

1 Introduction
Saponite belongs to the 2:1 clay mineral family with one octahedral sheet made of magnesium sandwiched between two tetrahedral sheets containing silicon partly substituted by aluminum. These structural substitutions induce a charge deficit that is compensated by counterions in the interlayer space (generally sodium Na+, calcium Ca2+, or potassium K+). In the case of organic–inorganic hybrids having a saponite-like structure, an organoalcoxysilane is used as a silicon source, by this way, an organic chain is covalently bonded to the silicon and thus to the inorganic framework. The general chemical formula of materials can be denoted as Cx[Mg3[(RSi)4−xAlx]O10(OH)2], where x represents the tetrahedral substitution rate and C the counterions used to compensate the charge deficit induced by the substitutions in the tetrahedral sheets [1–7], as shown in Fig. 1. Synthesis methods play an important role on the crystallinity, shape, and particle size of these compounds, so the preparation methods remain an important research subject in the field of layered materials [3–6]. Many methods have been developed for the synthesis of saponite over the last decades, but the main ones include coprecipitation, hydrothermal synthesis [3,5,8–11], and sol–gel method [12–18]. However, the morphology and crystallinity of such materials are related to the synthesis conditions.
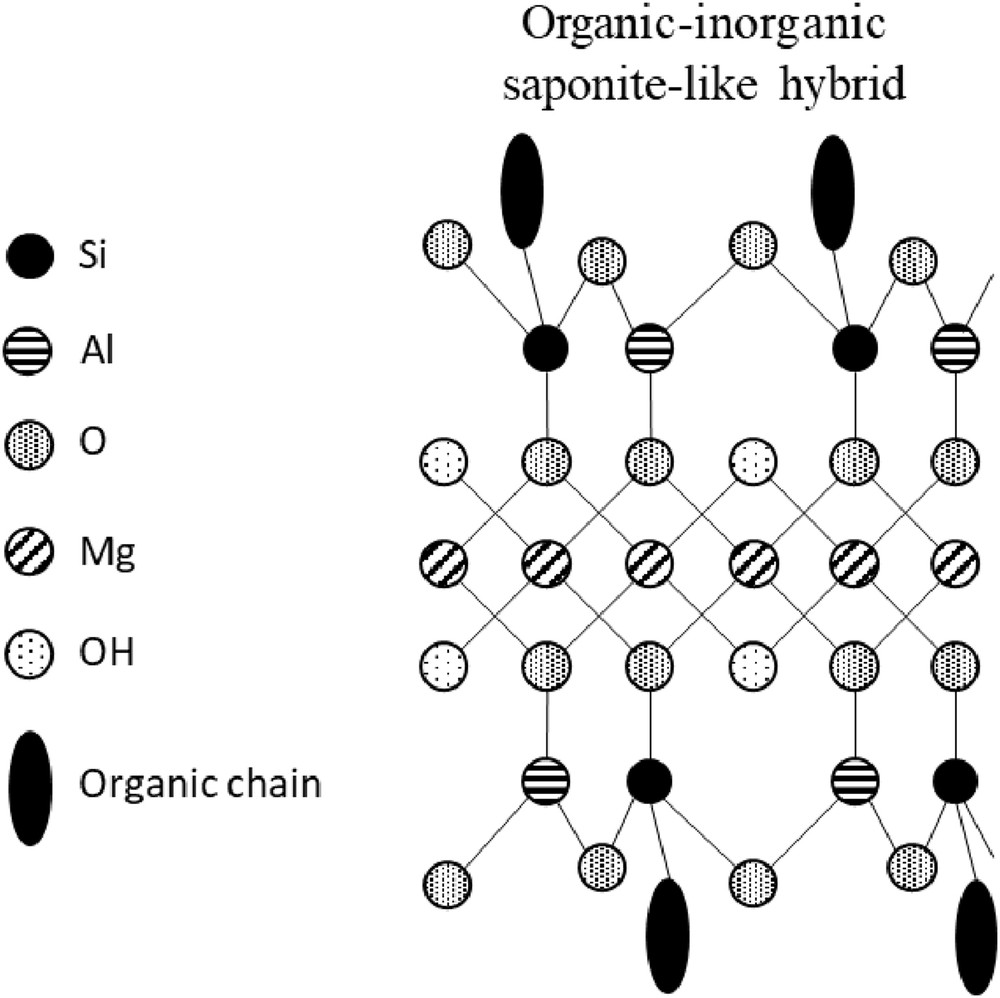
Schematic representation of organic–inorganic saponite-like hybrid.
