

1 Introduction
Pyrrolophenanthrolines have attracted attention in the past years mainly because of their biological [1–3], electrical [4–6], or optical [7] properties. Moreover, pyrrolo[1,2-a][1,10]phenanthrolines are compounds showing helical chirality similar to the helicene-type compounds [8].
Recently, our research group reported the synthesis of several pyrrolophenanthrolines showing antimycobacterial or anticancer properties [1,2]. Further to these results and taking into account the work we previously carried out in the field of pyrrolophenanthrolines (derived from 1,7-phenanthroline [1], 1,10-phenanthroline [4,5,9,10], and 4,7-phenanthroline [2,6]), this work envisaged the synthesis of new pyrrolophenanthroline derivatives bearing an amide group at the pyrrole ring. As the amide group is often found in pharmacologically active substances [11], we were interested to see how it will affect the biological properties of the newly synthesized compounds or simply to be used as a tool in further derivatization.
One of the most common strategies to obtain pyrroloheterocycles involves the 3 + 2 cycloaddition of cycloimmonium ylides to different dipolarophiles [12,13]. Therefore, we used a similar strategy in the synthesis of pyrrolophenanthroline derivatives by reacting 2-amino-2-oxoethyl-phenanthrolinium monoylides (derived from 1,7-, 4,7-, and 1,10-phenanthrolinium salts [14–16]) with activated alkynes and alkenes. Unexpectedly, in several reactions we obtained the desired pyrrolophenanthroline skeleton, but monosubstituted at the N-amide atom with an acrylate group. This occurred because the activated alkynes used in the synthesis (ethyl propiolate [EP] or dimethyl acetylenedicarboxylate [DMAD]) displayed both dipolarophile and N-amidation properties. Interestingly, in some reactions we have isolated other unexpected compounds, which will be further discussed.
2 Results and discussion
Our strategy was to initially study the reaction of 2-amino-2-oxoethyl-phenanthrolinium monoylides (derived from 1,7-, 4,7-, and 1,10-phenanthroline) with DMAD (used as symmetrically substituted dipolarophile) and EP (used as unsymmetrically substituted dipolarophile).
Thus, 1,7-phenanthrolinium ylide 1a, generated in situ (under treatment with triethylamine [TEA] as base) from the corresponding salt 1 [14], reacted with DMAD in a 3 + 2 dipolar cycloaddition reaction, leading to 8,9-dihydropyrrolo[1,2-i][1,7]phenanthroline derivative 2 in moderate yield (Schemes 1 and 2).
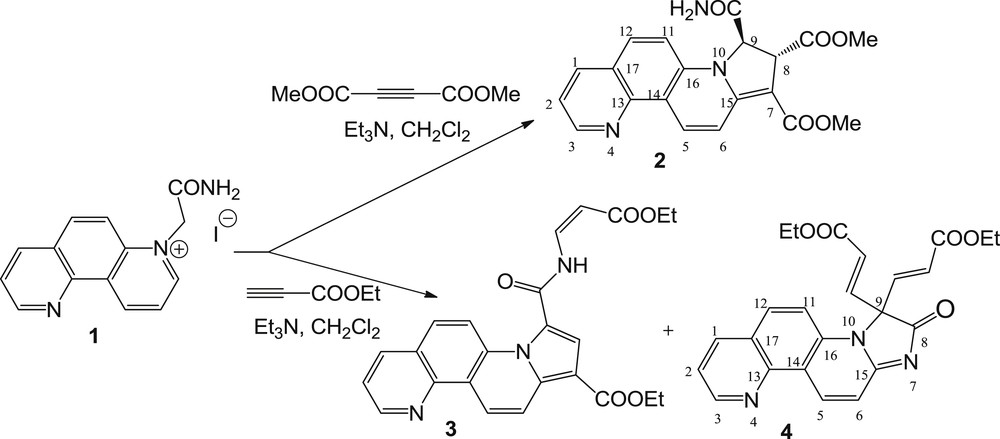
Reaction pathway of 2-amino-2-oxoethyl-[1,7]-phenanthrolinium ylide with DMAD and EP.
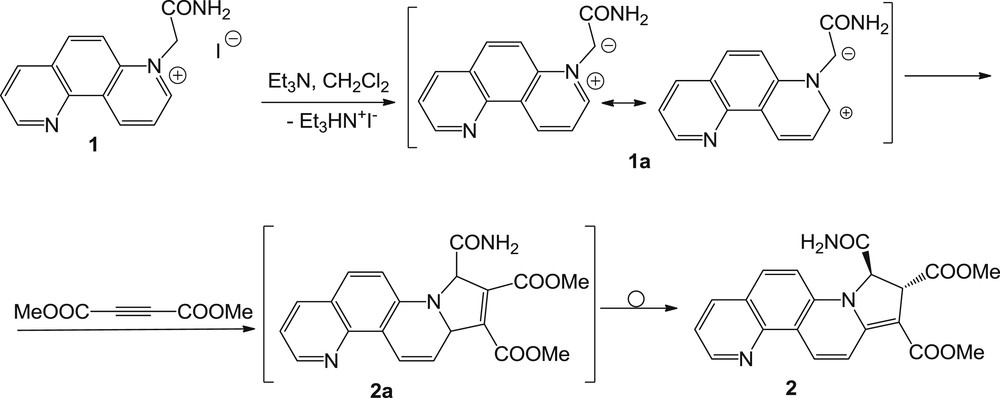
The mechanism proposed for the formation of adduct 2.
Interestingly, the reaction of 1,7-phenanthrolinium ylide 1a with EP occurred nonselective, leading to compound 3 as a major product. This product formed as a result of 3 + 2 dipolar cycloaddition and N-amidation, taking place simultaneously on the same substrate. Changing the conditions, in terms of molar ratio, temperature, and solvents, had no effect on the products formed, and the reaction occurred in the same manner, leading to fully aromatized cycloadduct 3 and the unexpected secondary compound 4 (Scheme 1). However, when the molar ratio salt 1/EP was changed from 1:1.1 (with the yields of 35% for 3 and 5% for 4) to 1:5, the yield in compound 3 increased to 73%, whereas the yield in compound 4 increased only slightly to 6%.
