intensification of the reforming process is emphasized by decreasing amounts of naphtenes.
The benzene formation occurs via two reaction sequences. One pathway is the direct dehydrocyclization to benzene. The second and the most likely way is the isomerization of hexane (formed by hydrocracking over the Ni–Mo catalyst) to methylpentane, followed by the cyclization to methylcyclopentane, isomerization to cyclohexane and, finally, dehydrogenation to benzene. Because the reaction product has a high content of aromatics but very low benzene concentration, it may be used as a gasoline component with high octane number.
3.5 LHSV effect
Two tests (no. 1 and 2) were conducted using LHSVs of 0.75 and 1.5 h−1 at the constant reaction conditions (360 °C and 90 bar) over (the Ni–Mo/γAl2O3 + Ni–La/Zn-ZSM-5–γAl2O3) double-layered catalyst (Fig. 7).

The distribution of the reaction products varying LHSV over (the Ni–Mo/γAl2O3 + Ni–La/Zn-ZSM-5–γAl2O3) double-layered catalysts. The experimental details are presented in Table 1.
The distribution of the reaction products is quite similar to those previously observed (Fig. 5), but the decrease in contact time between catalyst and feed has an unsatisfactory effect on the content of the linear saturated hydrocarbons in the reaction products.
It may be possible that the contribution of the dehydrogenation–dehydrocyclization reactions is less important than hydrocracking under these reaction conditions. The textural properties of catalysts (metal dispersion and surface area, especially) enhance the dehydrogenation–dehydrocyclization reactions, but too short a reaction time does not help completing the hydrocracking reaction.
3.6 The influence of n-octane
The addition of a solvent (e.g., n-octane) in the feed is investigated in this study. The molar MeO/n-octane of 1:1 ratio was used under a temperature of 360 °C and pressure of 90 bar. The results obtained were compared with the tests without the presence of n-octane in the feed (Fig. 8).
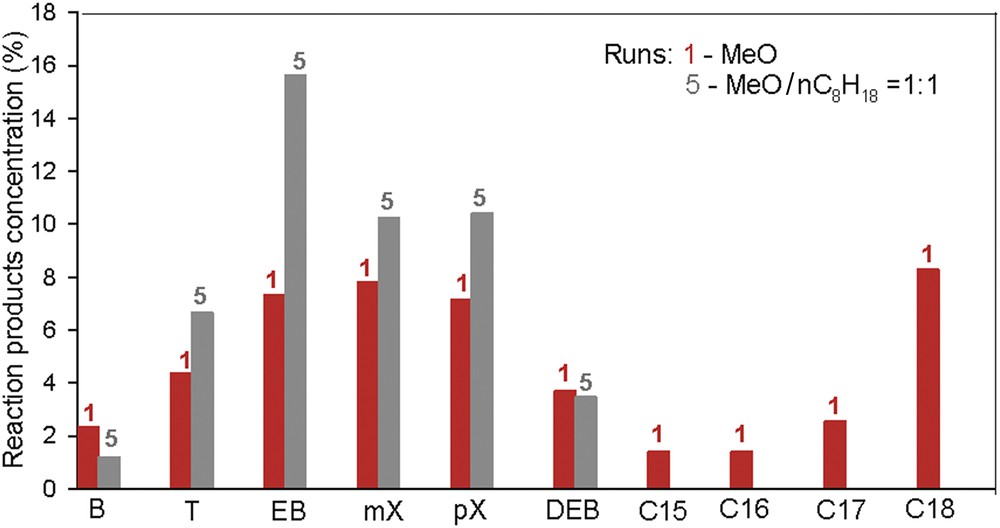
The composition of the reaction products by varying the feed composition over (Ni–Mo/γAl2O3 + Ni–La/Zn-ZSM-5–γAl2O3) double-layered catalysts. The experimental details are presented in Table 1.
Even if the use of a solvent lowers the viscosity of the feed, we found that the presence of n-octane inhibits the formation of linear or branched paraffin hydrocarbons.
As seen in Fig. 9, the chromatographic analysis of the reaction products contains only aromatic mono- and bicyclic hydrocarbons, predominantly substituted by one to four side chains containing one to four carbon atoms. The significant increase in the concentration of C8 aromatics in the presence of n-octane emphasizes that dehydrocyclization reactions occur faster than hydrocracking.
