

The search for efficient anti-Alzheimer drugs is at the moment the most difficult challenge in drug discovery because all clinical trials with drug candidates failed in the recent years [1]. These failures are due to several factors, one of them being the lack of a really predictive animal model. In fact, Alzheimer's disease (AD) is an aging disease related to many epigenetic factors, rather than simple mono- or digenic factors [2,3]. Consequently, the use of transgenic mice as a model of human AD is questionable.
Accumulation of metal ions has been evoked as a key parameter in the initiation of AD. A possible role for contribution of aluminum neurotoxicity to AD has been a matter of debate. However, there is actually no convincing causal link between aluminum and AD initiation or progression, despite five decades of controversial studies on this issue [4,5]. Beside zinc and copper [6], iron ions have been suspected to be damaging since the early 1950s [7,8]. As a matter of fact, the postmortem analysis of amyloid plaques has revealed accumulation of two redox metals ions, namely copper and iron with concentrations higher by a factor of ca. 5.6 and 2.7 times, respectively, with respect to the concentrations observed in normal brains [9]. One can consider that disruption of metal homeostasis in the brain of patients with AD is at the origin of the redox stress responsible for neuron death. In fact, in the presence of endogenous reductants, metal-loaded amyloids should trigger the reduction of dioxygen and produce reactive oxygen species (ROS) involved in neuron death [10–12]. Consequently, the use of metal chelators to regenerate normal trafficking of redox metal ions has been reported in recent articles [13–16]. Studies of such metal ligands have been largely devoted to copper chelators [17], despite putative iron–amyloid complexes may also be involved in ROS production (in medically focused publications, a similar phenomenon is frequently described as involved in inflammation processes).
To evaluate the putative role of iron associated with amyloids in the catalytic reduction of molecular oxygen and to compare this activity to that of copper–amyloids complex, we decided to investigate the in vitro oxidation of ascorbate by iron(III) or copper(II) in the presence or in the absence of amyloids. In fact, oxidation of ascorbate is considered as an indirect evaluation of Cu(II) and Fe(III) species to reduce molecular oxygen and, therefore, to induce catalytic reduction of dioxygen, and ROS production [18].
The assays were carried out in saline Hepes buffer 100 mM, pH 7.4, and the ascorbate consumption was measured by monitoring UV absorbance at 265 nm, over a period of 30 min. Final concentrations were as follows: [Aβ1-16] = 20 μM (where present), [FeIII] or [CuII] = 20 μM, and [ascorbate] = 100 μM. This [metal]/[ascorbate] ratio allows to evidence either stoichiometric or catalytic oxidation of ascorbate. Results are depicted in Fig. 1.
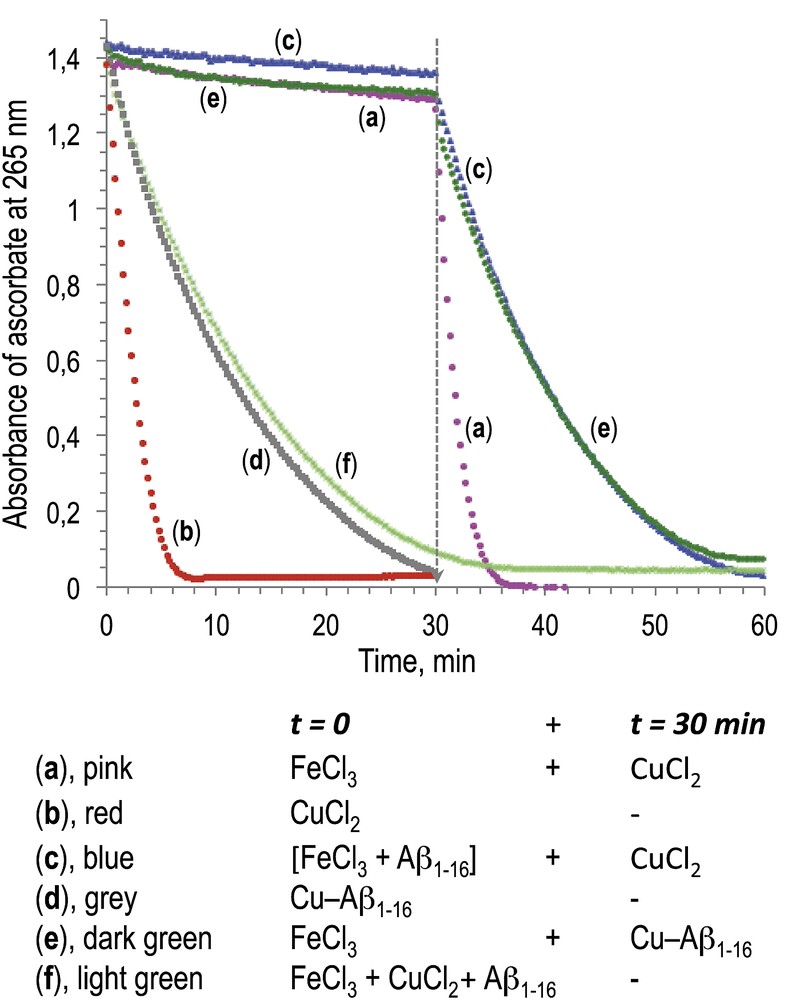