Regarding the mechanism for the synthesis of compound 2, we assume that after an initial 3 + 2 dipolar cycloaddition of ylide 1a to DMAD, the unisolable 9,15-dihydropyrrolo[1,2-i][1,7]phenanthroline intermediate 2a was generated and then in the presence of an excess of TEA, a stereo- and regioselective allylic prototropic rearrangement was induced, leading to compound 2, which is more thermodynamically stable (Scheme 2).
The structure including the position and the stereochemistry of the two protons on the dihydropyrrolo ring of compound 2 was established on the basis of the NMR data and by comparison with spectra of similar compounds obtained in our group [1].
The formation of pyrrolo[1,2-i][1,7]phenanthroline skeleton of cycloadduct 3 took place via a similar mechanism as shown in Scheme 2. An unstable intermediate similar to 1a was formed, which suffers an oxidative aromatization by losing two hydrogen atoms. The structure of compound 3 was also confirmed by NMR spectroscopy. Interestingly, compound 3 has a Z-enamide as substituent at position 9. Thus, in the 1H NMR spectrum, compound 3 showed a deshielded doublet at 11.36 ppm for the amidic proton, whereas the two alkene protons gave two signals, a doublet at 5.32 and a doublet of doublets at 7.79 ppm, with a common coupling constant of 9.0 Hz, confirming the Z-configuration at the level of CC double bond. This Z-form is stabilized by a hydrogen bond between NH and the oxygen of ester carbonyl function of ethyl acrylate moiety. This explains the Z-selectivity of the resulting enamide. The literature shows that similar Z-hydroamidation reaction of electron-deficient terminal alkynes by amides was only carried out in the presence of a Pd catalyst [17] and proved to be highly stereoselective (primary amides gave Z-enamide, whereas secondary amides gave E-enamides).
The unexpected (2E,2′E)-diethyl-3,3′-(2-oxo-2,3-dihydroimidazo[1,2-i][1,7]phenanthroline-3,3-diyl)diacrylate structure of compound 4 was fully accomplished using X-ray diffraction, as can be seen in Fig. 1. The terminal ethyl groups are disordered.
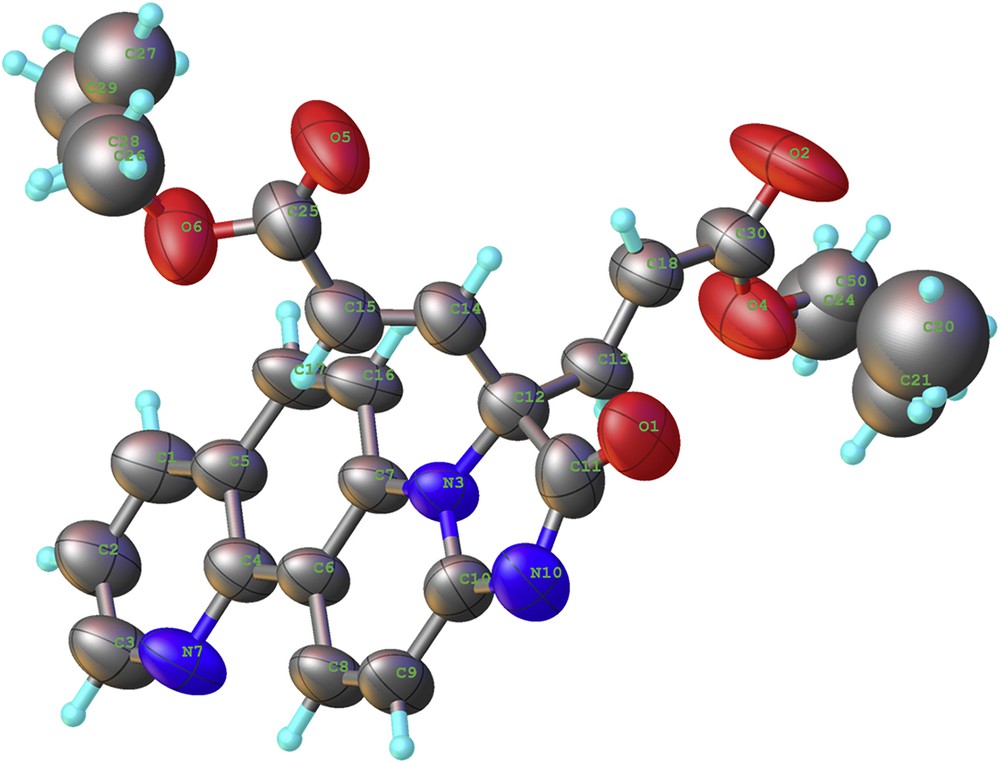
X-ray crystal structure of compound 4 with thermal ellipsoids at 50% probability level.
The 1H NMR spectrum of compound 4 also confirms the E-configuration at both CC double bonds of acrylate groups showing two doublets (J = 16.0 Hz) at 5.91 ppm and 7.27 ppm, respectively, for the alkene protons of two acrylate groups.
Regarding the mechanism for the formation of the unusual compound 4, it is very interesting to observe that EP has not acted as dipolarophile as usually does in this type of reactions but instead the cyclization assured by the amidic nitrogen occurred. The proposed mechanism for the formation of compound 4 is shown in Scheme 3.