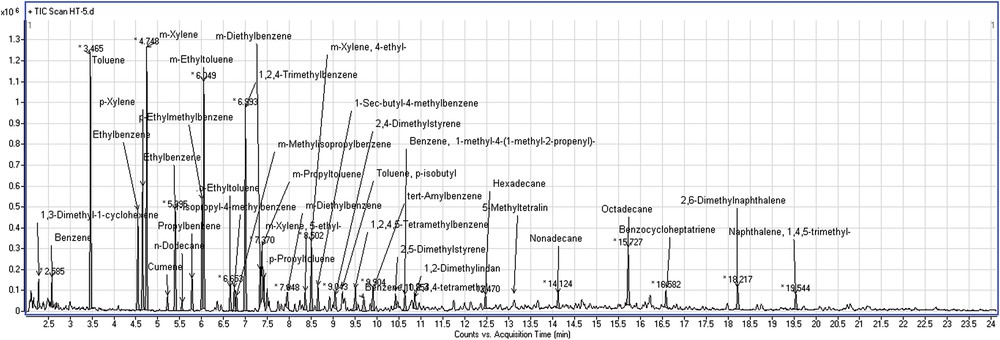
Chromatogram with the distribution of the reaction products during run 5.
3.7 The influence of catalytic layer disposal
To understand if the disposal of the catalysts into the reactor can influence the product composition, an experiment was performed using the same materials but the ordering of the catalytic layers into the reactor was different (e.g., 40 cm3 of Ni–La/Zn-ZSM-5–γAl2O3 + 40 cm3 of Ni–Mo/γAl2O3). This means that the feed was first contacted with the most acidic catalyst, that is, Ni–La catalyst. The experimental results were compared with those obtained for run 4, where the disposal of the materials was opposite (e.g., Ni–Mo/γAl2O3 + Ni–La/Zn-ZSM-5–γAl2O3).
It was found that the distribution of the hydrocarbon classes was deeply changed by altering the arrangement of the catalytic layers into the reactor.
In run 4, where the Ni–Mo catalyst was the first layer, the HDO was efficient and paraffins were formed as intermediary compounds. In the next step, the resulting paraffins were considered to be “feed” for the processes (e.g., dehydrocyclization), which were taking place over the second catalyst and aromatics C7C8 were produced in similar proportions.
Molybdenum located in the first catalytic layer provides the catalytic acid function that allows the hydrogenolysis and dehydration reactions to take place. The formed intermediate carbocations can be either saturated in a small amount, due to the hydrogenation function of Ni, or can be mostly isomerized and dehydrocyclized. The results of run 4 are in agreement with the aforementioned explanation.
In run 6, where the Ni–La catalyst is the first layer, the hydrogenolysis efficiency is negligible, and as a consequence, the dehydrocyclization does not occur. Instead, dehydroxygenation and partial hydrocracking occur over the second catalytic layer, Ni–Mo.
The GC analysis of the liquid organic product obtained from run 6 (see Fig. 11) indicates the presence of linear paraffinic hydrocarbons with carbon numbers between 15 and 18.
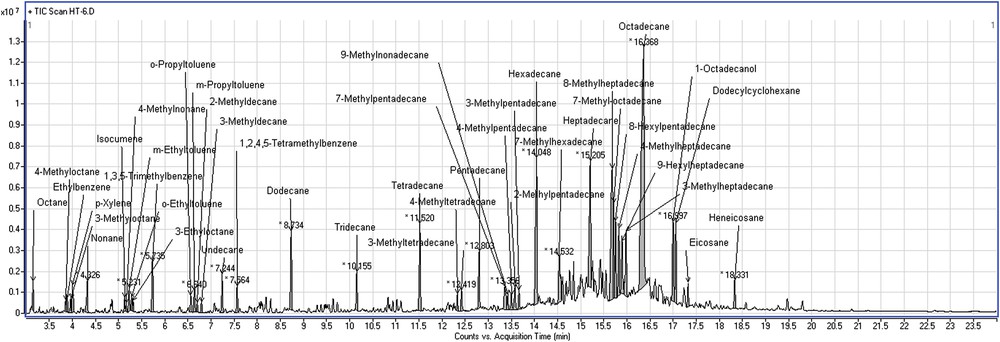
Chromatogram showing the formation of the reaction products for run 6.
In the case of Ni–La/Zn-ZSM-5–γAl2O3 used as the first exposed catalytic layer, hydrocracking and HDO reactions initially take place because of their higher acidity compared to their Ni–Mo/γAl2O3 counterpart catalyst. The formed intermediates will be rather hydrogenated, with the production of paraffinic hydrocarbons. As a consequence, the dehydrocyclization reactions will be low, as the results of run 6 shown in Fig. 10.
