

1 Introduction
The pharmacological activity of many drugs is hampered by low bioavailability, rapid metabolization, poor biodistribution into the pathological areas and/or limited intracellular penetration, leading to resistance to treatments. The drug encapsulation into nanocarriers has permitted to partly overcome some of these drawbacks. However, even if some nanomedicines have already reached the market, especially in oncology, their number remains rather low and only few are in advanced phase III clinical trials [1,2]. One of the reasons is the uncontrolled drug release, also called “burst effect”, resulting from the rapid leakage of the drug molecules, which are adsorbed at the surface of the nanocarriers rather than entrapped into their core. The poor drug loading (i.e., the weight of the drug molecule relative to the weight of the transporter material) is another major limitation to the use of nanotechnologies for the targeting of drugs. To face these problems, we have previously developed a nanoencapsulation methodology, which took advantage of the chemical linkage of the drug to the squalene (SQ), a natural and biocompatible lipid, triggering the constitution of nanoparticles (NPs) in aqueous media [3]. In fact, because of the dynamically folded conformation of the squalene, the drug–squalene bioconjugates were capable to self-assemble and to form nanosized supramolecular assemblies with a size of around 100–200 nm. This approach has resulted in the dramatical improvement of the drug loading and in the abolishment of the burst release. Moreover, it was demonstrated that the so-called “squalenoylation” technology could be applied to many drugs in various therapeutic areas. Indeed, the linkage of squalene to the anticancer drugs gemcitabine (Gem) [4], doxorubicin [5], and cisplatin [6] has resulted in decreasing the drug toxicity and increasing the anticancer activity in various experimental preclinical tumor models. Applications were also demonstrated in the treatment of spinal cord injury and brain ischemia when adenosine was conjugated with the squalene [7]. The treatment of resistant intracellular infections, either viral or bacterial, was also considered by the chemical linkage of nucleosides reverse transcriptase inhibitors or β-lactams to squalene [8]. Interestingly, it was recently observed that after intravenous administration, the squalenoylated NPs strongly interacted with cholesterol-rich lipoproteins (i.e., low-density lipoproteins (LDL) in humans and high-density lipoproteins (HDL) in rodents) and that the conjugation to squalene represented a clever way to exploit endogenous lipoproteins for the indirect targeting of cancer cells with a high expression of low-density lipoprotein receptors [9].
Because the squalene is a precursor of the cholesterol biosynthesis in mammalians, we investigated in the present study if the replacement of the squalene moiety by cholesterol could result in gemcitabine-loaded NPs with similar anticancer activity. The conjugation of gemcitabine to cholesterol was performed using a carbamate linkage (CholGem-carb), whereas the linkage to squalene used either a carbamate (SQGem-carb) or an amide bond (SQGem-amid) (Fig. 1). The anticancer activity of the three NPs was tested in vitro on the MDA-MB-231 human breast cancer cell line.
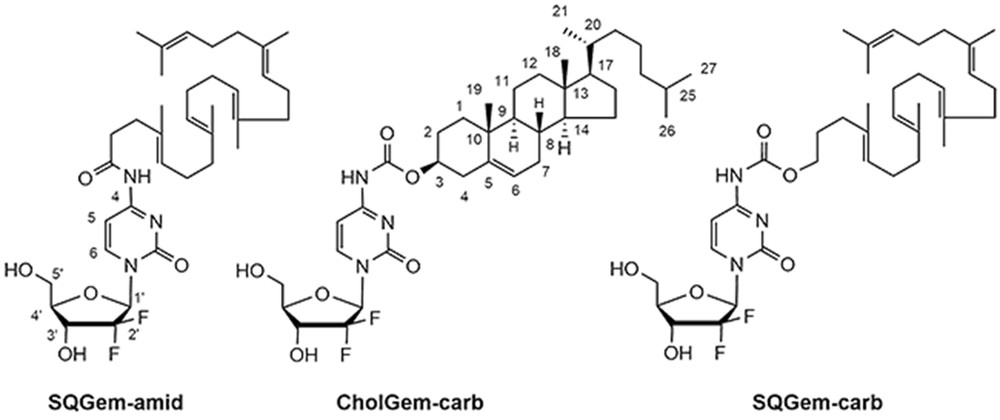
Chemical structure of the gemcitabine–cholesterol and gemcitabine–squalene bioconjugates.
2 Materials and methods
2.1 Chemicals and instruments
Gemcitabine base was obtained from Carbosynth Limited (UK). Cholesteryl chloroformate and diphosgene were purchased from Sigma–Aldrich Chemical Co (France). Chemicals obtained from commercial suppliers were used without further purification. Infrared (IR) spectra were obtained as solid using a Fourier Transform Bruker Vector 22 spectrometer. Only significant absorptions are listed. The 1H and 13C NMR spectra were recorded using Bruker Avance 300 (300 and 75 MHz, respectively) spectrometers. Recognition of methyl, methylene, methine, and quaternary carbon nuclei in 13C NMR spectra rests on the J-modulated spin-echo sequence. The 19F NMR spectrum was recorded using a Bruker Avance 400 spectrometer (376 MHz). Mass spectra were recorded using an LTQ-Velos Pro Thermofisher Scientific spectrometer. Analytical thin-layer chromatography was performed on Merck silica gel 60F254 glass precoated plates (0.25 mm layer). Column chromatography was performed on Merck silica gel 60 (230–400 mesh ASTM (American Standard Test Sieve Series)). Tetrahydrofuran (THF) was distilled from sodium/benzophenone ketyl. Pyridine was dried over CaH2 and distilled. All reactions involving air- or water-sensitive compounds were routinely conducted in glassware, which was flame-dried under a positive pressure of nitrogen or argon. SQGem-amid was obtained as previously described [3].