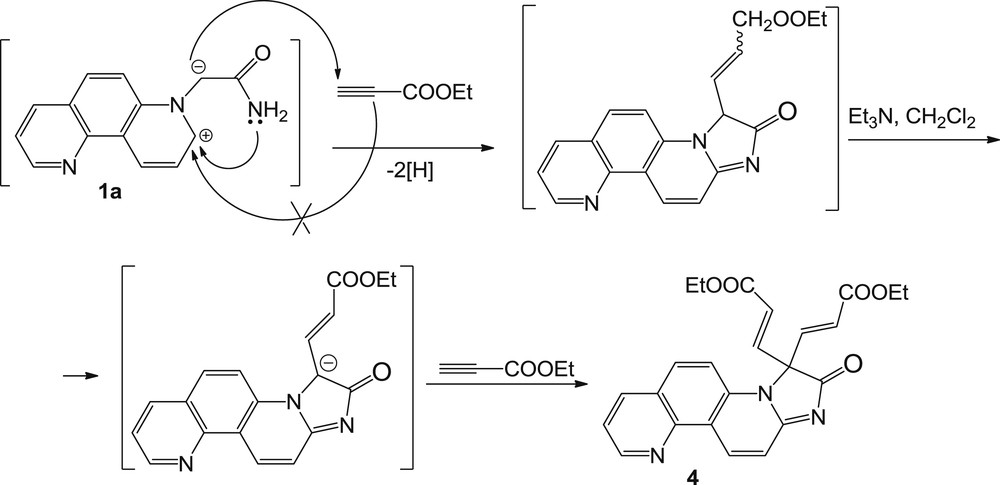
The mechanism proposed for the formation of adduct 4.
We continued our study using a similar 4,7-phenanthrolinium ylide as a substrate, using similar conditions to those in case of 1,7-phenanthrolinium ylide 1a, to see if this competition of cycloaddition versus amidation reactions has a general character in the series of 2-amino-2-oxoethyl-phenanthrolinium ylides.
The reactions of 4,7-phenanthrolinium ylide (generated in situ from the corresponding salt 5 [15], using TEA) with DMAD and EP occurred in the same manner, leading to 8,9-dihydropyrrolo[2,1-c][4,7]phenanthroline derivative 6 and the aromatized cycloadduct 7, having the amide group monosubstituted with an ethyl acrylate moiety (having Z-configuration at the CC double bond) (Scheme 4).
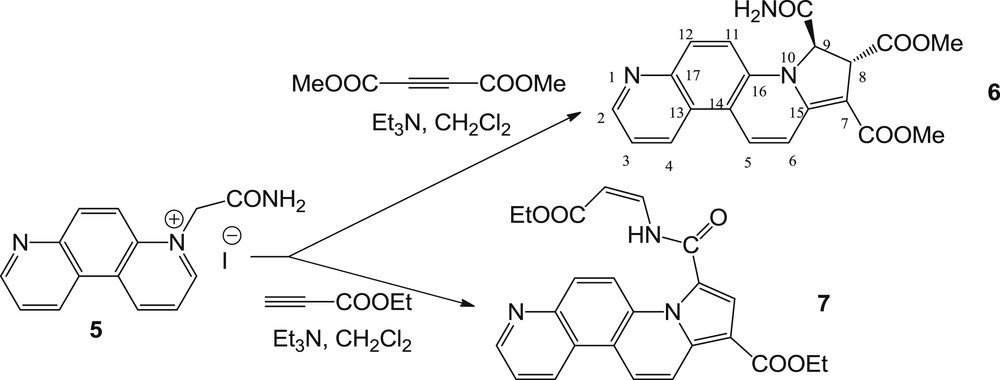
Reaction pathway of 2-amino-2-oxoethyl-[4,7]-phenanthrolinium ylide with DMAD and EP.
The study was then extended to the series of 1,10-phenanthrolinium ylides, which showed a different behavior. The reaction of the ylide corresponding to the salt 8 [16] with EP was leading to a very complex reaction mixture, from which it was impossible to isolate pure fractions. Similarly, the reaction of 1,10-phenanthrolinium ylides with DMAD led to a very complex reaction mixture. However, following laborious work, it was possible to separate one pure fraction, which proved to be compound 9 (Scheme 5). This compound with 10,11-dihydropyrrolo[1,2-a][1,10]phenanthroline structure was formed presumably from the intermediate 9a via a stereo- and regioselective allyl prototropic rearrangement, similarly to compound 2. As shown for compounds 3 and 7, compound 9 has also a Z-enamide substituent at position 11. The shifted signal (10.46 ppm) corresponding to the NH amidic hydrogen suggests an involving of that hydrogen atom in a hydrogen bond.
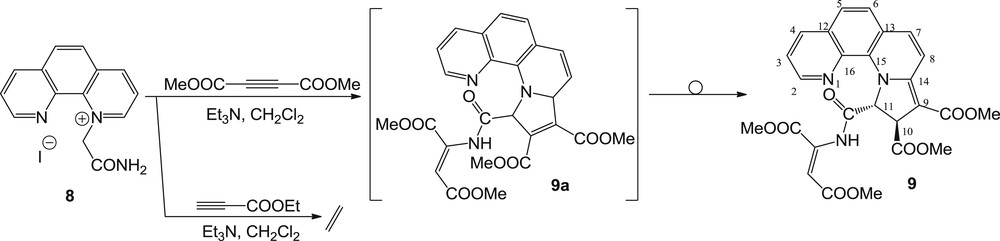
Reaction pathway of 2-amino-2-oxoethyl-[1,10]-phenanthrolinium ylide with DMAD and EP.
X-ray analysis helped elucidating the structure of compound 9 as (Z)-dimethyl 11-((1,4-dimethoxy-1,4-dioxobut-2-en-2-yl)carbamoyl)-10,11-dihydropyrrolo[1,2-a][1,10]phenanthroline-9,10-dicarboxylate (Fig. 2). The Z isomer is indeed stabilized by the intramolecular H-bond, which exists between HN-19 and O-25.