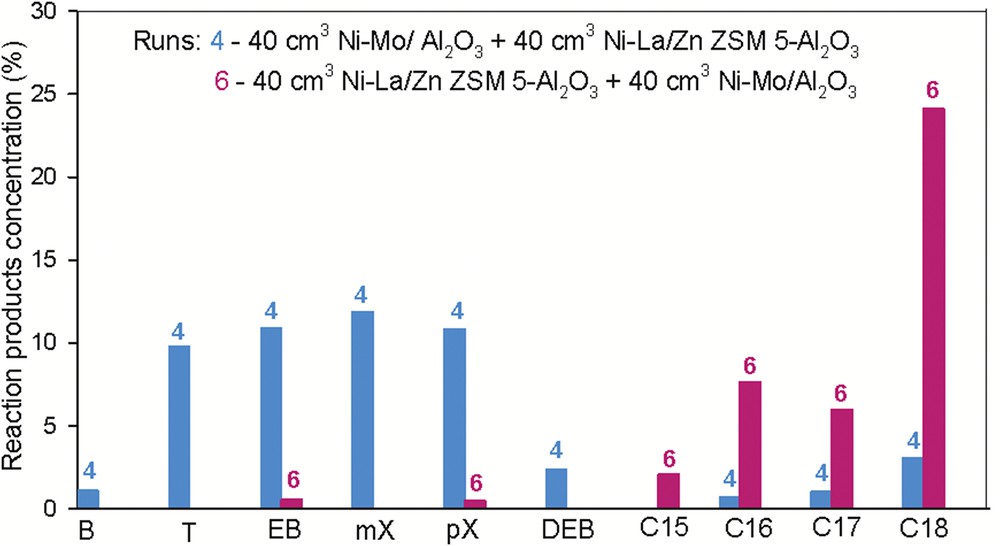
The behavior of the double-layered catalytic systems (Ni–Mo/γAl2O3 + Ni–La/Zn-ZSM-5–γAl2O3) and (Ni–La/Zn-ZSM-5–γAl2O3 + Ni–Mo/γAl2O3). Reaction conditions: t = 400 °C, p = 90 bar, and LHSV = 0.75 h−1.
It should be mentioned that a relatively high amount of 24.14% octadecane was produced during run 6. Isomerization reactions are relatively intense, emphasizing many saturated compounds with a short side chain, such as methyl. Instead, identified aromatic compounds are found to have fewer side chains, like methyl or ethyl. The product obtained can be used as a diesel fuel component, with a relatively high cetane number, allowing antiknocking combustion of fuel. However, some difficulties may be occurring due to the high freezing point of long-chain paraffins present in the reaction products.
3.8 Triple-layered catalytic system
To increase the amount of aromatics in the composition of the reaction products, we propose a new arrangement of the catalytic layers into the reactor system. First, a volume of 20 cm3 Ni–Mo/γAl2O3 catalyst was introduced into the reactor, followed by 40 cm3 of Ni–La/Zn-ZSM-5–γAl2O3 and 20 cm3 of Ni–Mo/γAl2O3. The reaction conditions were 400 °C and 90 bar. A comparative study of the experimental results for runs 4, 6, and 7 is presented in Fig. 12, and significant changes in the distribution of components are observed.