2.2 Synthesis and characterization of CholGem-carb
Pyridine (50 mg, 0.62 mmol) and in one portion 200 mg of cholesteryl chloroformate (0.44 mmol) were added to a suspension of gemcitabine base (99 mg, 0.37 mmol) in THF (5 mL). The resulting mixture was stirred at room temperature for 48 h and concentrated under reduced pressure. The residue was treated with aqueous 0.5 N HCl (10 mL) and the mixture was extracted with methylene chloride (3 × 15 mL). The combined extracts were washed with sodium bicarbonate (1 × 2 mL) and brine (2 × 2 mL), dried over magnesium sulfate, and concentrated under reduced pressure. The crude residue was chromatographed over silica gel eluting with cyclohexane/THF 1:1 to leave 190 mg white solid, which was recrystallized in EtOH/THF to give the title compound as colorless flakes (75% yield). Mp 220 °C (dec); [α]D +13.2 (c = 0.83, THF); IR (neat) ν 3500–3000 (br), 2940, 2915,1760, 1742, 1655, 1648, 1625, 1549, 1493, 1467, 1432, 1407, 1365, 1344, 1285, 1265, 1259, 1210, 1192, 1158, 1131, 1117, 1103, 1080, 1059, 1026, 1015, 999, 949, 919, 901, 827, 806, 784, 762, 722, 701, 690 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.75 (s, 1H, NH), 8.20 (d, J = 7.5 Hz, 1H, H-6gem), 7.08 (d, J = 7.5 Hz, 1H, H-5gem), 6.29 (d, J = 6.6 Hz, 1H, OH-3′), 6.56 (t, J = 7.3 Hz, 1H, H-1′), 5.39 (br s, 1H, H-6chol), 5.27 (t, J = 5.2 Hz, 1H, OH-5′), 4.55–4.40 (m, 1H, H-3′), 4.25–4.10 (m, 1H, HH-3chol), 3.95–3.75 (m, 2H, H-4′, H-5′), 3.70–3.55 (m, 1H, H-5′), 2.50–2.25 (m, 2H, H-4chol), 2.00–1.70 (m, 5H), 1.65–0.90 (m, 21H), 0.99 (s, 3H, H-19chol), 0.89 (d, J = 6.3 Hz, 3H, H-21chol), 0.84 (d, J = 6.3 Hz, 6H, H-26chol, H-27chol), 0.66 (s, 3H, H-18chol) ppm; 13C NMR (75 MHz, THF-d8) two carbons are hidden by the solvent signals δ 164.61 (C, C4gem), 155.13 (C, C2gem), 153.77 (C, NHCO2), 144.92 (CH, C6gem), 140.84 (C, C5chol), 124.04 (CF2, t, JCF = 262.4 Hz, C2′), 123.56 (CH, C6chol), 95.41 (CH, C5gem), 85.70 (m, CH, C1′), 82.68 (CH, C4′), 76.32 (CH, C3chol), 69.98 (CH, t, JCF = 22.6 Hz, C3′), 60.18 (CH2, C5′), 57.97 (CH, C14chol), 57.44 (CH, C17chol), 51.41 (CH, C9chol), 43.40 (CH2, C13chol), 40.98 (CH2, C12chol), 40.61(CH2, C24chol), 39.23 (CH2, C4chol), 38.04 (CH2, C1chol), 37.61 (C, C22chol), 37.34 (CH2, C10chol), 36.99 (CH, C20chol), 33.05 (CH, C8chol), 32.99 (CH2, C7chol), 30.91 (CH), 29.25 (CH2, C2chol), 29.13 (CH, C25chol), 28.91 (CH2, C16chol), 23.32 (CH3, C26chol), 23.08 (CH3, C27chol), 22.10 (CH2, C11chol), 19.81 (CH3, C19chol), 19.36 (CH3, C21chol), 12.43 (CH3, C18chol) ppm,; 19F NMR (376 MHz, THF-d8) 118.77 (d, J = 143.1 Hz, 1F), 119.75 (br d, J = 143.1 Hz, 1F); MS (ESI+) m/z(%) 698.3 (15) [M + Na]+, 676.3 (100) [M + H]+, 632 (10).
2.3 Synthesis and characterization of SQGem-carb
Pyridine (79 mg, 1.0 mmol) was added dropwise to a solution of diphosgene (361 mg, 1.82 mmol) in CH2Cl2 (2 mL) cooled at 0 °C. The mixture was stirred for 1 h and a solution of squalenol (338 mg, 0.87 mmol) in CH2Cl2 (1 mL) was added dropwise. The mixture was stirred while the temperature was gradually raised to room temperature. After 3 h, thin-layer chromatography analysis (cyclohexane/AcOEt, 4:1) indicated that the starting material was totally consumed. The mixture was concentrated under reduced pressure and the residue was taken into a mixture of THF (2 mL) and pyridine (0.2 mL) to give a white suspension. A solution of gemcitabine base (440 mg, 1.67 mmol) in DMF (5 mL) was then added and the resulting mixture was stirred at room temperature for 72 h. After being concentrated under reduced pressure, the residue was treated with aqueous 0.1 N HCl (10 mL) and the mixture was extracted with methylene chloride (3 × 15 mL). The combined extracts were washed with sodium bicarbonate (1 × 2 mL) and brine (2 × 2 mL), dried over magnesium sulfate, and concentrated under reduced pressure. The crude residue was chromatographed over silica gel eluting with cyclohexane/AcOEt 1:1 to give 266 mg (45% yield) of the title compound. [α]D = +32.7 (c = 0.88, EtOH); IR (neat) ν 3500–3100 (br), 2916, 2854, 1755, 1661, 1625, 1558, 1505, 1493, 1438, 1383, 1344, 1274, 1238, 1193, 1132, 1078, 1055, 994, 907, 807, 787, 734, 649 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.83 (s, NH), 8.21 (d, J = 7.5 Hz, 1H, H-6gem), 7.09 (d, J = 7.5 Hz, 1H, H-5gem), 6.31 (d, J = 6.3 Hz, 1H, OH), 6.16 (t, J = 7.5 Hz, 1H, H-1′), 5.27 (br. s, OH), 5.17–5.05 (m, 5 H, CH), 4.2–4.10 (m, H-3′), 4.06 (t, J = 6.6 Hz, OCOCH2), 3.88 (dt J = 8.1 Hz, J = 2.6 Hz, H-4′), 3.80 (d, J = 12.6 Hz, H-5′), 3.64 (d, J = 12.6 Hz, H-5′), 2.10–1.86 (m, 18H), 1.68 (t, J = 7.2 Hz, OCOCH2CH2), 1.62 (s, 3H, HC(CH3)2), 1.56 (s, 3H, HC(CH3)CH2), 1.54 (s, 12H, HC(CH3)CH2) ppm; 13C NMR (75 MHz, CDCl3) δ 163.32 (C, C4gem), 155.47 (C, C2gem), 152.67 (C, HNCO2), 145.11 (CH, C6gem), 135.23 (C, HCC(CH3)), 135.10 (C, HCC(CH3)), 135.01 (C, HCC(CH3)), 133.47 (C, HCC(CH3)), 131.34 (C, HCC(CH3)2), 125.46 (CH, HCC(CH3)), 124.51 (CH, HCC(CH3)), 124.45 (CH, HCC(CH3)), 124.37 (2CH, HCC(CH3)), 122.45 (CF2, t, JCF = 258.7 Hz, C2′), 96.42 (CH, C5gem), 85.5 (m, CH, C1′), 81.77 (CH, C4′), 69.05 (t, JCF = 22.3 Hz, C3′), 66.57 (CH2, OCOCH2CH2), 59.74 (CH2, C5′), 39.85 (3CH2, C(CH3)CH2), 39.79 (CH2, C(CH3)CH2), 35.63 (CH2, C(CH3)CH2CH2), 28.41 (2CH2, C(CH3)CH2CH2), 26.89 (3CH2, C(CH3)CH2CH2), 26.78 (CH2, C(CH3)CH2CH2), 25.81 (CH3, HC(CH3)2), 17.80 (CH3, HC(CH3)CH2), 16.17 (2CH3, HC(CH3)CH2), 16.12 (CH3, HC(CH3)CH2), 15.96 (CH3, HC(CH3)CH2) ppm; 19F NMR (188 MHz, DMSO-d6) −116.9 ppm; MS (ESI+) m/z(%) 698.3 (100) [M + Na]+, 676.3 (14) [M + H]+.
2.4 NP preparation and characterization
NPs were prepared in a single step according to the nanoprecipitation technique [10]. Briefly, SQGem-amid (4 mg/mL), SQGem-carb (4 mg/mL), or CholGem-carb (2 mg/mL) were dissolved in ethanol and the resulting organic solution was added dropwise under magnetic stirring into 1 mL of MilliQ water (ethanol/water 0.5:1 v/v). After solvent evaporation under vacuum using a Rotavapor (Buchi Sarl, France), an aqueous colloidal dispersion of NPs was obtained (final SQGem-amid and SQGem-carb concentration, 2 mg/mL; final CholGem-carb concentration, 1 mg/mL).
The NP diameter was measured by dynamic light scattering with a Nano ZS from Malvern (173° scattering angle) at 25 °C. The NP surface charge was investigated by ζ-potential measurement at 25 °C after dilution with 0.05 mM KCl solution applying the Smoluchowski equation and using the same apparatus. Measurements were carried out in triplicate. Colloidal stability was investigated by measuring the NP mean diameter over a period of 7 days. Stability was measured in (i) water at 4 °C, (ii) water at 37 °C, and (iii) cell culture medium (Dulbecco's Modified Eagle's Medium (DMEM); Lonza, Belgium) supplemented with 10% of fetal bovine serum (FBS) at 37 °C.