Sol–gel method is becoming a new way to synthesize hybrid organic–inorganic lamellar materials. It consists in dissolving inorganic salts containing divalent and trivalent cations, such as magnesium and aluminum salts, into a solvent and increasing the pH by adding NaOH to promote the condensation that occurs preferentially in basic media [12–18]. The solution is kept under constant stirring and fixed temperature, generally room temperature, for several hours. This process allows obtaining high purity materials in short times with low energy consumption, but the products generally present lower crystallinity than those obtained using coprecipitation or hydrothermal synthesis methods [3–6].
The novelty of this study stands on the sol–gel synthesis conditions and mechanisms bringing to the formation of different lamellar materials depending on the precursors, especially the use of different aluminum source. These materials were intended as materials presenting acidic properties, and a series of analysis has been performed (X-Ray diffraction [XRD], Fourier transform infrared [FT-IR], and solid-state nuclear magnetic resonance [NMR] spectroscopies) to characterize such materials.
2 Experimental section
2.1 Reactants
N-Propyltriethoxysilane C9H22O3Si (ABCR, 97 wt %), trimethoxyphenylsilane C9H14O3Si (TCI Chemicals, >98 wt %), magnesium chloride hexahydrate MgCl2·6H2O (Sigma–Aldrich, BioXtra, ≥99.0%), magnesium nitrate hexahydrate Mg(NO3)2·6H2O, aluminum acetylacetonate Al(C5H7O2)3 (Sigma–Aldrich, ReagentPlus, 99%), aluminum chloride hexahydrate AlCl3·6H2O (Fluka Analytical, ≥99.0%), ethanol CH3CH2OH (Carlo Erba Reagents, ≥97%), sodium hydroxide NaOH (Carlo Erba Reagents, 99.9%), and distilled water were used as received during the study.
2.2 Synthesis of the lamellar materials
The synthesis of organic–inorganic saponite-like hybrid materials involve magnesium chloride or magnesium nitrate, aluminum acetylacetonate or aluminum chloride, and N-propyltriethoxysilane or trimethoxyphenylsilane as sources of the hybrid material framework. The sol–gel synthesis process of hybrid organic–inorganic materials can be described as follow: the required amount of magnesium salt and aluminum source are, respectively, dissolved into ethanol, according to the desired Si/Al substitution rate in the saponite structural formula, Cx[Mg3[(RSi)4−xAlx]O10(OH)2] with 0.5 ≤x≤ 1.2, and their molar masses. A fixed quantity of organoalcoxysilane (2 g in the present study) was added under stirring, and then sodium hydroxide (NaOH) 1 M was added until the pH reaches 10. The amounts of the different precursors are detailed in Table 1. The solution is left under stirring at room temperature for 24 h, before being centrifuged at 11,000 rpm for 10 min, washed three times with 30 mL of ethanol, and dried for 24 h in an oven at 60 °C for further analysis. Samples are labeled as DCHxSy (for saponite-like hybrid samples) and DCHT00y (for talc-like hybrid samples), with x the theoretical Si/Al substitution rate according to the structural formula, and y the number of the sample.
Sol–gel synthesis parameters of the samples synthesized with aluminum chloride, trimethoxyphenylsilane, and magnesium chloride hexahydrate.
| Sample | Mg amount (g) | Al amount (g) | Si amount (g) | Si/Mg molar ratio | Si/Al molar ratio | Mg/Al molar ratio | pH | Mass obtained (g) |
| DCH0.5S014 | 1.757 | 0.352 | 1.998 | 1.2 | 6.9 | 5.9 | 11 | 1.42 |
| DCH0.75S014 | 1.893 | 0.560 | 2.003 | 11 | 4.3 | 4.0 | 10.5 | 1.38 |
| DCH1S014 | 2.05 | 0.811 | 2.004 | 1.0 | 3.0 | 3.0 | 11 | 1.32 |
| DCH1.2S014 | 2.195 | 1.042 | 1.999 | 0.9 | 2.3 | 2.5 | 10.5 | 1.28 |
2.3 Samples characterization
First, the structure of the as synthesized samples was investigated by RXRD using a Panalytical X'Pert PRO MPD with Cu Kα radiation (λ = 1.5418 Å), and performed from 2° to 70° 2θ with a step of 0.017° 2θ, on randomly oriented powder samples.