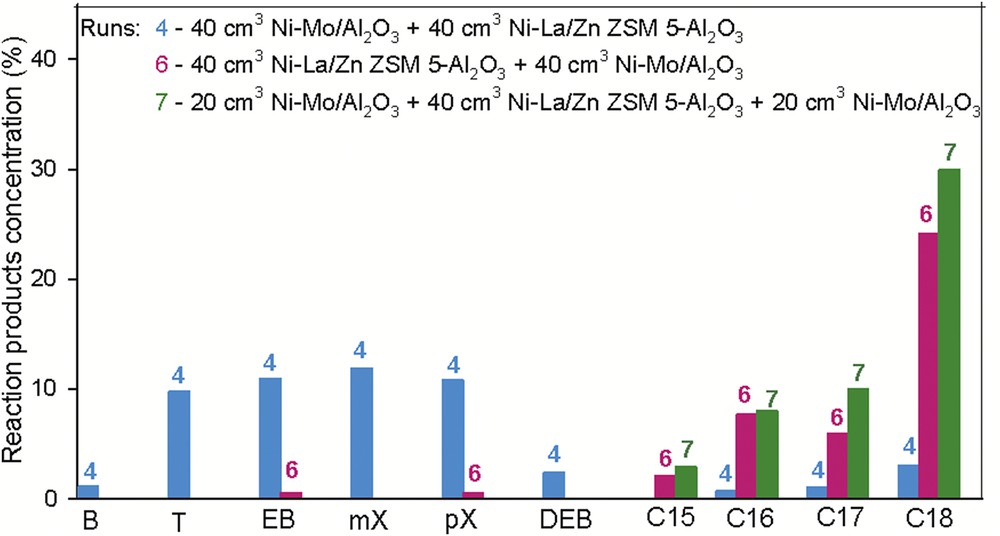
Comparative study between double- and triple-layered catalytic systems. Reaction conditions: t= 400 °C, p = 90 bar, and LHSV = 0.75 h−1.
The distribution of three catalytic layers providing dehydroxygenation brings important modification to the composition of the reaction product compared with the results performed over the double catalytic layer. The reaction product obtained in this catalytic settlement and under the aforementioned operating conditions is rich in n-paraffins with a high carbon number; the content of n-paraffins is higher than 50%. Between those, the C17 and C18 proportion is close to 40%. These compounds are important as diesel components because they burn smokelessly and yield a high cetane number value. This mixture has a low concentration of aromatics and those identified are mononuclear, with one to four methyl chains, helping the freezing properties as diesel components, but not in an adequate manner.
In this arrangement of three catalytic layers, it can be considered that in the first catalytic layer hydrogenolysis reaction occurs, but the catalytic volume of this layer was inadequate (20 cm3) and did not provide enough feed for the second layer to be dehydrocyclized. The hydrogenolysis and hydrocracking reactions started on the first layer were completed on the third layer. However, the second layer (40 cm3 of Ni–La/Zn-ZSM-5–γAl2O3) shows intense isomerization activity reflected by the presence of branched saturated compounds with one short side chain. The presence of compounds with tertiary carbons in their structures is desirable in fuel composition. They are more stable, thus lowering the danger of knocking combustion (Fig. 13).
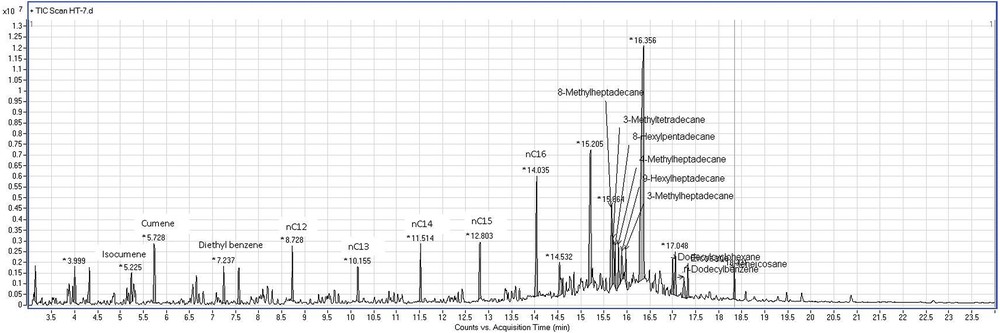
Chromatogram of the reaction products for run 7.
Due to the minor amounts of intermediary products obtained from the first layer, the Ni–La catalyst did not fulfill the dehydrocyclization role in this settlement.
3.9 The influence of LHSV
Two runs using the triple-layered catalytic system were conducted at various LHSVs from 0.38 to 0.75 h−1 at 400 °C and 90 bar (Fig. 14).