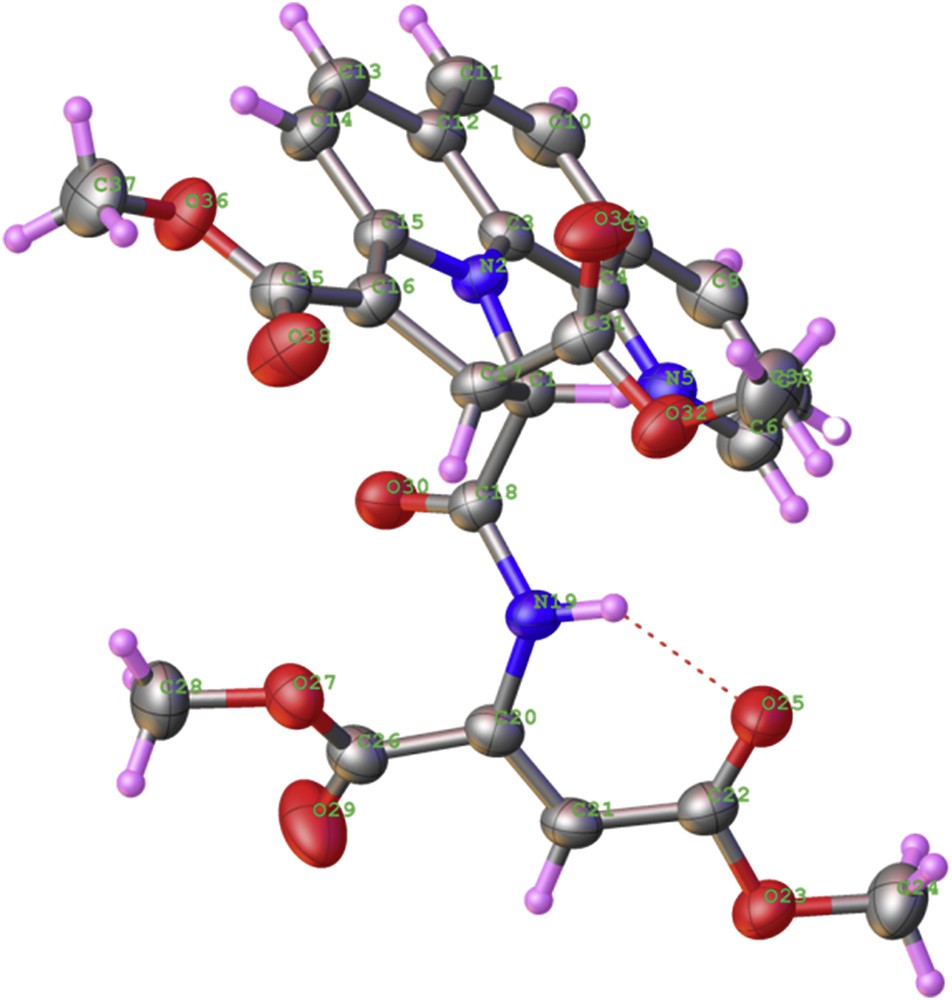
X-ray crystal structure of compound 9 (C26H23N3O9) with thermal ellipsoids at 50% probability level.
Three types of π–π interactions exist in the structure of the compound 9. The centroid to centroid distances are 3.821, 3.750, and 3.823 Å (Fig. 3).
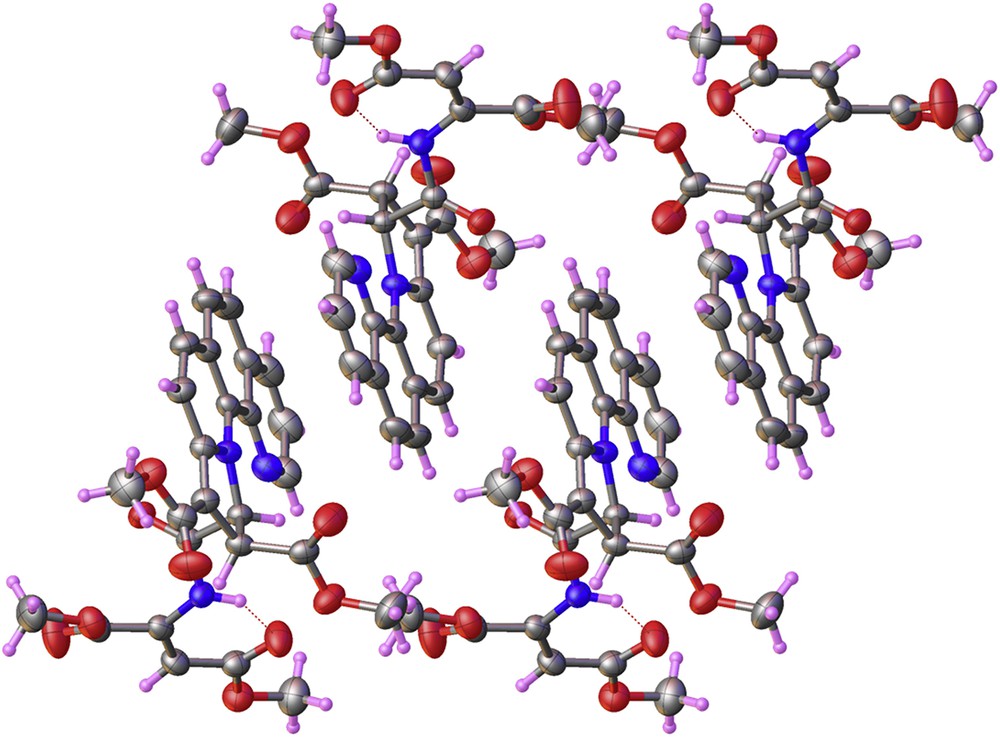
View of supramolecular architecture in the crystal structure of compound 9.
We then studied the reactions of 2-amino-2-oxoethyl-phenanthrolinium monoylides (derived from 1,7-, 4,7-, and 1,10-phenanthroline) with activated alkenes as dipolarophiles, to see if a similar behavior as in the alkyne series will occur. Acrylonitrile was used as an electron-deficient terminal alkene. Regardless of the conditions used in these reactions, amidation reaction did not occur, instead Huisgen 3 + 2 dipolar cycloadditions took place. Thus, the reactions of 1,7- and 4,7-phenanthrolinium ylides with acrylonitrile resulted in dihydropyrrolo derivatives 10 and 11, respectively (Scheme 6). As previously encountered, the formation of dihydropyrrolo cycloadducts could be explained by an oxidative dehydrogenation under the reaction conditions of the unstable intermediate 10a and 11a, respectively (Scheme 6).