Potassium bromide (KBr) pellets were prepared to perform FT-IR spectroscopy, made with 5 mg of the samples mixed with 100 mg of KBr. The measurements were performed in the 400–4000 cm−1 region using a Bruker Equinox 55 spectrometer with a DTGS detector. The number of scans was fixed at 100 scans with a resolution of 4 cm−1.
Thermogravimetric analysis (TGA) of the synthesized samples was performed using a Mettler-Toledo TGA/DSC1 LF1100 apparatus under argon, in alumina sample holders, with a flow rate of about 100 mL min−1 from 25 to 600 °C and a heating rate of 5 °C min−1. An empty sample holder was recorded as reference to correct baseline deviation.
29Si and 1H decoupled 29Si magic angle spinning (MAS and single pulse MAS, respectively) NMR spectroscopy spectra were recorded using a Bruker AVANCE II 300 MHz spectrometer at 59.6 MHz. Samples were analyzed in a 7-mm diameter cylindrical rotor at a spinning frequency of 4 kHz and recorded for 24 h. 1H decoupled 29Si NMR experiments were performed with a π/4 pulse duration of 2.57 μs with a recycle time of 90 s. 27Al MAS NMR spectroscopy spectra were recorded using a Bruker AVANCE II 400 MHz spectrometer at 104.3 MHz. Samples were analyzed in a 2.5-mm diameter cylindrical rotor at a spinning frequency of 25 kHz and recorded for 8 h. 27Al spectrum was recorded with a delay time of 1 s by using 4 μs single pulse, corresponding to a pulse angle of π/12.
3 Results and discussion
3.1 Syntheses with aluminum chloride
Syntheses with aluminum chloride have first been performed during this study. The compositions and molar ratios of the synthesized samples are reported in Table 1, with experimental Si/Al, Al/Mg, and Si/Mg molar ratios calculated according to the molar masses of the different precursors.
Diffraction patterns of lamellar materials generally show (00l) reflections because of their two-dimensional structure, which allows us to estimate the c-cell parameter (thickness of the lamellar material) by adding the interlayer distance and the thickness of a single layer. Fig. 2 displays the XRDR patterns of the samples synthesized in the presence of aluminum chloride. Four main reflections are observed: 5.53°–6.42°, 20.1°, 36°, and 60.6° 2θ, which correspond to distances of 16–13.5 (d001), 4.4 (d110,020), 2.5 (d130,200), and 1.53 Å (d060), respectively, according to the Bragg law:
| 2dhkl.sin(θ) = nλ |
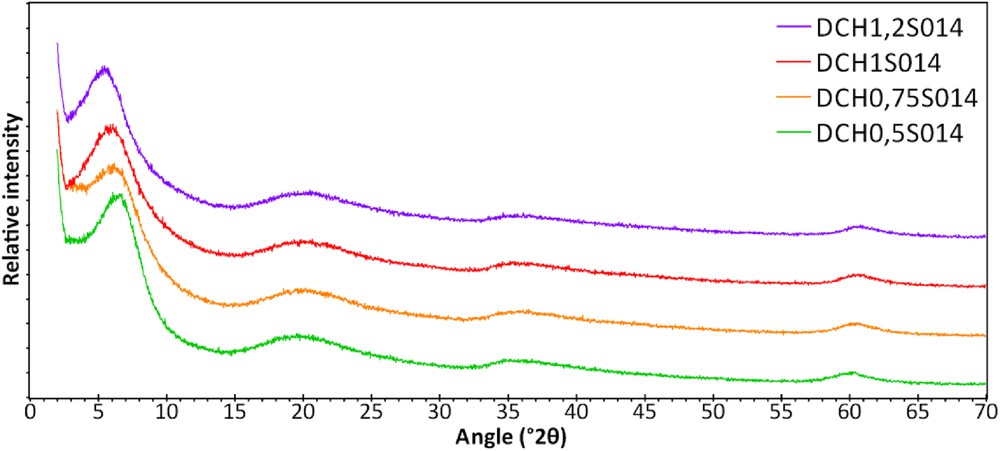
XRD patterns of the samples S014 synthesized with aluminum chloride by sol–gel method, washed three times with ethanol.