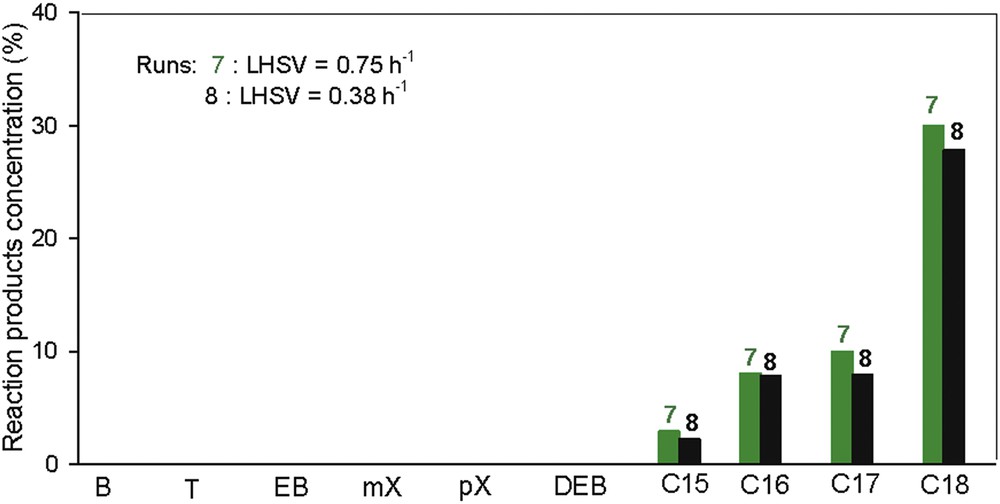
The reaction products obtained over the triple-layered catalytic system at different LHSVs from 0.38 to 0.75 h−1. Reaction conditions: t= 400 °C, p = 90 bar.
Fig. 15 shows that the reaction products contain only aromatics (Ar) and paraffins (P) under the target reaction conditions (400 °C and 90 bar).

Distribution of hydrocarbon classes (P and Ar) at various LHSVs (0.38 and 0.75 h−1) over triple-layered catalytic systems.
As mentioned previously, the catalytic settlement in the triple-layered catalytic system limited the dehydrocyclization of intermediary compounds and therefore does not lead to the formation of aromatics. However, a low LHSV value of 0.38 h−1 exhibits a longer contact time between feed and each catalytic layer, with a small intensification of dehydrocyclization reaction, reflected by lower C18 proportion and higher aromatics content in the final product composition.
Hydrotreating MeO under these conditions leads to the formation of large amounts of C18 and significant proportions of C16 and C17. Along with these compounds, a reaction product mixture also contains saturated linear inferior terms, C8–C12, in proportions between 1.55% and 3%, and ring aromatic compounds, with one to three short side chains in the proportion of up to 3.03%.
This low content of inferior paraffins can be explained by the value of applied pressure. At high pressure, advanced breaks are hindered, with only one CC bond break taking place. For this reason, a straight chain is obtained, the structure of which is similar to the compounds found in classical diesel fuel. From this point of view, the product obtained can be used successfully as a component in diesel fuel formulation, featuring a high cetane number and antiknocking combustion.
3.10 Effect of temperature
Two experiments were conducted at different temperatures of 420 and 445 °C, at a lower pressure (50 bar) and low LHSV (0.38 h−1) over triple catalytic layers. The resulting data are presented in Fig. 16.
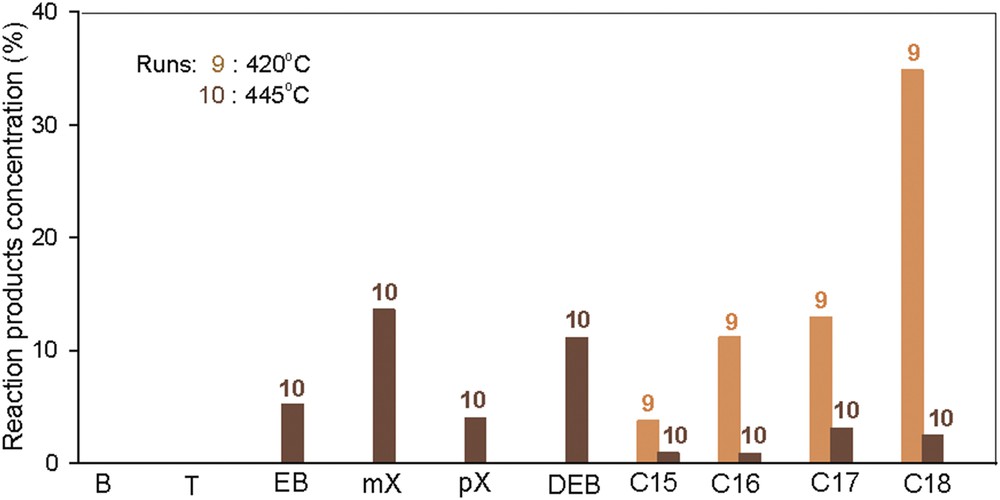
The reaction products obtained over the triple-layered catalytic system at 420 and 445 °C. Reaction conditions: p = 50 bar and LHSV = 0.38 h−1.
