

1 Introduction
Catalytic hydrogenation of unsaturated compounds is indeed very important for organic synthesis as well as chemical and pharmaceutical industry [1,2], and homogeneous hydrogenation catalyzed by bifunctional metal-ligand–cooperated catalysts is one of the most promising pathways [3–25]. Pioneering studies of bifunctional catalysts were reported by Shvo and co-workers and Noyori and co-workers [4,5,26–30]. Noyori's theoretical study on the highly efficient bifunctional chiral RuII amido catalyst revealed that hydrogenation of unsaturated functional groups CX (XO, N) takes place by metal–ligand cooperation in an outer sphere manner via a six-membered transition state [3–5].
On the basis of metals and ligands, there are two types of bifunctional catalysts [31]. One undergoes the aromatization and dearomatization interconversion, such as the Shov catalysts via OH bond and the Milstein catalysts via CH bond [26,27,32,33]. Another one involves the metal–ligand cooperation, such as the Noyori, Morris, and Beller catalysts via NH bond [3–5,34,35]. Among these catalysts, the pincer type complexes by Milstein and Beller have attracted much attention because such tridentate ligands can cover much of the coordination sphere and offer control over the vacant coordination sites with enhanced activity and stability [6].
Because the catalytic properties of a catalyst are generally determined by its electronic structure, design and screen of new catalysts are an integrated molecular approach [4,36]. The nature of noninnocent pincer ligands facilitating the hydride and proton transfer in the secondary sphere manner allows for fine-tuning electronic structure of metal individually without significant change in the coordination geometry [23,24,37]. Several pincer-type complexes on the basis of metal substitution have been prepared and applied for the hydrogenation of nitriles, alkynes, alkenes, aldehydes, ketones, and esters as well as transfer hydrogenation [6–25]. Examples are the complexes of Mo and W [19,20,24], Mn [17,18], Fe [15,16], Ru [35,38], Os [21,22], Co [39,40], and Ir [23]. As metal screening and mechanism elucidation are limited by considerable amount of experimental work, electronic structure calculations are playing an important role in the design of new catalysts [23,36,37] and excellent pioneer work has been done [41–44]. There are also recent corresponding theoretical studies about the detailed mechanisms of hydrogenation and dehydrogenation as well as transfer hydrogenation [38,43,45–51].
In our previous work, we explored the hydrogenation of CH3CN, Ph-CO2CH3, and Ph-CHO by using the d8-MII PNP pincer complexes (M = Fe, Ru, and Os). For the d5 metals, to the best of our knowledge, only the [NbIVCl3(N(CH2CH2PiPr2)2)] and [NbIIICl2(N(CH2CH2PiPr2)2)] complexes were prepared [52], but neither testing nor characterization of their catalytic activity was reported experimentally and theoretically. Herein, we explored the hydrogenation of phenyl-substituted CN, CN, CC, CC, and CO functional groups by d5-MI PNP pincer complexes by means of density functional theory computation.
2 Computational details
2.1 Method
In our previous work, we found that B3PW91 gas phase calculations give results in excellent agreement with the experimentally observed stability and reactivity of the amido (M-PNP) and amino (H-M-PNHP) complexes (M = Fe, Ru, Os, Ir, Mn, Mo, and W); the reaction energies as calculated from other methods, which include solvation effects, do not account for the possible equilibrium under the reaction conditions [23,38,45,53]. Because there are no experimental results available to validate the computational methods for these d5 metal complexes, we used the gas phase B3PW91 method in our calculations by using the Gaussian 09 program [54]. All structures were optimized at the B3PW91 [55] level with the TZVP [56] basis set (LANL2DZ [57] for metals). All optimized structures were characterized either as energy minimums without imaginary frequencies or transition states with only one imaginary mode by frequency calculations, and the imaginary model connects the initial and the final states. The thermal correction to Gibbs free energy at 298 K from the frequency analysis was added to the total electronic energy. Natural atomic orbital and natural bond orbital analysis were carried out on the B3PW91 optimized structures with the natural bond orbital method [58,59]. The computed energetic data and Cartesian coordinates are listed in the Supplementary data.