2.5 Molecular modeling
2.5.1 Simulation details
All quantum chemical calculations were performed using Gaussian 09 [11] at the B3LYP/6-311+G(d,p) level of theory with the integral equation formalism of polarizable continuum model (IEFPCM) of the solvent to account for the surrounding aqueous solution. All molecular dynamics (MD) simulations were performed using GROMACS 5.1.2 software [12]. The Slipids force field [13] was used for lipids and the AMBER99SB force field for water and ions. Simulations were performed maintaining constant the number of particles, pressure, and temperature (i.e., NPT conditions) using the Berendsen barostat [14] at 1 atm and velocity rescale thermostat [15] at 320 K. An integration step of 2 fs was used [16]. All bonds were treated as rigid constraints. Long-range electrostatics was computed with the particle mesh Ewald (PME) method [17]. Preparation of the systems and data analysis was performed using the Pteros 2.0 molecular modeling library [18,19]. VMD 1.9.2 was used for visualization [20].
2.5.2 Model membrane
We used the model of the plasma membrane of mammalian cells with asymmetric lipid content in the outer and inner monolayers, which corresponds to the well-established composition of mammalian erythrocyte membranes [21]. Phosphatidylinositol was not included into the simulations because of its low concentration, which results in about one molecule per system. The composition of the model system is shown in Table 1.
Lipid content (absolute number of molecules) of the monolayers of the simulated membrane.
| Component | Outer monolayer | Inner monolayer |
| SM (sphingomyelin) | 41 | 12 |
| PC (1,2-dioleoyl-sn-glycero-3-phosphocholine) | 47 | 12 |
| PE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) | 11 | 27 |
| PS (1,2-dioleoyl-sn-glycero-3-phospho-l-serine) | 0 | 43 |
| Cholesterol | 30 | 25 |
| Water | 8930 | |
| Na+ | 43 |
This planar bilayer membrane was used as a reference in our recent work [22] and was pre-equilibrated for 200 ns. In this study, we used the final state of the pre-equilibrated system as an initial structure (Fig. 2).
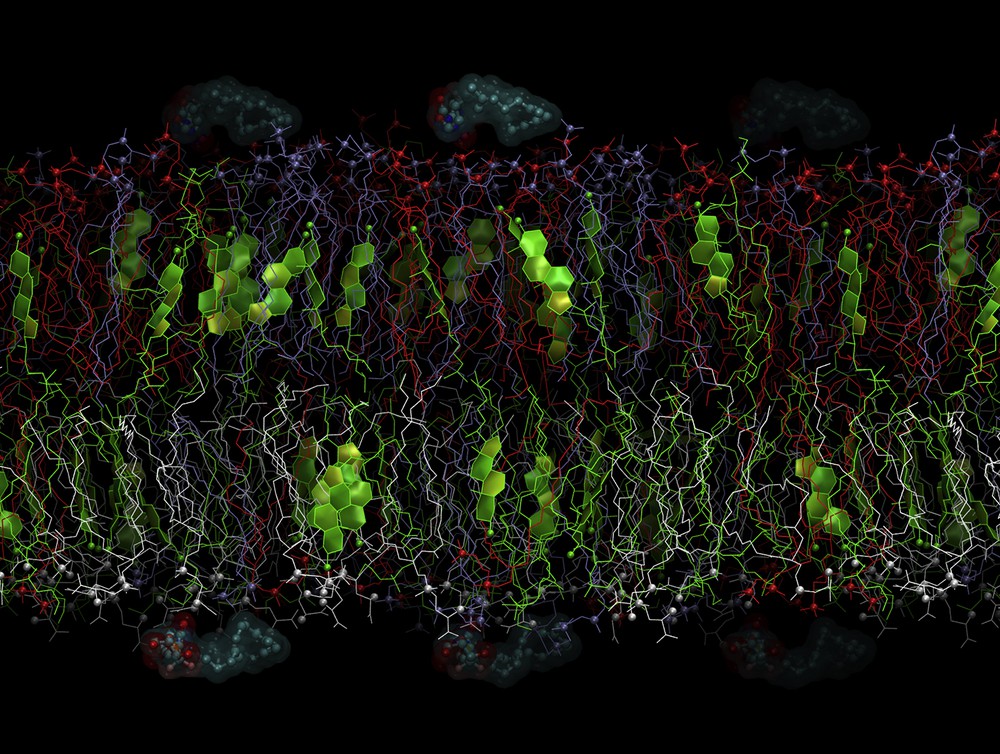
Initial state of the system with SQGem-carb. The lipids are shown in the following colors: PC (1,2-dioleoyl-sn-glycero-3-phosphocholine), red; SM (sphingomyelin), blue; PE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine), green; and PS (1,2-dioleoyl-sn-glycero-3-phospho-l-serine), gray. P and N atoms of the lipid heads are shown as small spheres. Cholesterol rings are shown as colored planes. SQGem-carb molecules are shown as balls and sticks surrounded by semitransparent molecular surfaces. Water is removed for clarity. The outer membrane leaflet is on top.
2.5.3 Ligand models
The geometries of SQGem-carb and CholGem-carb were optimized using quantum chemistry methods. Structural parameters (bond lengths, angles, and dihedrals) together with electrostatic features such as the electrostatic potential (ESP)-derived partial charges were extracted. Noteworthy is that several conformers were found for each molecule in the course of geometry optimization, but the structural parameters and partial atomic charges of the carbamate linker do not differ significantly between these conformations. For the sake of simplicity, we will only discuss the results of the most stable conformers, based on computed electronic energy.
The effect of squalene and cholesterol moieties on the carbamate bond reactivity with the hydroxide was also investigated with truncated systems, which only contain the chemical groups of interest. It was hypothesized that removing parts of the molecule that are located more than seven atoms away from the carbamate bond did not affect significantly the relevance of the models. On the gemcitabine part, the difluorinated ribofuranose ring was removed. On the lipid moiety, we only conserved the eight closest carbon atoms to the carbamate bond in the case of squalene (12 carbon atoms for cholesterol). The dangling bonds formed by the truncations were capped by hydrogen atoms. We used these truncated models of SQGem-carb and CholGem-carb to calculate the Gibbs free energy barriers of activation and reaction energies of their respective hydrolyses by a hydroxide anion. Practically, we scanned the CO bond distance between the oxygen atom of the carbamate linker and the carbon atom belonging to squalene or cholesterol groups and searched for the transition state along this reaction coordinate. This distance of interest will be referred as C2O1 in the following. The nature of the found transition state was further checked by frequency and intrinsic reaction coordinate (IRC) calculations.
The ESP charges of the atoms of the carbamate linker were assigned to its initial topology generated by the Acpype topology generator [23]. This resulted in two different topologies of the linker used for SQGem-carb and CholGem-carb topologies, respectively. The topology of SQGem-carb was created from existing topology of SQGem-amid used in our previous work [9] by manually substituting amid linker with the carbamate one. The topology of CholGem-carb was composed by fusing the topology of Gem, from our previous work [9], and the topology of the carbamate linker and the topology of cholesterol from the Slipids force field [13]. Slight manual fine-tuning of the linker charges was performed to achieve electroneutrality of both molecules. Acpype topology generator was used to generate missed parameters for bonds, angles, and dihedrals, which surround the carbamate linker.
2.5.4 Simulations in bulk water
Simulations of SQGem-carb and CholGem-carb in bulk water were performed in cubic periodic cell with the size of 3.9 nm, which contained single ligand and ∼1800 water molecules. Isotropic pressure coupling was used. The systems were simulated for 20 ns. All other parameters were the same as in membrane simulations. Then, the number of the hydrogen bonds formed by the linker was computed using Gromacs command “hbond”. The solvent accessible area was computed by VMD.