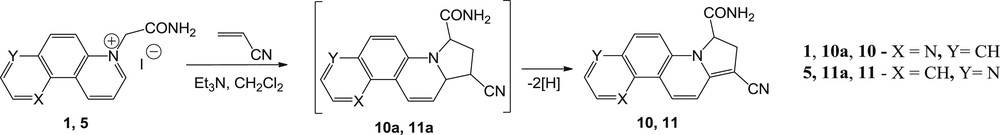
Reaction pathway of 1,7- and 4,7-phenanthrolinium ylides with acrylonitrile.
1,10-Phenanthrolinium ylide generated from salt 8 under similar conditions proved to be once more a very tough substrate in this type of reaction, leading to a complicated reaction mixture. The only pure fraction possible to isolate was the unexpected 4-cyano-4-(1,10-phenanthrolin-2-yl) butanoic acid 12 (Scheme 7).
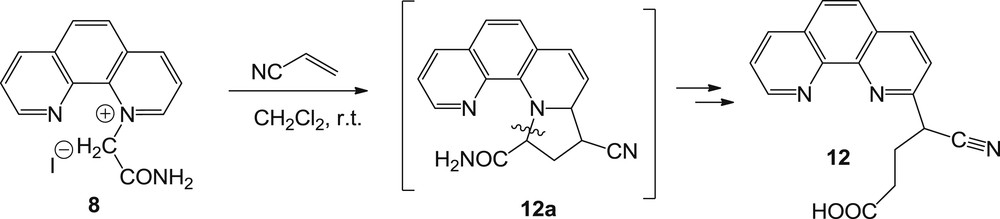
Reaction pathway of 1,10-phenanthrolinium ylides with acrylonitrile.
The formation of compound 12 could be explained by an initial 3 + 2 cycloaddition that led to the tetrahydropyrrolo intermediate 12a, which then has undertaken a ring-opening reaction followed by a hydrolysis of the amide group (that occurs probably under the given reaction conditions or during the column chromatography).
3 Conclusions
An unusual behavior in cycloaddition reactions was found for 2-amino-2-oxoethyl-phenanthrolinium ylides in some 3 + 2 cycloaddition reactions to electron-deficient terminal activated alkynes. The terminal alkyne was proved to be reactive enough to act as both dipolarophile and N-amidation reagent. Thus, in many cases the amide group of the substrates suffered subsequent enamidation with the alkynes used as dipolarophiles in 3 + 2 cycloaddition reactions. The reaction is stereoselective: monoenamidation leading to a Z-conformation at the level of an acrylate double bond. The Z-conformation of the monosubstituted amides can be explained via stabilization given by an intramolecular H-bond of the NH proton and the carbonyl oxygen atom of ethyl acrylate moiety. In similar conditions, electron-deficient terminal activated alkenes gave only 3 + 2 cycloadditions without being attacked by the amide groups.
4 Experimental section
4.1 Chemistry
Melting points were recorded using an A. Krüss Optronic Melting Point Meter KSPI and are uncorrected. Proton, carbon, and bidimensional (COSY, HMQC, HMBC) NMR spectra were recorded using a Bruker Avance III 500 MHz spectrometer. All chemical shifts are quoted on the δ-scale in ppm. Coupling constants are given in Hz. IR spectra were recorded using an FTIR Shimadzu or Jasco 660 plus FTIR spectrophotometer. Thin-layer chromatography was carried out on Merck silica gel 60F254 plates. Column chromatography was carried out on silica gel (Roth 60, 0.04–0.063 mm). Visualization of the plates was achieved using a UV lamp (λmax = 254 or 365 nm). All commercially available products were used without further purification unless otherwise specified.
4.2 X-ray crystallography
Crystallographic measurements for compounds C24H21N3O5 and C26H23N3O9 were carried out using a SuperNova, Dual, Cu at zero, Eos diffractometer. The crystals were kept at 294 K during data collection. The unit cell determination and data integration were carried out using the CrysAlisPro package of Oxford Diffraction. Using Olex2 [18], the structures were solved with the Superflip [19] structure solution program using Charge Flipping and refined with the ShelXL [20] refinement package using least-squares minimization.
The main crystallographic data together with refinement details are summarized in Table 1. CCDC 1577836 and 1538093 contain the full crystallographic data. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Crystal data and structure refinement for compound 4 and 9.
| Compound | 4 | 9 |
| Empirical formula | C24H21N3O5 | C26H23N3O9 |
| Formula weight | 431.44 | 521.47 |
| Temperature/K | 293(2) | 293(2) |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21/c | P-1 |
| a/Å | 9.1475(11) | 8.7626(4) |
| b/Å | 24.477(2) | 9.7291(6) |
| c/Å | 10.1642(15) | 15.3392(6) |
| α/° | 90 | 92.145(4) |
| β/° | 109.132(13) | 104.536(4) |
| γ/° | 90 | 105.764(5) |
| V/Å3 | 2150.1(5) | 1210.39(11) |
| Z | 4 | 2 |
| Dcalc/mg/mm3 | 1.333 | 1.431 |
| μ/mm−1 | 0.783 | 0.927 |
| Crystal size/mm3 | 0.05 × 0.02 × 0.02 | 0.50 × 0.22 × 0.20 |
| θmin, θ max(o) | 10.23–133.19 | 2.99–70.72 |
| Reflections collected | 7457 | 7775 |
| Independent reflections | 3760[R(int) = 0.0840] | 4520[R(int) = 0.0220] |
| Data/restraints/parameters | 3760/4/291 | 4520/0/347 |
| R1a(I > 2σ(I)) | 0.0816 | 0.0477 |
| wR2b (all data) | 0.2379 | 0.1378 |
| GOFc | 0.955 | 1.033 |
| Largest diff. peak/hole/e Å−3 | 0.23/−0.32 | 0.34/−0.28 |
a R1 = Σ||Fo| − |Fc||/Σ|Fo|.
b wR2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2.
c GOF = {Σ[w(Fo2 − Fc2)2]/(n − p)}1/2, where n is the number of reflections and p is the total number of parameters refined.
General procedure for the synthesis of compounds 2, 3, 4, 6, 7, 9, 10, 11, and 12
All three monoquaternary phenanthrolinium iodides we used as precursors of the N-ylides were prepared following our reported procedures [14–16].