2.2 Model
To establish the formal 18 valence electron complexes of the amido (M-PNP) complex (1M) and amino (H-M-PNHP) complex (2M), two NO ligands are coordinated to the metal center along with the PNP ligand (Scheme 1). Benchmark calculations show that the amido complex (1M) in C1 symmetry due to different orientations of the isopropyl groups is more stable than the symmetrical isomer (Cs), and the amino (H-M-PNHP) complex (2M) in Cs symmetry is more stable than the C1 isomer (Table S1). Therefore, we used these more stable isomers for our energetic discussion and comparison (Scheme 1).
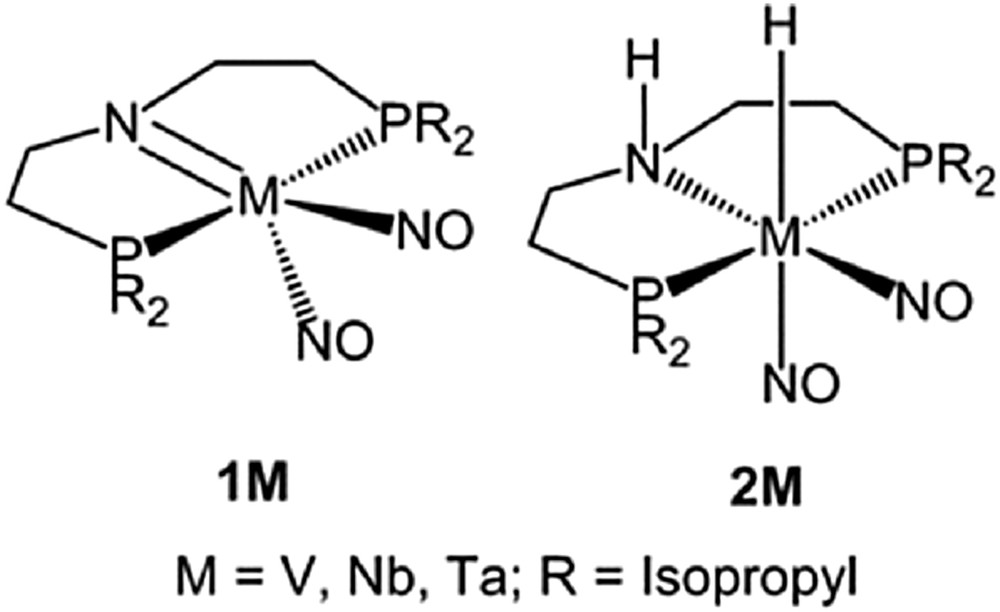
Amido (1M) and amino (2M) complexes.
3 Results and discussion
3.1 Bonding of M-PNP (1M) and H-M-PNHP (2M)
The B3PW91 computed bond distances of the distorted trigonal-bipyramid 1M and distorted octahedral 2M are given in Table S2. It shows that the M−N, M−P and M−NO bond lengths increase from V to Nb and then decrease to Ta, and this can be ascribed to the hybridization of metal and ligands [60]. Compared with the M−N distance in 1M, the M−N distance in 2M is elongated by 13.8, 11.9, and 12.3% for 2V, 2Nb, and 2Ta, respectively, revealing extra bonding interaction in 1M. In 2M, the NH bond length changes hardly, and the M−H bond length varies strongly, for example, the VH bond (1.750 Å) is much shorter than the NbH (1.928 Å) and TaH (1.912 Å) bonds. In addition to these bond distances, we computed the NO vibrational frequencies. On the basis of frequency calculations, the symmetrical and asymmetrical NO stretching frequencies are 1605/1548, 1589/1515, and 1570/1500 cm−1 for 1V, 1Nb, and 1Ta, respectively, as well as 1628/1532, 1594/1510, and 1576/1492 cm−1 for 2V, 2Nb, and 2Ta, respectively.