2.5.5 Binding of the ligands to the membrane
To study spontaneous binding and insertion of SQGem-carb and CholGem-carb into the membrane, six ligand molecules (either SQGem-carb or CholGem-carb) were placed in the water phase parallel to the membrane. Three of them were placed at the distance of ∼0.3 nm from the outer monolayer and the other three at the same distance from the inner monolayer. The energy of the system was minimized to remove possible sterical clashes of the ligands with water molecules and the lipid head groups. Lateral diffusion of the ligands was restricted by weak harmonic potentials with the force constant 200 kJ mol−1 nm−2 centered at their initial centers of masses. These potentials prevented ligands from aggregating in the water phase and ensured their independent binding to the membrane. The diffusion of the ligands into bulk water was restricted by applying semiharmonic potential with the force constant 1000 kJ mol−1 nm−2, which only acts if the ligand moves for more than 2.6 nm from the center of the membrane.
Such restricting potential effectively prevented the ligands from detaching from the membrane and diffusing into the bulk water. Being kept close to the membrane surface, the ligands bind to it and penetrate into the corresponding membrane leaflet at the time scale of tens of nanoseconds. This characteristic time is much shorter than in real system and could only be used to compare the relative propensities of SQGem-carb and CholGem-carb to penetrate into the membrane. Penetration of the ligands into the membrane was monitored by recording the distance between the center of the membrane and the “head” and “tail” regions of the ligand along Z axis. The “heads” of the ligands were defined as the centers of masses of Gem moiety. The “tails” were defined as three most distal carbon atoms of either squalene of cholesterol tails.
2.6 Cell culture
Human breast basal epithelial cells (MDA-MB-231) were obtained from the American Type Culture Collection (ATCC, France) and maintained as recommended. Briefly, cells were cultured in Leibovitz's L15 medium supplemented with 15% (v/v) FBS, glutamine (2 mM), and sodium hydrogen carbonate (20 mM). Medium was further supplemented with 50 U/mL penicillin and 50 μg/mL streptomycin (Lonza, France). Cells were maintained in a humid atmosphere at 37 °C with 5% CO2. Cells were used between passage 3 and 12 after thawing.
2.7 Cytotoxicity studies
NP cytotoxicity was measured by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] viability test. Briefly, 5000 cells per well were incubated in 100 μL of complete culture medium in 96-well plates for 24 h before treatment with dilutions of NPs (i.e., SQGem-amid NPs, SQGem-carb NPs, or CholGem-carb NPs) in culture medium. At the end of the incubation period (72 h), 20 μL of a 5 mg/mL MTT (Sigma Aldrich, Germany) solution in phosphate-buffered saline was added to each well. After 3 h of incubation, the culture medium was gently aspirated and replaced by 200 μL of dimethylsulfoxide to dissolve the formazan crystals. The absorbance of the solubilized dye was measured spectrophotometrically with a microplate reader (LAB System Original Multiscan MS, Finland) at 570 nm. The fraction of viable cells was calculated from the absorbance ratio between the treated cells and the average absorbance of the untreated ones. All experiments were repeated at least three times.
3 Results and discussion
3.1 Synthesis of bioconjugates
The CholGem-carb conjugate was simply prepared in 75% yield by direct reaction of gemcitabine base without any protection, with commercially available cholesteryl chloroformate using pyridine as base. Similarly, SQGem-carb was prepared in 45% yield by condensation of gemcitabine base with the squalenyl chloroformate derived from the known 1,1′,2-trisnorsqualenol. The synthesis of the trisnorsqualenol was carried out from squalene via 1,1′,2-trisnorsqualenic aldehyde according to previously reported methods [24,25]. Activation into chloroformate was achieved upon treatment of squalenol with diphosgene in the presence of pyridine and the obtained product was used directly in the condensation with gemcitabine base without further purification [26]. The obtained carbamate conjugates were purified by flash chromatography on silica. Chemoselectivity of the process for the formation of the N-4 carbamate was ascertained by the presence of the bands at 1760–1755 in the IR spectra and by the observation of NH signals at 10.75 and 10.83, respectively in the NMR spectra of both conjugates. Furthermore, the chemical shift of the two 5′CH2 protons was found unchanged in respect of the parent gemcitabine, definitively ruling out the possible reaction on the sugar moiety (Scheme 1).
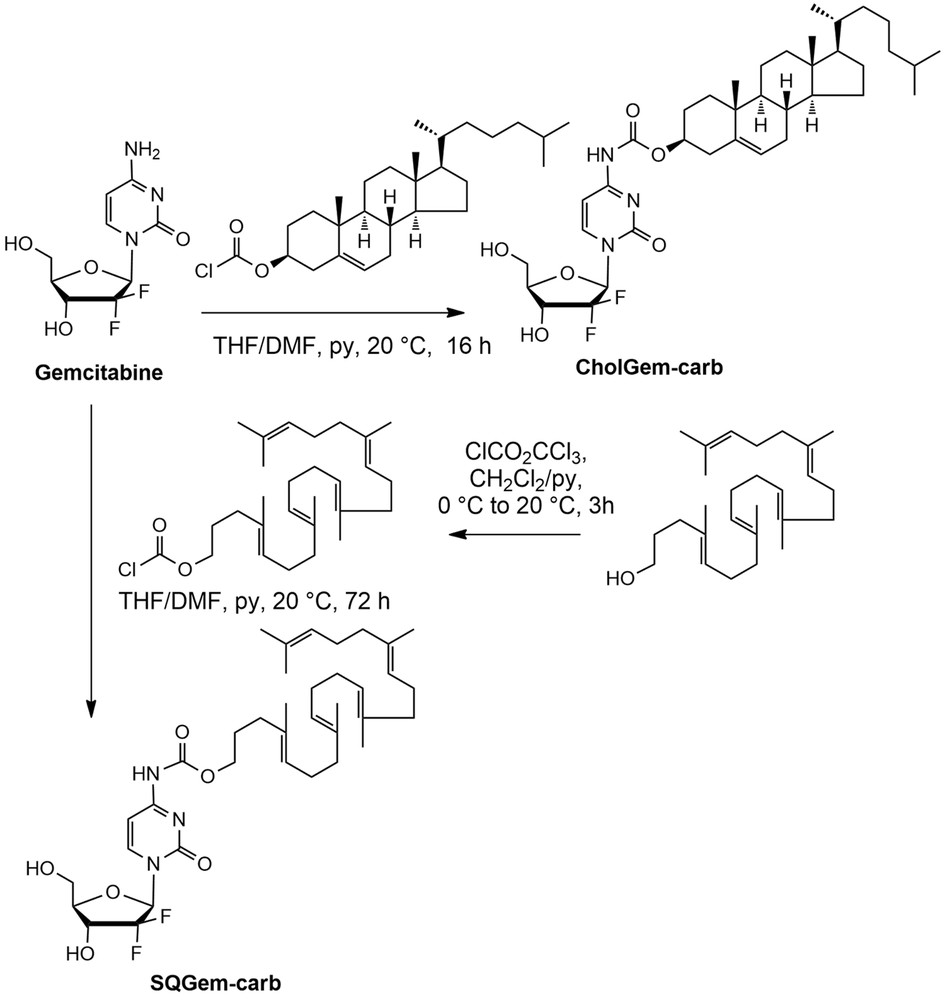
Synthetic scheme of CholGem-carb and SQGem-carb conjugates.