The reaction products obtained in run 9 consist mainly of saturated hydrocarbons with a large number of carbons in the molecule, between 12 and 18, with straight or one short side chain. The operating conditions designed are beneficial for the HDO reaction, but the hydrocracking reaction is limited. Thus, the second catalytic layer has not contributed to produce intermediary compounds for dehydrocyclization into aromatics.
At higher temperatures, hydrocracking reactions, which occur on the first catalytic layer, are more efficient, providing the raw material for the second catalytic layer. There are favored dehydrocyclization reactions of C8–C10 hydrocarbons, which are probably the main compounds formed in the hydrocracking step. The third catalytic layer is not efficient, probably making a small contribution to the final composition of the reaction product.
4 Conclusions
Classical diesel fuels are mainly composed of up to 70% straight, branched, and cyclic saturated hydrocarbons, 20%–25% aromatics, whereas the balance comprises small amounts of olefins and heterocompounds of oxygen and sulfur [27,58]. Because of straight paraffins C17 and C18 and isoparaffins with the same carbon number in its molecule, which has a single side methyl chain exhibiting a high cetane number and good rheological behavior, the amount of these compounds formed after the hydrotreating process represents the main criterion for assessing the performance of the process [59,60].
The Ni–Mo/γAl2O3 and Ni–La/Zn-ZSM-5–γAl2O3 catalysts were prepared by impregnation and further characterized by atomic absorption spectrometry, BET, TPR, and SEM. The BET measurements reveal a high surface area of Ni–Mo/γAl2O3, whereas a low surface area of Ni–La/Zn-ZSM-5–γAl2O3 was found.
A low aromatic proportion resulting from the settlement of the Ni–La/Zn-ZSM-5–γAl2O3 layer in the triple-layered catalyst system confirms the inconsistency between the particle size and pore diameter. Some pores of the sample are blocked with the metal species leading to the decrease in surface area.
The acid strength of the materials reveals a noticeable presence of weak acidic centers in the range of 44%–53%, whereas a relatively equal distribution of medium and strong acid centers is evidenced.
The H2-TPR profile the Ni–Mo/γAl2O3 catalyst presents three main reduction peaks (Fig. 3a). The large peak, which is attributed to the maximum temperature of 298 °C, can be assigned to the reduction of molybdenum from 6+ to 4+ for the octahedral Mo species, whereas the broad peak, which may include two small peaks at the temperature range of 440–525 °C, indicates the presence of MoO3 on alumina support. The presence of lanthanum in the Ni-La–based catalyst shifted the reduction peaks to the lower temperatures of 390–430 °C.
The effect of various reaction conditions (e.g., temperature, pressure, and liquid hourly space velocity) on the catalytic performance was investigated. The increase in reaction temperature favors the hydrocracking of cyclohexane and methylcyclopentane, which competes with isomerization, leading to the decrease in benzene yield. It should be noticed that no unsaturated hydrocarbons were produced in the tests performed.
The variation in benzene concentration with temperature argues that the mechanism toward benzene occurs through the isomerization reactions of hexane to methylpentane, followed by cyclization to methylcyclopentane, isomerization to cyclohexane, and dehydrogenation to benzene.
The LHSV effect is evidenced and it was found that lower values of LHSV have a minor effect on the dehydrocyclization, emphasized by a low C18 amount and a high aromatics content.
The presence of n-octane in the feed favors the increase in C8 aromatics and dehydrocyclization occurs faster than hydrocracking.
Another focus of the present study was the effect of the disposal of catalysts into the reactor system and double- and triple-layered catalytic systems were applied.
Double-layered catalytic systems involve the HDO reaction with paraffin formation as intermediary compounds when Ni–Mo/γAl2O3 is first disposed into the reactor, whereas the hydrogenolysis is negligible and the dehydrocyclization does not occur when Ni–La/Zn-ZSM-5–γAl2O3 is first loaded. Triple-layered catalytic systems enhance n-paraffin formation, mainly C17 and C18, which are considered significant as diesel components. The settlement in the triple-layered catalytic system inhibits dehydrocyclization of intermediaries and does not lead to the formation of aromatics.
Acknowledgments
The study was performed at the Petroleum-Gas University of Ploiesti, Romania. The authors are gratefully acknowledging the help of the National Institute for Research Development for Chemistry and Petrochemistry-ICECHIM Bucharest for the SEM and BET measurements.