In addition, natural localized molecular orbital analysis reveals an MN double bond (one σ bond and one π bond) in 1Nb and 1Ta, and an MN single bond in 2Nb and 2Ta (Table S3). The computed MN Wiberg bond index in 1Nb and 1Ta is about double of that in 2Nb and 2Ta (Table S4), respectively. For the MNPNP σ bond in 1Nb and 1Ta, the NPNP atom is sp hybridized and contributes more strongly to the MNPNP σ bond than the metal atom, and for the MNPNP π bond, the NPNP atom has pure p character and has stronger contribution to the bond than the metal atom. The Nb atom in 1Nb is slightly negatively charged, whereas the Ta atom in 1Ta is slightly positively charged. In 2Nb and 2Ta, the metal atom is much more negatively charged, and the NPNP atom is sp3 hybridized and contributes more strongly to the MNPNP σ bond than the metal atom. All these show the polarization of the NM and NM bonds. In 1Nb and 1Ta, the MNO bond also has double bond character (one σ bond and one π bond), and the metal atom contributes more strongly than that of the N atom of the NO ligand.
However, different binding properties in 1V and 2V are found. In 1V, there is no σ bond between the V atom and the N atom of the PNP ligand, but there is a perpendicular π bond, mainly contributed by the N atom. Furthermore, there is a VN triple bond between the V atom and the N atom of the NO ligand, and the V metal atom contributes more strongly than the N atom to the σ bond and the two π bonds. In 2V, there is also no σ bond between the V atom and the N atom of the PNP ligand, but a VN triple bond between the V atom and the N atom is found, and the V metal atom contributes more strongly than that of the N atom to the σ bond and the two π bonds. In 1V and 2V, the V atom is more negatively charged than the N atom of the PNP ligand.
Despite these bonding differences, it is noted that in 2M the hydrogen atom (HM) to metal is negatively charged, whereas the hydrogen atom (HN) to the nitrogen atom is positively charged. This might fulfill the criteria of bifunctional hydrogenation catalysts. Therefore, we are interested in their stability and hydrogenation performances.
3.2 Catalysts interconversion (1M + H2 = 2M)
At first, we calculated the catalyst interconversion between 1M and 2M by heterolytic H2 addition [24,45]. The full potential energy surface is shown in the Supplementary data (Scheme S1). For the heterolytic H2 addition, we located a concerted transition state in Cs symmetry. In the transition state of 2V, 2Nb, and 2Ta, the forming MH/NH distances are 1.914/1.508, 2.103/1.494, and 2.072/1.496 Å, and the breaking HH distances are 0.916, 0.934, and 0.937 Å, respectively. As shown on the simplified potential energy surface (Fig. 1), 1M to 2M interconversion has barrier of 102.5, 94.8, and 99.4 kJ/mol as well as is endergonic by 28.5, 30.4, and 28.7 kJ/mol for 2V, 2Nb, and 2Ta formation, respectively. The endergonic property indicates that the heterolytic H2 addition is less favored kinetically and thermodynamically, in turn, the heterolytic H2 elimination is more favored. Thus, 2M can be only stable under high H2 pressure and removal H2 from the system should shift 2M back to 1M. This is very important for reactions with even higher barriers, because reactions with higher barriers can be kinetically hindered, although they are favorable thermodynamically (Fig. 1).

Potential energy surface for M-PNP (1M) and H-M-PNHP (2M) interconversion.