3.2 NP preparation and stability
All NPs were prepared by nanoprecipitation in MilliQ water of the ethanolic solution of the bioconjugates followed by solvent evaporation under reduced pressure. The resulting dispersion contained NPs with a mean diameter of 100–140 nm, a narrow size distribution and negative surface charge (Table 2). Of note, the mean diameter of CholGem-carb NPs was slightly larger than that of SQGem NPs.
Characterization of formulated NPs.
| NPs | Mean diametera (nm) (±SD) | PdIa | ζ-Potential (mV) (±SD) |
| SQGem-amid NPs | 122 ± 8 | 0.11 | −22 ± 5 |
| SQGem-carb NPs | 101 ± 1 | 0.04 | −61 ± 1 |
| CholGem-carb NPs | 136 ± 15 | 0.10 | −48 ± 7 |
a Determined by dynamic light scattering.
NP colloidal stability was dependent on the storage conditions. For SQGem-amid NPs, a fast size increase (two times) was observed in water at 4 °C after 24 h only, whereas constant mean diameter values were measured up to 4 days at 37 °C (Fig. 3). On the contrary, SQGem-carb NPs and CholGem-carb NPs displayed longer stability in both conditions up to 7 days (Fig. 3). Probably a consequence of the interaction with the serum proteins, incubation in complete cell culture medium led to an increase in the mean diameter for all studied NPs (Fig. 3e). In general, NP sizes remained rather homogenous as shown by polydispersity index (PdI) (Table 2 and Fig. 3b and d).
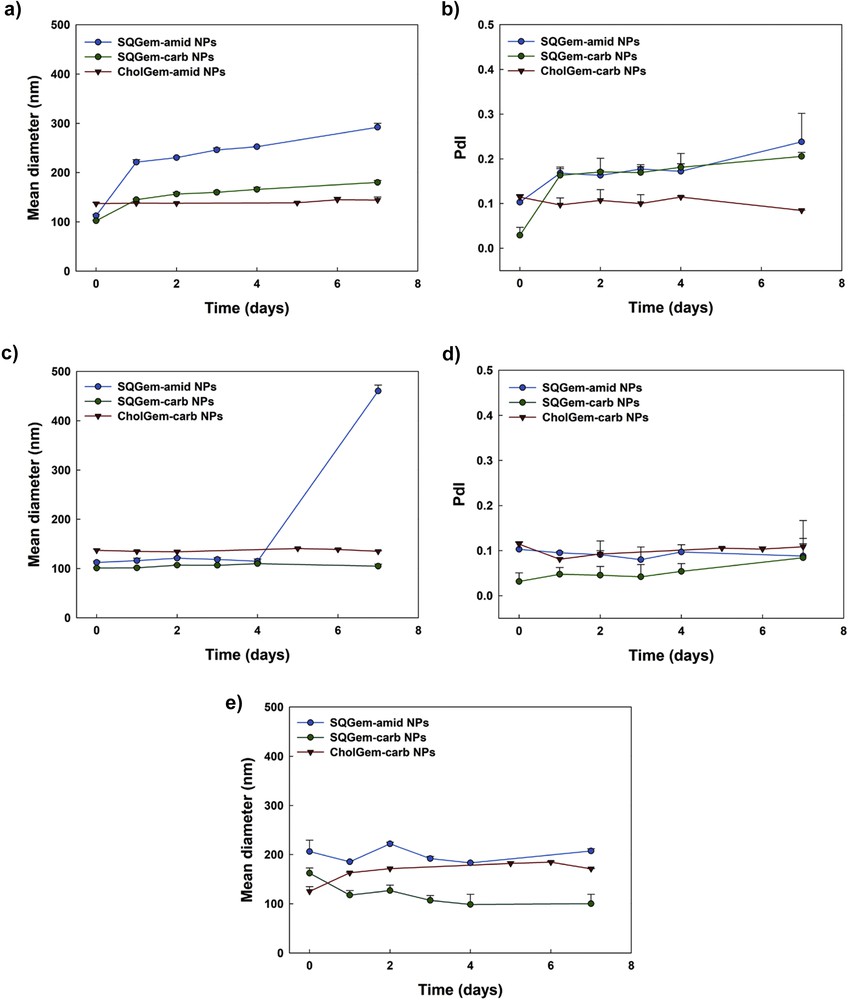
Evolution over time of SQGem-amid NPs, SQGem-carb NPs, and CholGem-carb NPs' mean diameter and PdI after incubation in water at 4 °C (a and b); water at 37 °C (c and d), and cell culture medium supplemented with 10% of FBS (e). Values are represented as mean ± standard deviation.
3.3 Cytotoxicity
After incubation of CholGem-carb NPs with MDA-MB-231 cancer cell line, the absence of cytotoxicity was observed and 70% of the cells remained alive even after 72 h of incubation with NPs at 80 μM concentration. On the contrary, when cells were exposed to either SQGem-amid NPs or SQGem-carb NPs, complete cell death was observed already at the concentration of 25 μM (Fig. 4).
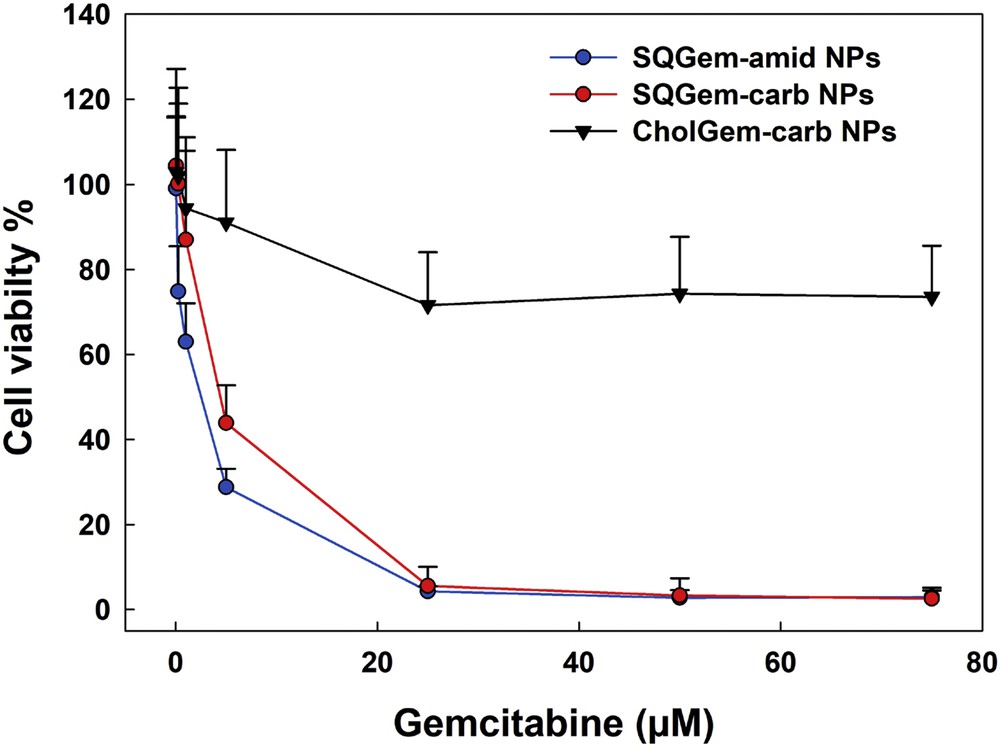
Viability of MDA-MB-231 cells after 72 h exposure to increasing concentrations of SQGem-amid NPs, SQGem-carb NPs, or CholGem-carb NPs. Values are represented as mean ± standard deviation.