The cycloimmonium salt (1 mmol, 1 equiv) and dipolarophile (DMAD, EP, or acrylonitrile (1.1 mmol, 1.1 equiv); in some of the reactions the quantity of dipolarophile was increased up to 5 equiv) were added to 5 mL of dichloromethane and the obtained suspension was stirred at room temperature (rt) under N2 atmosphere. TEA (3 mmol, 3 equiv) was added dropwise over a period of 1 h under magnetic stirring, and the resulting mixture was then stirred for 24 h at rt or reflux. Methanol (5 mL) was added and the resulting mixture was kept for 24 h without stirring. When a precipitate was formed, it was collected by filtration as a powder and was washed with a few milliliters of methanol. The product was then crystallized from an appropriate solvent or purified by column chromatography. The reactions that did not form a precipitate, the reaction mixture was separated using column chromatography using a mixture of solvents as indicated for each of the compounds.
Dimethyl-9-carbamoyl-8,9-dihydropyrrolo[1,2-i][1,7]phenanthroline-7,8-dicarboxylate (2)
Obtained by general procedure, at rt. Crystallized from methanol–chloroform 1:1 (v/v). Yield: 91%, orange powder, mp = 223–225 °C. IR (KBr), ν(cm−1): 3286, 1735, 1686, 1655, 1620, 1219, 1155, 1116, 1072, 944. 1H NMR (500 MHz, DMSO-d6): 3.60 (s, 3H, H-19), 3.69 (s, 3H, H-21), 3.85 (d, J = 4.0 Hz, 1H, H-8), 5.35 (d, J = 4.0 Hz, 1H, H-9), 7.18 (d, J = 9.0 Hz, 1H, H-5), 7.48 (dd, J = 8.0, 4.5 Hz, 1H, H-2), 7.54 (s, 1H, NH), 7.60 (d, J = 9.5 Hz, 1H, H-12), 7.90 (s, 1H, NH), 8.03 (d, J = 9.5 Hz, 1H, H-6), 8.30 (ad, J = 8.0 Hz, 1H, H-1), 8.67 (d, J = 9.5 Hz, 1H, H-11), 8.92 (ad, J = 4.0 Hz, 1H, H-3). 13C NMR (125 MHz): 49.9C-19, 50.5C-8, 52.5C-21, 64.8C-9, 88.9C-7, 113.9C-5, 116.7C-14, 117.0C-12, 120.4C-2, 122.8C-17, 131.6C-6, 131.9C-11, 136.5C-1, 139.2C-16, 143.9C-13, 151.2C-3, 152.4C-15, 164.5C-18, 169.4C-22, 172.5C-20. Anal. Calcd. for C20H17N3O5: C, 63.32; H, 4.52; N, 11.08. Found: C, 63.39; H, 4.47; N, 11.07.
(Z)-Ethyl-9-((3-ethoxy-3-oxoprop-1-en-1-yl)carbamoyl)pyrrolo[1,2-i][1,7]phenanthroline-7-carboxylate (3)
Obtained by general procedure, at rt. Purified by column chromatography: CH2Cl2 → CH2Cl2: MeOH (98:2, v/v), then crystallized from methanol–chloroform 1:1 (v/v). Yield: 73%, brown powder, mp = 207–209 °C. IR (KBr), ν(cm−1): 1699, 1680, 1625, 1237, 1208, 1175, 1083, 766. 1H NMR (500 MHz, CDCl3): 1.36 (t, J = 7.0 Hz, 3H, CH3), 1.47 (t, J = 7.0 Hz, 3H, H-20), 4.27 (q, J = 7.0 Hz, 2H, CH2), 4.45 (q, J = 7.0 Hz, 2H, H-19), 5.32 (d, J = 9.0 Hz, 1H, CHCO), 7.57 (dd, J = 8.0, 4.0 Hz, 1H, H-2), 7.79 (dd, J = 9.0, 11.0 Hz, 1H, NHCH), 7.96 (ad, J = 11.0 Hz, 2H, H-12, H-8), 8.26–8.29 (m, 2H, H-11, H-1), 8.58 (d, J = 9.5 Hz, 1H, H-6), 9.08 (dd, J = 3.5, 1.5 Hz, 1H, H-3), 9.19 (d, J = 9.5 Hz, 1H, H-5), 11.36 (d, J = 11.0 Hz, 1H, NH). 13C NMR (125 MHz): 14.5 CH3, 14.8C-20, 60.4C-19, 60.5 CH2, 97.3CHCO, 107.3C-7, 118.4C-6, 120.0C-11, 121.7C-9, 122.1C-2, 123.0C-14, 124.2C-5, 124.7C-8, 125.4C-17, 127.9C-12, 133.6C-16, 136.2C-1, 138.5NHCH, 140.6C-15, 145.3C-13, 150.6C-3, 159.5C-21, 164.1C-18, 169.6CO. Anal. Calcd. for C24H21N3O5: C, 66.81; H, 4.91; N, 9.74. Found: C, 66.83; H, 4.85; N, 9.79.
(2E,2′E)-diethyl-3,3'-(2-oxo-2,3-dihydroimidazo[1,2-i][1,7]phenanthroline-3,3-diyl)diacrylate (4)
Obtained by general procedure, at rt. Purified by column chromatography: CH2Cl2 → CH2Cl2: MeOH (98:2, v/v), then crystallized from methanol–chloroform 1:1 (v/v). Yield: 7%, yellow powder, mp = 216–217 °C. IR (KBr), ν(cm−1): 2924, 1726, 1637, 1260, 1072, 1019, 799. 1H NMR (500 MHz, CDCl3): 1.27 (m, 6H, 2× CH3), 4.20 (m, 4H, 2× CH2), 5.91 (d, J = 16.0 Hz, 2H, 2× CH-CO), 7.27 (d, J = 16.0 Hz, 2H, 2× CCH), 7.33 (d, J = 9.0 Hz, 1H, H-11), 7.53 (d, J = 9.5 Hz, 1H, H-6), 7.60 (dd, J = 8.0, 4.0 Hz, 1H, H-2), 8.01 (d, J = 9.0 Hz, 1H, H-12), 8.25 (dd, J = 8.0, 1.5 Hz, 1H, H-1), 9.09 (dd, J = 4.0, 1.5 Hz, 1H, H-3), 9.60 (d, J = 9.5 Hz, 1H, H-5). 13C NMR (125 MHz): 14.3 2× CH3, 61.4 2× CH2, 73.3C-9, 116.2C-11, 120.6C-14, 122.6C2, 124.5 2× CHCO, C-17, 131.1C-6, 133.3C-12, 136.6C-1, 137.3C-16, 139.9C-5, 140.0 2× CCH, 145.3C-13, 152.0C-3, 164.8 2× COOEt, 168.8C-15, 181.5C-8. Anal. Calcd. for C26H23N3O5: C, 68.26; H, 5.07; N, 9.19. Found: C, 68.27; H, 5.05; N, 9.28.