3.3 Hydrogenation of benzonitrile and imine
For Ph-CN hydrogenation to phenylmethanimine (Ph-CHNH), a two-step asynchronous ionic mechanism [61] for the hydride (HM) and proton (HN) transfer was identified. The first transition state (TS-HM) is the HM transfer to the carbon atom of CN, resulting in the formation of the ionic intermediate (PhCHN−, 2M-int). In TS-HM, the breaking MH distances are 1.822, 1.983, and 1.976 Å, and the forming CH distances are 1.726, 1.741, and 1.664 Å, for 2V, 2Nb, and 2Ta, respectively. The barriers of HM transfer are 74.8, 66.1, and 68.9 kJ/mol for 2V, 2Nb, and 2Ta, respectively, and the formation of 2M-int is endergonic by 64.4, 55.2, and 63.7 kJ/mol for 2V, 2Nb, and 2Ta, respectively. The second transition state (TS-HN) is the HN transfer from NPNP to NPhCN, and the breaking/forming NH distances are 1.214/1.382, 1.211/1.392, and 1.203/1.400 Å for 2V, 2Nb, and 2Ta, respectively. Relative to 2M and Ph-CN, the barriers of HN transfer are 59.5, 50.2, and 59.6 kJ/mol for 2V, 2Nb, and 2Ta, respectively. On the simplified potential energy surface (Fig. 2), TS-HN is more stable than M-int by 4.9, 5.0, and 4.1 kJ/mol for 2V, 2Nb, and 2Ta, respectively. The whole hydrogenation is exergonic by 45.3, 47.2, and 45.5 kJ/mol for 2V, 2Nb, and 2Ta, respectively. Starting from the amido complex (1M), the apparent free energy barriers for the hydrogenation of Ph-CN are 103.3, 96.5, and 97.7 kJ/mol for 1V, 1Nb, and 1Ta, respectively (Fig. 2).

Potential energy surface of benzonitrile and phenylmethanimine hydrogenation.
For Ph-CHNH hydrogenation to phenylmethanamine (Ph-CH2NH2), a one-step mechanism is identified. This step has barrier of 99.5, 89.2, and 93.4 kJ/mol as well as is exergonic by 76.8, 78.7, and 77.1 kJ/mol for 2V, 2Nb, and 2Ta, respectively. In the transition state for 2V, 2Nb, and 2Ta, the breaking MH/NH distances are 1.865/1.056, 2.023/1.056, and 2.018/1.064 Å, respectively, and the forming CH/NH distances are 1.586/1.770, 1.604/1.776, and 1.545/1.735 Å, respectively. The transition state corresponds mainly to HM transfer and followed by the subsequent HN transfer without energy barrier. Compared to Ph-CN hydrogenation, Ph-CHNH hydrogenation has higher barrier by 24.7, 23.1, and 24.5 kJ/mol for 2V, 2Nb, and 2Ta, respectively, and Ph-CHNH hydrogenation should be more difficult than Ph-CN hydrogenation. Starting from the amido complex (1M), the apparent free energy barriers for the hydrogenation of Ph-CHNH are 128.0, 119.6, and 122.1 kJ/mol for 1V, 1Nb, and 1Ta, respectively.
As a competitive reaction to Ph-CHNH hydrogenation, depending on the reaction kinetics, the formed Ph-CHNH and Ph-CH2NH2 can condense to Ph-CHNCH2-Ph by NH3 release. Therefore, we computed the hydrogenation of Ph-CHNCH2-Ph to Ph-CH2NHCH2-Ph and located one transition state for HM transfer with the subsequent barrier-less HN transfer. In the transition state for 2V, 2Nb, and 2Ta, the breaking M-H/NH distances are 1.897/1.051, 2.023/1.045, and 2.019/1.051 Å, respectively, and the forming CH/NH distances are 1.551/1.835, 1.668/1.904, and 1.599/1.862 Å, respectively. As shown in Fig. 3, the reaction has barrier of 141.4, 125.3, and 130.8 kJ/mol as well as is exergonic by 63.3, 65.2, and 63.5 kJ/mol for 2V, 2Nb, and 2Ta, respectively. The energy barriers of Ph-CHNCH2-Ph hydrogenation are 41.9, 36.2, and 37.5 kJ/mol higher than that of Ph-CHNH for 2V, 2Nb, and 2Ta, respectively. Starting from the amido complex (1M), the apparent free energy barriers for the hydrogenation of Ph-CHNCH2-Ph are 170.0, 155.7, and 159.6 kJ/mol for 1V, 1Nb, and 1Ta, respectively (Fig. 3).

Potential energy surface of PhCHNCH2Ph and PhCHNPh hydrogenation.