It was concluded that when gemcitabine was chemically linked to cholesterol instead to squalene, the anticancer activity completely vanished. Noteworthy is that gemcitabine is a nucleoside analogue, which inserts into DNA and competitively inhibits DNA chain elongation, leading to DNA fragmentation and cell death. When conjugated to squalene or cholesterol, the gemcitabine remains inactive as long as it is not released from the bioconjugates. Thus, gemcitabine in free form is mandatory for anticancer activity to occur. As shown previously, the amide linker in SQGem-amid NPs was subject to hydrolysis by intracellular proteases, such as the cathepsins B and D, triggering the fast release of gemcitabine intracellularly, which explains the observed cytotoxicity of these NPs [3]. Carbamate has also been used as a key structural motif in many approved prodrugs [27]. Although amide resonance in carbamates has been shown to be about 3–4 kcal/mol lower than those of amides, owing to the steric and electronic perturbations because of the additional oxygen, carbamates still remain sensitive to enzymatic hydrolysis [28]. This may explain why SQGem-carb, being capable of gemcitabine release, displayed similar anticancer activity to SQGem-amid.
Very surprisingly, although CholGem-carb bioconjugate was synthesized with a similar carbamate linker than SQGem-carb, it behaved very differently because the CholGem-carb NPs were unable to display any cytotoxic activity. It was hypothesized that because of differences in molecular conformation of CholGem-carb versus SQGem-carb, the enzymatic accessibility was hindered when the squalene moiety was replaced by the cholesterol.
3.4 Analysis of the carbamate linker properties
To compare the effect of the lipid part (cholesterol or squalene) on the properties of the carbamate linker the optimized structures of CholGem-carb and SQGem-carb molecules were compared (Fig. 5). The computed partial atomic charges, bond lengths, and angles of interest are shown in Tables 3 and 4.
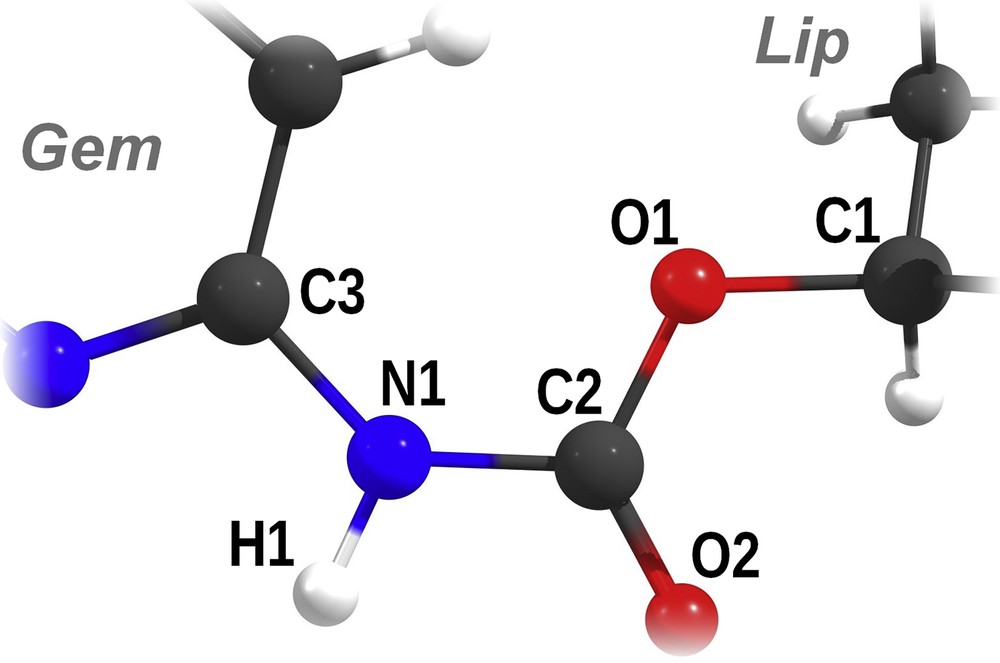
Scheme of the carbamate bond with atom names used in Tables 3 and 4. Gem represents the gemcitabine moiety, and Lip represents the cholesterol or squalene moiety.
ESP partial atomic charges of the atoms involved in the carbamate bond, calculated at the B3LYP/6-311+G(d,p) level of theory.
| Atom | CholGem-carb | SQGem-carb |
| O1 | −0.64 | −0.51 |
| C2 | +1.10 | +1.07 |
| O2 | −0.65 | −0.62 |
| N1 | −0.89 | −0.91 |
| H1 | +0.44 | +0.42 |
| C3 | +1.04 | +1.08 |
Structural parameters of the bonds (in angströms) and selected angles (in degrees) of the carbamate bond, calculated at the B3LYP/6-311+G(d,p) level of theory.
| CholGem-carb | SQGem-carb | ||
| Bonds | C1O1 | 1.464 | 1.455 |
| O1C2 | 1.345 | 1.347 | |
| O2C2 | 1.207 | 1.206 | |
| C2N1 | 1.394 | 1.392 | |
| N1C3 | 1.382 | 1.382 | |
| N1H1 | 1.011 | 1.011 | |
| Angles | O1C2N1 | 126.9 | 127.1 |
| C2N1C3 | 129.7 | 129.6 |
Most atoms involved in the carbamate bond display similar partial charges (Table 3), with the notable exception of the oxygen atom linking the carbamate linker to either cholesterol or squalene (O1). In the latter case, the charge difference is 0.13e, which could possibly lead to a difference in reactivity between SQGem-carb and CholGem-carb. This atom is indeed targeted by the hydrogen atom of hydroxide during the most likely process of the first step of the hydrolysis [29]. The bond lengths involving O1, however, are extremely similar (first two rows of Table 4), which prevents from drawing strong conclusions in terms of reactivity difference. Thus, we have also evaluated the respective strength of the carbamate bond for the SQGem-carb and CholGem-carb in the course of hydrolyses by a hydroxide anion. The two respective transition states display almost identical geometries (Fig. 6) and the activation barriers are unsurprisingly very similar (12.0 kcal mol−1 in the case of SQGem-carb and 12.5 kcal mol−1 for CholGem-carb). The energy difference of 0.5 kcal mol−1 was not significant with respect to the chemical accuracy of the density functional theory method and therefore insufficient to explain the observed decrease in the anticancer activity in the case of CholGem-carb. The products of hydrolysis were equally stable as well (Gibbs free energies of the reaction are −35.5 kcal mol−1 for the SQGem-carb model and −35.8 kcal mol−1 for the CholGem-carb model). Overall, these quantum chemistry calculations strongly suggest that the absence of the anticancer effect in the case of CholGem-carb can neither be explained by a difference in the electronic structure of the carbamate linker nor by a difference in reactivity regarding the hydrolysis of the carbamate bond.
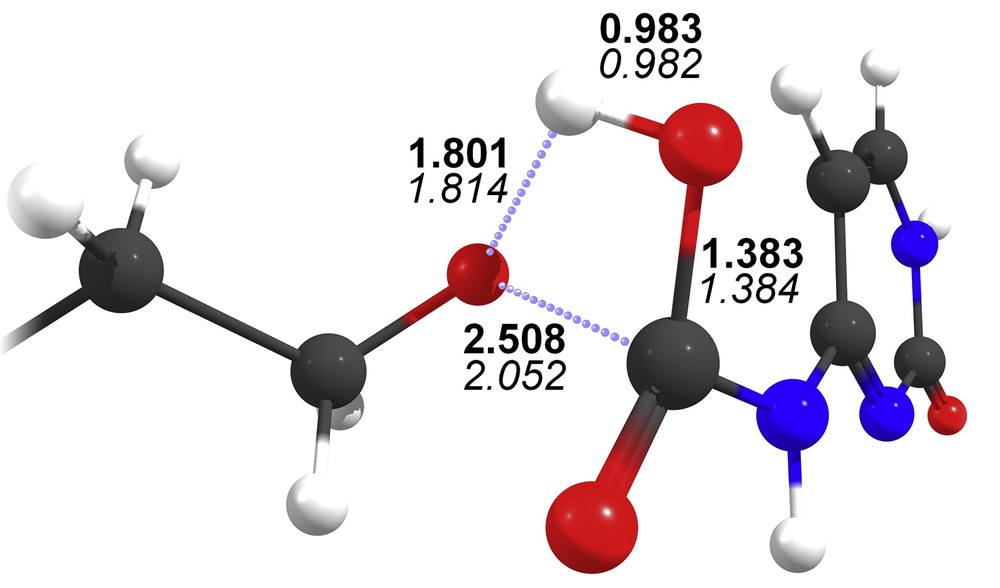
Transition state of the hydrolysis of the truncated model of SQGem-carb, optimized at the B3LYP/6-311+G(d,p) level of theory. Bond lengths (in angstroms) are shown in bold, with the values of the respective transition state of CholGem-carb in italics for comparison.