Dimethyl-9-carbamoyl-8,9-dihydropyrrolo[2,1-c][4,7]phenanthroline-7,8-dicarboxylate (6)
Obtained by general procedure, at rt. Crystallized from methanol–chloroform 1:1 (v/v). Yield: 93%, orange powder, mp = 277–279 °C. IR (KBr), ν(cm−1): 2955, 1752, 1694, 1669, 1556, 1286, 1170, 799. 1H NMR (500 MHz, DMSO-d6): 3.60 (s, 3H, H-19), 3.69 (s, 3H, H-21), 3.83 (d, J = 4.0 Hz, 1H, H-8), 5.40 (d, J = 4.0 Hz, 1H, H-9), 7.35 (d, J = 9.0 Hz, 1H, H-12), 7.58 (s, 1H, NH), 7.60 (d, J = 10.0 Hz, 1H, H-5), 7.64 (dd, J = 8.5, 4.0 Hz, 1H, H-3), 7.94 (s, 1H, NH), 8.11 (d, J = 9.0 Hz, 1H, H-11), 8.46 (d, J = 10.0 Hz, 1H, H-6), 8.83 (ad, J = 4.0 Hz, 1H, H-2), 8.85 (ad, J = 8.5 Hz, 1H, H-4). 13C NMR (125 MHz): 50.1C-19, 50.5C-8, 52.3C-21, 65.0C-9, 88.6C-7, 115.3C-13, 116.8C-12, 117.4C-5, 123.0C-3, 124.7C-14, 130.3C-4, 137.1C-16, 131.9C-6, 133.3C-11, 143.7C-17, 149.0C-2, 152.5C-15, 164.6C-18, 169.5CO, 172.6C-20. Anal. Calcd. for C20H17N3O5: C, 63.32; H, 4.52; N, 11.08. Found: C, 63.31; H, 4.47; N, 11.18.
(Z)-Ethyl-9-((3-ethoxy-3-oxoprop-1-en-1-yl)carbamoyl)pyrrolo[2,1-c][4,7] phenanthroline-7-carboxylate (7)
Obtained by general procedure, at rt. Crystallized from methanol–chloroform 1:1 (v/v). Yield: 56%, brown powder, mp = 183–185 °C. IR (KBr), ν(cm−1): 2980, 1717, 1680, 1622, 1244, 1196, 1148, 1094. 1H NMR (500 MHz, CDCl3): 1.37 (t, J = 7.0 Hz, 3H, CH3), 1.46 (t, J = 7.0 Hz, 3H, H-20), 4.28 (q, J = 7.0 Hz, 2H, CH2), 4.45 (q, J = 7.0 Hz, 2H, H-19), 5.34 (d, J = 9.0 Hz, 1H, CHCO), 7.63 (dd, J = 8.5, 4.5 Hz, 1H, H-3), 7.79 (dd, J = 9.0, 11.0 Hz, 1H, NHCH), 7.95 (s, 1H, H-8), 8.25 (d, J = 10.0 Hz, 2H, H-11), 8.41 (at, J = 9.0 Hz, 2H, H-12, H-6), 8.57 (d, J = 9.5 Hz, 1H, H-5), 8.89 (ad, J = 8.5 Hz, 1H, H-4), 9.03 (ad, J = 4.5 Hz, 1H, H-2), 11.38 (d, J = 11.0 Hz, 1H, NH). 13C NMR (125 MHz): 14.3 CH3, 14.6C-20, 60.3C-19, 60.4 CH2, 97.4 CHCO, 107.3C-7, 118.6C-5, 120.7C-13, 121.6C-6, 121.8C-3, C-9, 122.7C-12, 124.3C-8, 124.9C-14, 130.5C-11, 131.1C-4, 131.5C-16, 138.3 NHCH, 139.5C-15, 146.2C-17, 150.5C-2, 159.2 CONH, 163.8C-18, 169.5 COOEt. Anal. Calcd. for C24H21N3O5: C, 66.81; H, 4.91; N, 9.74. Found: C, 66.85; H, 4.86; N, 9.81.
(Z)-Dimethyl-11-((1,4-dimethoxy-1,4-dioxobut-2-en-2-yl)carbamoyl)-10,11-dihydropyrrolo[1,2-a][1,10]phenanthroline-9,10-dicarboxylate (9)
Obtained by general procedure, at rt. Crystallized from methanol–chloroform 1:1 (v/v). Yield: 35%, red crystals, mp = 239–241 °C. IR (KBr), ν(cm−1): 3258, 3001, 2955, 1745, 1726; 1633; 1454, 1289, 1227, 1103, 1030. 1H NMR (500 MHz, CDCl3): 3.62, 3.66, 3.74, 3.76 (4 × s, 4 × 3H, H-18, H-20, H-25, H-27), 4.24 (s, 1H, H-10), 5.55 (s, 1H, H-23), 6.59 (s, 1H, H-11), 7.29 (br s, 4H, H-5, H-3, H-8, H-6), 7.77 (ad, J = 6.5 Hz, 1H, H-4), 7.97 (ad, J = 6.5 Hz, 1H, H-7), 8.62 (br s, 1H, H-2), 10.46 (s, 1H, NH). 13C NMR (125 MHz): 50.3C-10, 50.6, 52.1, 52.9, 53.2C-20, C-18, C-27, C-25, 70.5C-11, 88.8C-9, 102.8C-23, 119.8C-4, 121.3C-8, 122.1C-3, 126.7C-6, 130.4C-13, 131.0C-12, 135.7C-15, 136.5C-7, C-5, 138.2C-16, 143.9C-22, 147.4C-2, 154.9C-14, 163.9; 165.9, 167.9, 168.3, 173.2C-21, C-24, C-23, C-19, C-17. Anal. Calcd. for C26H23N3O9: C, 59.88; H, 4.45; N, 8.06. Found: C, 59.90; H, 4.47; N, 8.08.