For comparison, we computed trans-Ph-CHN-Ph hydrogenation and found only one transition state corresponding mainly to HM transfer. In the transition state for 2V, 2Nb, and 2Ta, the breaking MH/NH distances are 1.860/1.036, 2.018/1.037, and 2.009/1.042 Å, respectively, and the forming CH/NH distances are 1.736/1.943, 1.780/1.947, and 1.723/1.903 Å, respectively. As shown in Fig. 3, trans-Ph-CHN-Ph hydrogenation has barrier of 131.8, 114.8, and 120.4 kJ/mol for 2V, 2Nb, and 2Ta, respectively, lower than that of Ph-CHNCH2-Ph hydrogenation by 9.7, 10.5, and 10.4 kJ/mol for 2V, 2Nb, and 2Ta, respectively. Starting from the amido complex (1M), the apparent free energy barrier for the hydrogenation of trans-Ph-CHN-Ph is 160.3, 145.2, and 149.2 kJ/mol for 1V, 1Nb, and 1Ta, respectively.
From Figs. 2 and 3, it is clearly seen that Ph-CN can be most easily hydrogenated to Ph-CHNH, whereas hydrogenation of imines needs higher barrier and is more exergonic. For the hydrogenation of imines, Ph-CHNH hydrogenation has the lowest barrier, followed by Ph-CHN-Ph hydrogenation, whereas Ph-CHNCH2-Ph hydrogenation has the highest barrier. In addition, the CN hydrogenation and the formation of 2M have very close barriers (102.5 kJ/mol for 2V formation vs 103.3 kJ/mol for CN hydrogenation, 94.8 kJ/mol for 2Nb formation vs 96.5 kJ/mol for CN hydrogenation, and 99.4 kJ/mol for 2Ta formation vs 97.7 kJ/mol for CN hydrogenation). For CN hydrogenation, the second step of CN hydrogenation is the rate-determining step for 2V, 2Nb, and 2Ta. Starting from the amido complex (1M), the apparent barriers of the hydrogenation of Ph-CN, Ph-CHNH, and Ph-CHN-Ph are much higher than the barrier of H2 elimination from 2M to 1M, high H2 pressure is needed for the stability of 2M and the effective hydrogenation.
3.4 Hydrogenation of phenylacetylene and styrene
Apart from hydrogenation of Ph-CN, we computed the sequential hydrogenation of phenylacetylene (Ph-CCH). The full potential energy surface is shown in the Supplementary data (Scheme S6) and the simplified one is used for discussion and comparison (Fig. 4). In contrast to Ph-CN, there are two C atoms in Ph-CCH and therefore two possibilities for HM transfer. For the HM transfer to CH and the HN transfer to Ph-C, we located one-step transition state, and the barrier is 84.6, 84.8, and 92.0 kJ/mol for 2V, 2Nb, and 2Ta, respectively. Starting from the amido complex (1M), the apparent free energy barrier for the hydrogenation of Ph-CCH is 113.1, 115.2, and 120.8 kJ/mol for 1V, 1Nb, and 1Ta. In the transition state for 2V, 2Nb, and 2Ta, the breaking MH/NH distances are 1.825/1.024, 2.004/1.025, and 2.000/1.027 Å, respectively, and the forming CH/NH distances are 1.669/2.346, 1.661/2.359, and 1.602/2.302 Å, respectively. For HM transfer to Ph-C and HN transfer to CH, alternatively, we located a two-step asynchronous ionic mechanism with two transition states (Scheme S7). For the first transition state, the barriers of HM transfer are 117.6, 111.1, and 114.0 kJ/mol for 2V, 2Nb, and 2Ta, respectively. For the second transition state, the barriers of HN transfer are 116.0, 111.6, and 120.9 kJ/mol for 2V, 2Nb, and 2Ta, respectively. It clearly shows that HM transfer to CH and HN transfer to Ph-C are more favored kinetically than HM transfer to Ph-C and HN transfer to CH by 33.0, 26.3, and 22.0 kJ/mol for 2V, 2Nb, and 2Ta, respectively. Starting from the amido complex (1M), the apparent free energy barriers for the hydrogenation of Ph-CCH are 103.1, 115.2, and 120.7 kJ/mol for 1V, 1Nb, and 1Ta, respectively (Fig. 4).