3.5 Estimation of the dehydration cost of the carbamate linker
Because it is unknown which enzymes facilitate cleavage of the carbamate linker of SQGem-carb and CholGem-carb intracellularly, the direct modeling of this process was currently not possible. However, it was possible to make an indirect estimate of the dehydration energy cost for carbamate linker during its enzymatic cleavage. The average number of hydrogen bonds (Nhb), formed by the linker with surrounding water molecules and its solvent accessible surface area (SASA), correlated with the number of water molecules that form the linker hydration shell. Usually, the active sites of enzymes are narrow and the ligand has to dehydrate completely or partially before entering it. The energy cost of dehydration could contribute significantly to the barrier of the reaction. Thus, it is expectable that ligands with a larger shell of coordinated water around the linker would be cleaved by the intracellular enzymes with more difficulty. Table 5 shows Nhb and SASA for C and N atoms, which form the bond to be cleaved in SQGem-carb and CholGem-carb. The values were computed for single ligand molecules in bulk water over MD trajectories of 20 ns.
Average number of hydrogen bonds with water molecules (Nhb) and the SASA of C and N atoms of the carbamate linker in SQGem-carb and CholGem-carb.
| Property | SQGem-carb | CholGem-carb |
| Nhb | 0.054 | 0.18 |
| SASA (Å2) | 10.5 | 13.1 |
It is clearly seen from Table 5 that both parameters are larger for CholGem-carb, which means that it possesses a larger shell of coordinated water, and therefore, it is expected to display a higher energy barrier of dehydration during enzymatic cleavage of the carbamate linker.
3.6 Incorporation into the membranes
In addition to different rates of enzymatic cleavage inside the cells, the difference concerning the pharmacological efficacy of SQGem-carb and CholGem-carb could also result from the different propensity of those bioconjugates to translocate through the cell membrane. The transport of squalene conjugates through the membrane is a complex process. In this study, we only considered one stage of this process, namely the spontaneous binding of SQGem-carb and CholGem-carb to the cell membrane and their incorporation into the membrane leaflets.
MD simulations showed that SQGem-carb and CholGem-carb spontaneously incorporated into both outer and inner monolayers of the model bilayer; however, the details of these events were different for the two compounds. The mean time of incorporation into the membrane appeared to be different for SQGem-carb and CholGem-carb and dependent on the membrane leaflet (Table 6).
Mean time of incorporation of SQGem-carb and CholGem-carb into inner and outer leaflets of the membrane.
| Leaflet | CholGem-carb (ns) | SQGem-carb (ns) |
| Outer | 34.0 ± 12.5 | 13.0 ± 4.5 |
| Inner | 17.6 ± 2.5 | 7.0 ± 1.0 |
Despite large uncertainties caused by the small number of observed incorporation events (three per simulation in each monolayer), there was a pronounced difference between inner and outer leaflets of the membrane. Both ligands incorporated slower into the outer leaflet, which is enriched in sphingomyelin (SM) and cholesterol. This difference is also evident from Figs. 7 and 8 in which the evolution of the tail positions for both types of ligands is visualized. Such behavior is expectable because SM-rich outer leaflet is known to be much more ordered and stiff in comparison to the more fluid inner one.
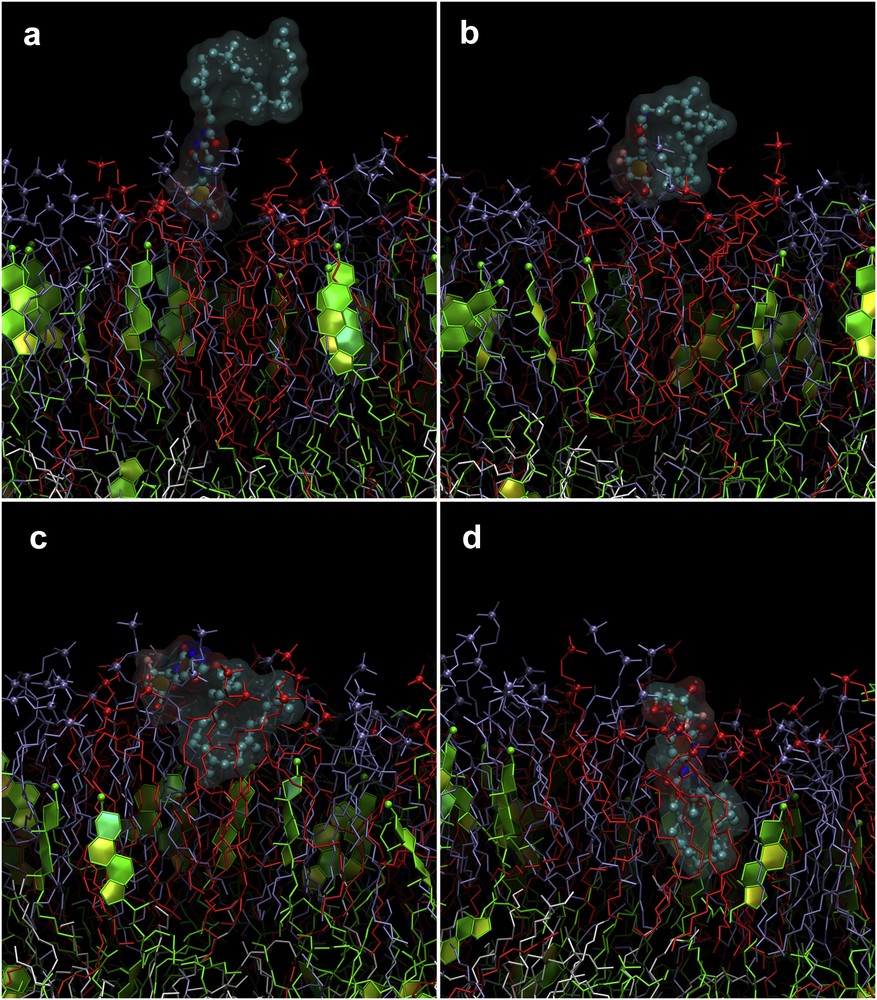
Sequence of events during the penetration of SQGem-carb into the outer leaflet of the model membrane. Only the outer leaflet of the membrane is shown in the figure. Colors are the same as in Fig. 2. Gem moiety binds to the lipid head groups (a). SQ tail bends and binds to the membrane (b). SQ tail starts entering the membrane in folded conformation (c). Complete embedding into the membrane while SQ tail remains partially folded (d).