Oxidation of ascorbate measured by UV–visible absorbance at 265 nm. Final concentrations are [ascorbate] = 100 μM, [Aβ1-16] = 20 μM (where present), and [FeIII] or [CuII] = 20 μM.
First of all, FeCl3 alone was unable to efficiently oxidize ascorbate (only 8% after 30 min, trace a), whereas the oxidation of ascorbate was complete in the presence of CuCl2 (trace b) during the same interval of time. A similar trend was observed in the presence of Aβ1-16 associated with metal ions (M/Aβ1-16 molar ratio = 1/1): oxidation of ascorbate was only marginal in the presence of FeCl3/Aβ1-16 (5%, trace c), whereas it was complete after 30 min in the presence of CuCl2/Aβ1-16 (trace d). Consistently, it has been previously reported that Fe(NO3)3 associated with Aβ1-16 was 10 times less efficient than CuCl2/Aβ1-16 to oxidize ascorbate [19]. Moreover, after 30 min of incubation of FeCl3 or FeCl3/Aβ1-16, respectively, we added 1 mol equiv of CuCl2 (traces a and c, respectively, 30–60 min). In both cases, the oxidation of ascorbate occurred upon addition of CuCl2 with a similar kinetic as observed when CuCl2 was added at t = 0 (traces a compared to b, and c compared to d). When CuCl2/Aβ1-16 was added at 30 min in the mixture containing FeCl3 and ascorbate, the consumption of ascorbate was the same as that in the presence of Cu/Aβ1-16 alone (trace e compared to d). In addition, when FeCl3, CuCl2, and Aβ1-16 were incubated together, oxidation of ascorbate was the same as in the presence of Cu/Aβ1-16 alone (trace f compared to d).
These results clearly evidence a much lower reductive activation of dioxygen induced by Fe(III) associated with ascorbate as compared to Cu(II)/ascorbate, either in the absence or in the presence of Aβ1-16, under the same conditions, close to biological ones (aqueous medium pH 7.4, presence of chloride as potential counter ion) [20]. These results suggest that the deregulation of the homeostasis of copper ions in AD brains and accumulation of this redox ion in amyloids are more relevant in terms of oxidative damage created on neurons than the loading of iron ions on amyloids.
However, an indirect role of iron ions in AD cannot be ruled out, because the metabolism of iron is linked to proteins involved in AD. As an example, like the copper-carrier ceruloplasmin, the Amyloid Precursor Protein (APP) protein interacts with ferroportin, oxidizes Fe(II), and loads Fe(III) into transferrin, thus taking part in iron regulation [21].
In conclusion, the reported data indicate that the reductive activation of copper–amyloid complexes by a biological reductant is much more critical in terms of oxidative damage than that of the iron–amyloid complexes. Consequently, these results support the necessity to design specific copper chelators as potential drug candidates for the treatment of the early stages of AD, to avoid the continuous destruction of neurons by an oxidative stress created by the reductive activation of copper–amyloid complexes.
Acknowledgments
This work was supported by the CNRS and partially by GDUT (M.H., Y.L., and B.M.).
Vous devez vous connecter pour continuer.
S'authentifier