Potential energy surface of phenylacetylene and styrene hydrogenation.
As the counterpart of Ph-CHNH, we computed styrene (Ph-CHCH2) hydrogenation into ethylbenzene (Ph-CH2CH3). In contrast to Ph-CHNH hydrogenation, we found a two-step ionic mechanism. The simplified potential energy surface is shown in Fig. 4, and the full potential energy surface is shown in the Supplementary data (Scheme S8). The first step is the HM transfer to the CH2 carbon (β-carbon) and the second step is HN transfer from NPNP to the Ph-CH carbon (α-carbon). In the first transition state (TS-HM), the breaking MH distances are 1.855, 2.033, and 2.030 Å, and the forming CH distances are 1.617, 1.613, and 1.556 Å, for 2V, 2Nb, and 2Ta, respectively. In the second transition state (TS-HN), the breaking/forming NH distances are 1.196/1.558, 1.178/1.597, and 1.158/1.634 Å for 2V, 2Nb, and 2Ta, respectively. As shown in Fig. 4, the barriers of HM transfer to the CH2 carbon are 103.8, 102.3, and 105.9 kJ/mol as well as the barriers of HN transfer are 87.2, 80.6, and 95.2 kJ/mol for 2V, 2Nb, and 2Ta, respectively. For Ph-CHCH2 hydrogenation, the reaction is exergonic by 111.5, 113.4, and 111.7 kJ/mol for 2V, 2Nb, and 2Ta, respectively. Starting from the amido complex (1M), the apparent free energy barriers for the hydrogenation of Ph-CHCH2 are 132.3, 132.7, and 134.6 kJ/mol for 1V, 1Nb, and 1Ta, respectively.
Fig. 4 shows clearly that Ph-CCH hydrogenation has lower barrier and is more exergonic than Ph-CHCH2 hydrogenation. In addition, CC hydrogenation is the rate-determining step. On the basis of the whole potential energy surface, the hydrogenation of CC and CC bonds is the rate-determining step for metals of V, Nb, and Ta. Because the apparent barriers of the hydrogenation of Ph-CCH, Ph-CHCH2 starting from the amido complex (1M) are much higher than the barrier of H2 elimination from 2M to 1M, high H2 pressure is needed to enhance the stability of 2M for the hydrogenation reactions.
3.5 Hydrogenation of benzaldehyde and acetophenone
For benzaldehyde (Ph-CHO) hydrogenation, a two-step asynchronous ionic mechanism is identified (Fig. 5). The full potential energy surfaces are shown in the Supplementary data (Scheme S9). Initially, a transition state for HM transfer to the carbon atom of CHO corresponding to the MH bond breaking and the CH bond forming was located (TS-HM). HM transfer results in the formation of the ionic intermediate (Ph-CH2O−, M-int). Subsequently, a transition state for HN transfer (TS-HN) corresponding to NPNPH bond breaking and H bond forming is located. In the first transition state (TS-HM), the breaking M-H distances are 1.825, 1.983, and 1.972 Å, and the forming CH distances are 1.842, 1.902, and 1.819 Å for 2V, 2Nb, and 2Ta, respectively. In the second transition state (TS-HN), the breaking/forming NH distances are 1.246/1.211, 1.182/1.328, and 1.085/1.542 Å for 2V, 2Nb, and 2Ta, respectively. As shown in Fig. 5, the barriers of HM transfer are 62.9, 50.8, and 55.0 kJ/mol for 2V, 2Nb, and 2Ta, respectively, and the formation of the M-int is endergonic by 28.5, 18.8, and 28.2 kJ/mol for 2V, 2Nb, and 2Ta, respectively. In addition, the barriers of HN transfer are 24.5, 14.7, and 43.7 kJ/mol, which is 38.3, 36.0, and 11.3 kJ/mol lower than that of HM transfer for 2V, 2Nb, and 2Ta, respectively. Starting from the amido complex (1M), the apparent free energy barriers for the hydrogenation of Ph-CHO are 91.4, 81.2, and 83.8 kJ/mol for 1V, 1Nb, and 1Ta, respectively (Fig. 5).