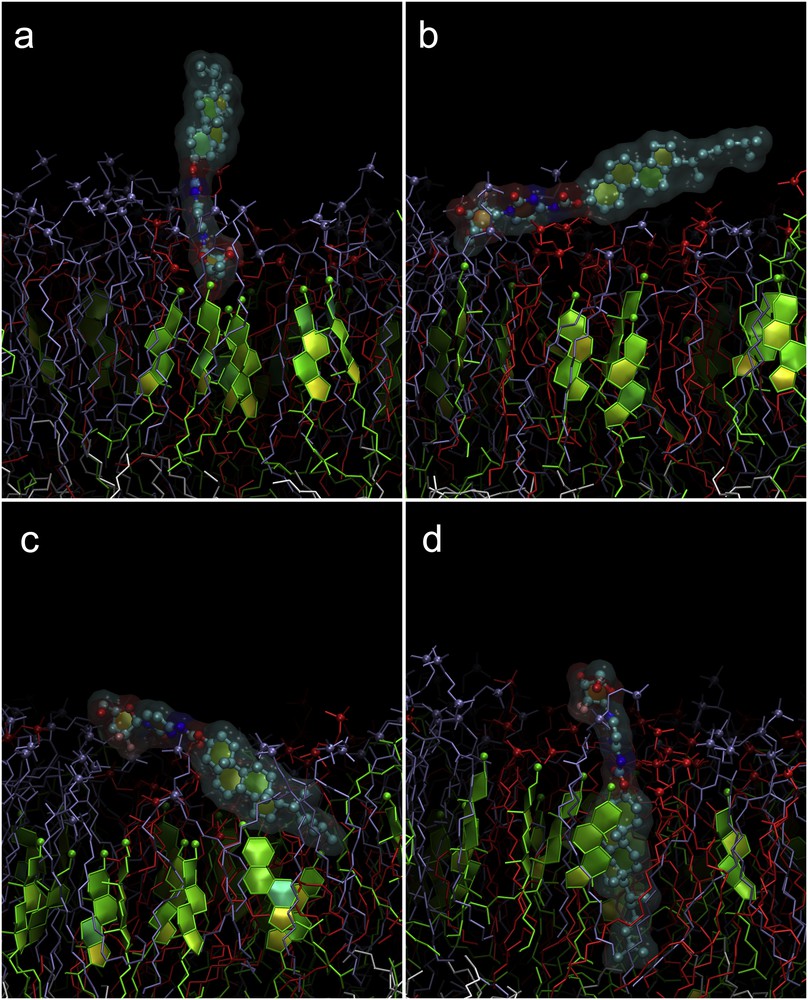
Sequence of events during the penetration of CholGem-carb into the outer leaflet of the model membrane. Colors are the same as in Fig. 2. Only the outer leaflet of the membrane is shown. Gem moiety binds to lipid head groups (a). The whole molecule rotates parallel to the membrane (b). The rotation continues and the cholesterol moiety enters the membrane (c). The molecule is embedded completely in “vertical” orientation (d).
Nevertheless, SQGem-carb incorporates into the membrane faster than CholGem-carb, regardless of the membrane leaflet. It is believed that this difference was caused by different mechanism of insertion of SQGem-carb and CholGem-carb, which was elucidated from our MD trajectories. In the case of SQGem-carb, the molecule first binded to the lipid head groups by its Gem moiety, whereas the SQ tail remained in the water phase (Fig. 7a). On the next stage, flexible SQ tail folded and binded to the membrane by its distal part (Fig. 7b). The SQ tail gradually submerged into the hydrophobic core of the membrane, whereas the Gem moiety remained anchored at the level of lipid head groups (Fig. 7c and d). The SQ tail remained partially folded during penetration.
The behavior of CholGem-carb was quite different. In a first step, CholGem also binded to the lipid head groups by its Gem moiety, whereas the cholesterol stretched into the water phase (Fig. 8a). However, the cholesterol moiety is very rigid and could not bend toward the membrane surface. Instead, the whole molecule rotated first to become parallel to the membrane (Fig. 8b) and then to flip and submerge the cholesterol moiety into it (Fig. 8c). Finally, the molecule aligned almost perpendicular to the membrane with the cholesterol in the hydrophobic core of the lipid monolayer, whereas the Gem anchored at the level of lipid head groups (Fig. 8d).
The evolution of the positions of SQGem-carb and CholGem-carb tails during MD simulations is shown in Fig. 9, whereas equilibrium positions of their tails and heads in the membrane are shown in Table 7.

Evolution of the position of the tails of SQGem-carb and CholGem-carb in the course of MD simulations with k = 1000 kJ mol−1 nm−2. Each curve corresponds to an individual molecule. Molecules 1–3 are bound to the outer leaflet and are represented by black, red, and blue curves, whereas the molecules 4–6 are bound to the inner leaflet and shown as magenta, green, and purple.
Average positions of the heads and tails of SQGem-carb and CholGem-carb after their complete incorporation into the model membrane.
| CholGem-carb (nm) | SQGem-carb (nm) | |
| Inner leaflet | ||
| Heads | 2.0 ± 0.2 | 1.8 ± 0.1 |
| Tails | 0.1 ± 0.2 | 0.9 ± 0.2 |
| Outer leaflet | ||
| Heads | 2.0 ± 0.2 | 1.6 ± 0.1 |
| Tails | 0.3 ± 0.1 | 0.8 ± 0.1 |
It is evident that SQ tails of SQGem-carb inside the membrane were coiled as observed in Fig. 7d and almost never adopted a stretched conformation. SQGem-carb ends resided in the middle of the corresponding monolayer quite far (∼0.9 nm) from the center of the membrane. In contrast, CholGem-carb molecules were straight and almost perpendicular to the membrane surface (Fig. 8d) with the ends located almost in the membrane midplane (only ∼0.1–0.3 nm from its center). The data of MD simulations confirmed an assumption that SQGem-carb and CholGem-carb molecules possessed different molecular conformations both in solution and inside the membrane. CholGem-carb molecules behave as rigid rods in water and incorporated into the membrane without any significant bending or other conformational changes. In contrast, SQGem-carb molecules were highly flexible, their SQ tails were quite dynamic and adopted a folded conformation in water to minimize the area of hydrophobic mismatch. This flexibility made the process of penetration into the membrane significantly faster than in the case of CholGem-carb. SQ tails of SQGem-carb remained disordered and partially coiled inside the membrane as well, which was different from the strict vertical orientation of CholGem-carb molecules.
These observed differences in mechanism and speed of binding and incorporation into the plasma membrane between SQGem-carb and CholGem-carb may contribute to their different intracellular accessibility and concentration. These effects might be synergetic to the higher dehydration energy of the carbamate linker in CholGem-carb in the course of enzymatic hydrolysis. On the whole, these observations may explain the different anticancer activity of CholGem-carb versus SQGEM-carb in cell culture.
4 Conclusions
Although it was demonstrated that the CholGem-carb bioconjugate was also able to self-assemble as NP, it did not display any advantage compared to the SQGem NPs because of the absence of anticancer activity. Numerical simulations suggest (1) higher dehydration energy of the CholGem-carb carbamate linker in the course of its enzymatic cleavage and (2) slower incorporation of CholGem-carb into the cell membrane compared to SQGem, which may justify its lower anticancer activity.
Acknowledgments
The research leading to these results has received funding from the ERC under the European Community's Seventh Framework Programme FP7/2007–2013 (no. 249835). The CNRS and the French Ministry of Research are also warmly acknowledged for financial support. S.Y., B.C., and C.R. were supported by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 690853 and the NATO Science for Peace and Security programme under the project SPS 985291. The computational studies work was performed using HPC resources from GENCI-[TGCC/CINES/IDRIS] (Grants 2015-[c2016077586] and 2016-[A0020707586]), Mésocentre de calcul de Franche-Comté, and Centre de Calcul de Champagne–Ardenne ROMEO.