7-Cyano-8,9-dihydropyrrolo[1,2-i][1,7]phenanthroline-9-carboxamide (10)
Obtained by general procedure, at rt. Purified by column chromatography: CH2Cl2 → CH2Cl2: MeOH (98:2, v/v), then crystallized from methanol–chloroform 1:1 (v/v). Yield: 15%, brown powder, mp = 318–320 °C. IR (KBr), ν(cm−1): 2928, 2184, 1678, 1630, 1553. 1H NMR (500 MHz, DMSO-d6): 2.82 (dd, J = 15.0, 5.0 Hz, 1H, H-8a), 3.90 (dd, J = 15.0, 13.0 Hz, 1H, H-8b), 5.27 (dd, J = 13.0, 5.0 Hz, 1H, H-9), 6.81 (d, J = 9.5 Hz, 1H, H-5), 7.03 (d, J = 9.5 Hz, 1H, H-12), 7.42 (s, 1H, NH), 7.45 (dd, J = 8.0, 4.5 Hz, 1H, H-2), 7.87 (s, 1H, NH), 7.99 (d, J = 9.5 Hz, 1H, H-6), 8.28 (dd, J = 8.5, 2.0 Hz, 1H, H-1), 8.46 (d, J = 9.5 Hz, 1H, H-11), 8.90 (dd, J = 4.5, 2.0 Hz, 1H, H-3). 13C NMR (125 MHz): 33.4C-8, 61.6C-9, 65.8C-7, 114.1C-12, 115.5C-5, 116.0CN, 119.8C-14, 120.2C-2, 122.6C-17, 130.9C-11, 131.4C-6, 136.5C-1, 140.1C-16, 143.8C-13, 151.3C-3, 154.6C-15, 170.9CO. Anal. Calcd. for C17H12N4O: C, 70.82; H, 4.20; N, 19.43. Found: C, 70.85; H, 4.14; N, 19.49.
7-Cyano-8,9-dihydropyrrolo[2,1-c][4,7]phenanthroline-9-carboxamide (11)
Obtained by general procedure, at reflux. Crystallized from methanol–chloroform 1:1 (v/v). Yield: 45%, orange powder, mp = 183–185 °C. IR (KBr), ν(cm−1): 2936, 2172, 1690, 1630, 1535. 1H NMR (500 MHz, DMSO-d6): 2.81 (dd, J = 14.5, 4.0 Hz, 1H, H-8a), 3.35 (overlapped signal, 1H, H-8b), 5.28 (dd, J = 13.0, 4.0 Hz, 1H, H-9), 6.80 (d, J = 10.0 Hz, 1H, H-6), 7.17 (d, J = 10.0 Hz, 1H, H-12), 7.43 (s, 1H, NH), 7.58 (dd, J = 8.0, 4.0 Hz, 1H, H-3), 7.89 (s, 1H, NH), 8.05 (d, J = 9.0 Hz, 1H, H-11), 8.21 (d, J = 10.0 Hz, 1H, H-5), 8.76 (overlapped signals, 2H, H-4, H-2). 13C NMR (125 MHz): 33.4C-8, 61.7C-9, 65.5C-7, 114.4C-14, 115.9C-6, 116.9C-12, 119.9CN, 122.9C-3, 124.7C-13, 130.0C-4, 130.6C-5, 132.8C-11, 137.9C-16, 143.4C-17, 148.6C-2, 154.3C-15, 170.9CO Anal. Calcd. for C17H12N4O: C, 70.82; H, 4.20; N, 19.43. Found: C, 70.85; H, 4.14; N, 19.49.
4-Cyano-4-(1,10-phenanthrolin-2-yl)butanoic acid (12)
Obtained by general procedure, at rt. Purified by column chromatography: CH2Cl2 → CH2Cl2: MeOH (98:2, v/v), then crystallized from methanol–chloroform 1:1 (v/v). Yield:15%, brown semisolid. IR (KBr), ν(cm−1): 3398; 2921; 2240; 1705; 1507, 1238. 1H NMR (500 MHz, CDCl3): 2.19–2.23 (m, 1H, H-16a), 2.47–2.53 (m, 1H, H-17a), 2.72–2.76 (m, 1H, H-16b), 3.00–3.07 (m, 1H, H-17b), 6.16 (dd, J = 9.0, 3.0 Hz, 1H, H-15), 7.46 (d, J = 9.5 Hz, 1H, H-4), 7.68 (dd, J = 4.5 Hz, 8.0 Hz, 1H, H-8), 7.78 (as, 1H, H-5, H-6), 8.12 (d, J=9.0 Hz, 1H, H-3), 8.31 (ad, J = 8.5 Hz, 1H, H-7), 9.19 (dd, J = 4.0; 1.0 Hz, 1H, H-9). 13C NMR (125 MHz): 12.0C-17, 28.1C-16, 65.1C-15, 117.4C-4, 118.9CN, 123.1C-11, 123.9C-8, 124.6C-6, 127.2C-5, 130.7C-12, 134.8C-14, 136.8C-7, 137.9C-13, 142.2C-3, 150.0C-9, 169.8C-2, 188.8C-18. Anal. Calcd. for C17H13N3O2: C, 70.09; H, 4.50; N, 14.42. Found: C, 70.07; H, 4.47; N, 14.45.
Acknowledgments
The authors thank the POSCCE-O 2.2.1, SMIS-CSNR 13984-901, Project no. 257/28.09.2010, CERNESIM, for the NMR and XRD experiments.