Potential energy surface of benzaldehyde and acetophenone hydrogenation.
For the hydrogenation of acetophenone (Ph-COCH3), we found a one-step mechanism. In the transition state for 2V, 2Nb, and 2Ta, the breaking MH/NH distances are 1.889/1.036, 2.031/1.035, and 2.016/1.038 Å, respectively, and the forming CH/NH distances are 1.770/1.780, 1.816/1.790, and 1.749/1.760 Å, respectively. The barrier is 104.8, 86.0, and 89.7 kJ/mol for 2V, 2Nb, and 2Ta, respectively (Scheme S10), and the reaction is exergonic by 34.7, 36.7, and 35.0 kJ/mol for 2V, 2Nb, and 2Ta, respectively. Most importantly, the barriers of benzaldehyde hydrogenation are lower than those of Ph-COCH3 hydrogenation, and this indicates that Ph-CHO hydrogenation is more favored kinetically than Ph-COCH3 hydrogenation. Furthermore, Ph-CHO hydrogenation is more favored thermodynamically than Ph-COCH3 hydrogenation. Compared with the barrier of H2 elimination from 2M to 1M, which is lower than that of acetophenone hydrogenation and higher than that of benzaldehyde hydrogenation, high H2 pressure is needed to enhance the stability of 2M for the hydrogenation. In addition, catalyst hydrogenation is the rate-determining step for Ph-CHO hydrogenation, whereas acetophenone hydrogenation is the rate-determining step. Starting from the amido complex (1M), the apparent free energy barriers for the hydrogenation of Ph-COCH3 are 133.3, 116.4, and 118.5 kJ/mol for 1V, 1Nb, and 1Ta. On the basis of the whole potential energy surface, 2M formation is the rate-determining step for CHO hydrogenation for V, Nb, and Ta, whereas C(CH3)O hydrogenation is the rate-determining step for V, Nb and Ta. Because the apparent barrier of the hydrogenation of Ph-CHO, Ph-COCH3 starting from the amido complex (1M) is much higher than the barrier of H2 elimination from 2M to 1M, high H2 pressure can enhance the stability of 2M for the hydrogenation reactions.
In addition, we compared the structures and stability of the Fe-amido and Fe-amino [Fe-PNP(CO)(H) and HFePNHP(CO)(H)] complexes as well as their hydrogenation for Ph-CN, Ph-CCH, and Ph-CHO with those of the d5 metal complexes (Table 1). For the stepwise reactions, we used the effective barriers. Starting from the amido complex (1M), the formation of the amino complex (2M) of the d5 metals (M = V, Nb, and Ta) needs higher barrier (102.5, 94.8, and 99.4 kJ/mol, respectively) and is stronger endergonic (28.5, 30.4, and 28.7 kJ/mol, respectively) than that of the corresponding Fe complexes (82.8 vs 2.4 kJ/mol). These reveal that the 2M complexes of the d5 metals are less stable than the corresponding Fe complex, and even higher H2 pressure is needed to maintain the stability of the d5 amino complexes. On the basis of the barrier of 2M dehydrogenation, the corresponding amino Fe complex should be more stable than 2V, 2Nb, and 2Ta under the same hydrogen atmosphere (Table 1).
Gibbs free energy barriers (ΔG≠, kJ/mol) and effective barriers (ΔG≠eff, kJ/mol) for 2M formation as well as Ph-CN, Ph-CCH, and Ph-CHO hydrogenation.
| Metals | 1M + H2 = 2M | Ph-CN+ H2 = Ph-CHNH | Ph-CCH + H2= Ph-CHCH2 | Ph-CHO + H2= Ph-CH2OH | |||
| ΔG≠ | ΔG≠ | ΔG≠eff | ΔG≠ | ΔG≠eff | ΔG≠ | ΔG≠eff | |
| V | 102.5 [74.0]a | 74.8 | 103.3 | 84.6 | 113.1 | 62.9 | 102.5 |
| Nb | 94.8 [64.4]a | 66.1 | 96.5 | 84.8 | 115.2 | 50.8 | 94.8 |
| Ta | 99.4 [70.7]a | 68.9 | 99.4 | 92.0 | 120.8 | 55.2 | 99.4 |
| Fe | 82.8 [80.4]a | 77.0 | 82.8 | 85.4 | 87.8 | 56.7 | 82.8 |
a The barrier of the reverse reaction is given in square bracket.
For the hydrogenation of Ph-CN, Ph-CCH, and Ph-CHO starting from the amino complexes (2M) of the d5 metals, the effective barriers of the hydrogenation of Ph-CN and Ph-CHO are higher than that of Ph-CCH hydrogenation by about 10 kJ/mol for 2V as well as by about 20 kJ/mol for 2Nb and 2Ta, indicating that Ph-CN and Ph-CHO can be more easily hydrogenated than Ph-CCH. The same trend is found for the corresponding Fe complex, although the effective barrier differences are 5 kJ/mol.
For the hydrogenation of Ph-CN and Ph-CHO, the effective barrier has the decreasing order of V > Ta > Nb > Fe. For the hydrogenation of Ph-CCH, the effective barrier has the decreasing order of Ta > Nb > V > Fe. This clearly shows that the Fe complexes are the most effective catalysts for the hydrogenation of Ph-CN, Ph-CCH, and Ph-CHO.
4 Conclusion
In this work, we computed the structures and stability of the amido (M-PNP) (1M) and amino (H-M-PNHP) (2M) complexes (M = V, Nb, and Ta) as well as their hydrogenation mechanisms for the phenyl-substituted CN, CN, CC, CC, and CO functional groups at the B3PW91 level of density functional theory.
On the basis of the computational results, we can make some predicative judgment about the hydrogenation of these V, Nb, and Ta PNP complexes. At first, the interconversion from 1M to 2M complex is via heterolytic addition of H2. The endergonic property reveals that 2M complexes can only be stable under H2 environment.
For all these hydrogenation reactions by using these d5 PNP complexes, we found the same reaction mechanisms, that is, the hydrogenation of Ph-CN, Ph-CHCH2, and Ph-CHO undergoes a two-step mechanism, where the first step is the MH transfer, followed by an ionic intermediate, and the second step is the NH transfer, whereas that of Ph-CCH, Ph-CHNH, Ph-CHNH-Ph, Ph-CHNCH2Ph, and Ph-COCH3 prefers a one-step mechanism, and only transition state of MH transfer could be located. The barrier has the increasing order of Ph-CN < Ph-CHNH < Ph-CHN-Ph < Ph-CHNCH2-Ph. The hydrogenation of CC and CN bonds has lower barrier than that of CC and CN bonds, respectively. In addition, the barrier of CC and CC bonds is lower than that of CN and CN bonds. Furthermore, Ph-COCH3 hydrogenation has higher barrier than Ph-CHO hydrogenation. Under high H2 pressure, all these d5 complexes which exhibit similar intrinsic energy barrier and higher effective energy barrier can be as effective catalysts for the hydrogenation reactions as the well-known corresponding Fe complexes. All of these provide the basis for experimental proofs.
Acknowledgments
This work was supported by the state of Mecklenburg-Vorpommern and the Leibniz Association (Leibniz Competition, SAW-2016-LIKAT-1).
Appendix A Supplementary data
The following is the supplementary data related to this article:
Tables listing test results of solvent effect, Relative energies of M-PNP (1M) and H-M-PNHP (2M), computed bond lengths, natural charge (δ/e), natural bond orbitals, atomic hybrid contributions and Wiberg bonding indexes of M-PNP (1M) and H-M-PNHP (2M). Figures showing potential energy surface for M-PNP and H-M-PNHP interconversion as well as Ph-C≡N, Ph-CH=NH, Ph-CH=NH-Ph, Ph-CH=N-CH2Ph, Ph-C≡CH, Ph-CH=CH2, Ph-CHO and Ph-CO-CH3 hydrogenation. Tables listing Cartesian coordinates for all optimized structures and the energetic data.