The broadness of these peaks indicates a low organization of the structure (crystallinity) because of the synthesis process and the presence of an organic chain linked to the organoalcoxysilane, but these positions are typical from a trioctahedral 2:1 clay-like structure according to the positions of (001) and (060).
FT-IR has been performed to detect typical bonds between aluminum, magnesium, and silicon. The results are shown in Fig. 3, with a large wide band around 3500 cm−1 corresponding to the presence of water in the structure. However, the sample DCH0.5S014 (with the lower amount of aluminum in the structure) shows two vibrational bands at 3715 and 3620 cm−1 corresponding to stretching SiAl. These bands tend to disappear with increasing aluminum content into the samples, so they correspond to stretching SiAl bonds. The two small bands at 3072 and 3049 cm−1 correspond to the CH stretching vibrational bands, and the presence of stretching SiC around 1151 cm−1 indicates the silicon precursor has efficiently been incorporated in the structure with the organic chain. Additional small bands between 1300 and 1200 cm−1 prove the presence of magnesium into the structure of the materials, but also aluminum with AlOH vibration band at 860 cm−1. Moreover, two intense combination bands at 1967 and 1760 cm−1 are visible in these spectra and are related to the incorporation of aluminum and magnesium into the structure.
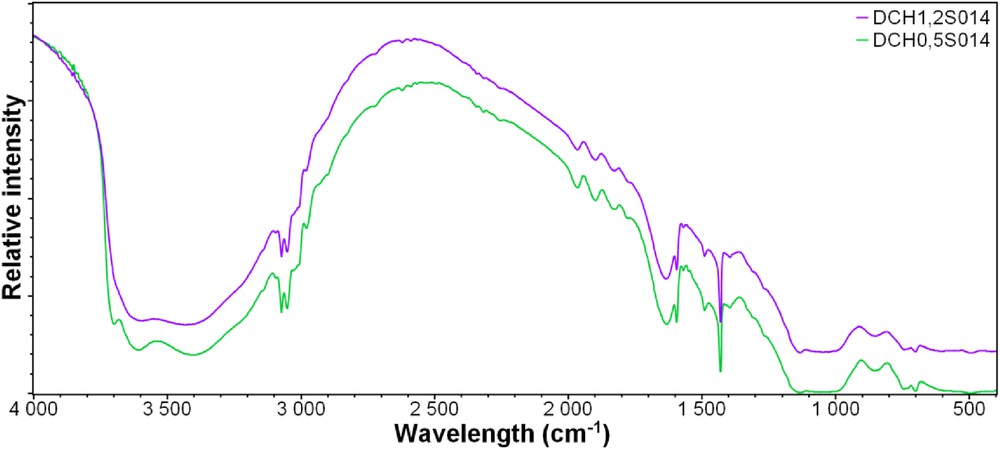
FT-IR spectra of the samples S014 recorded from 400 to 4000 cm−1.
To deeply analyze the samples, solid-state NMR spectroscopy has been performed to probe the local environment of 27Al and 29Si into the samples. Regarding 29Si NMR (cross polarized [CP]MAS and single pulse MAS), clay-like materials generally exhibit three main resonances between −40 and −80 ppm, called Tn environments where T stands for trifunctional silane and n the number of oxygen bonded to the probed silicon atom, according to the formula of the silicon part RSi(OM)n(OH)3−n with M corresponding to silicon or magnesium. Thus, T3 environments correspond to very well condensed silicon atoms without surrounding OH groups and generally located at the center of the tetrahedral sheets (RSi(OM)3), whereas T2 and T1 are related to silicon atoms at the border of these sheets, with respectively one and two surrounding OH groups [4–6,14,17]. Fig. 4(a) shows that the samples exhibit these three main features: around −76, −67, and −60 ppm corresponding, respectively, to T3, T2, and T1 environments. It also shows an evolution of the condensation of silicon into the tetrahedral sheet: T3 environment tends to decrease with increasing aluminum amount into the samples, which induces more defects into the structure. 27Al MAS NMR has also been performed to probe the position of this element into the structure, which depends on its coordination state. According to Fig. 4(b), two resonances can be seen around 8.5 and 53 ppm corresponding, respectively, to aluminum into octahedral (Al VI) and tetrahedral (Al IV) sheets. As the four samples exhibit these positions, aluminum is located in both sheets, which does not correspond to pure theoretical saponite. However, the area of the first peak around 53 ppm seems higher than the second one, which may indicate a more important presence of aluminum into the tetrahedral sheet.
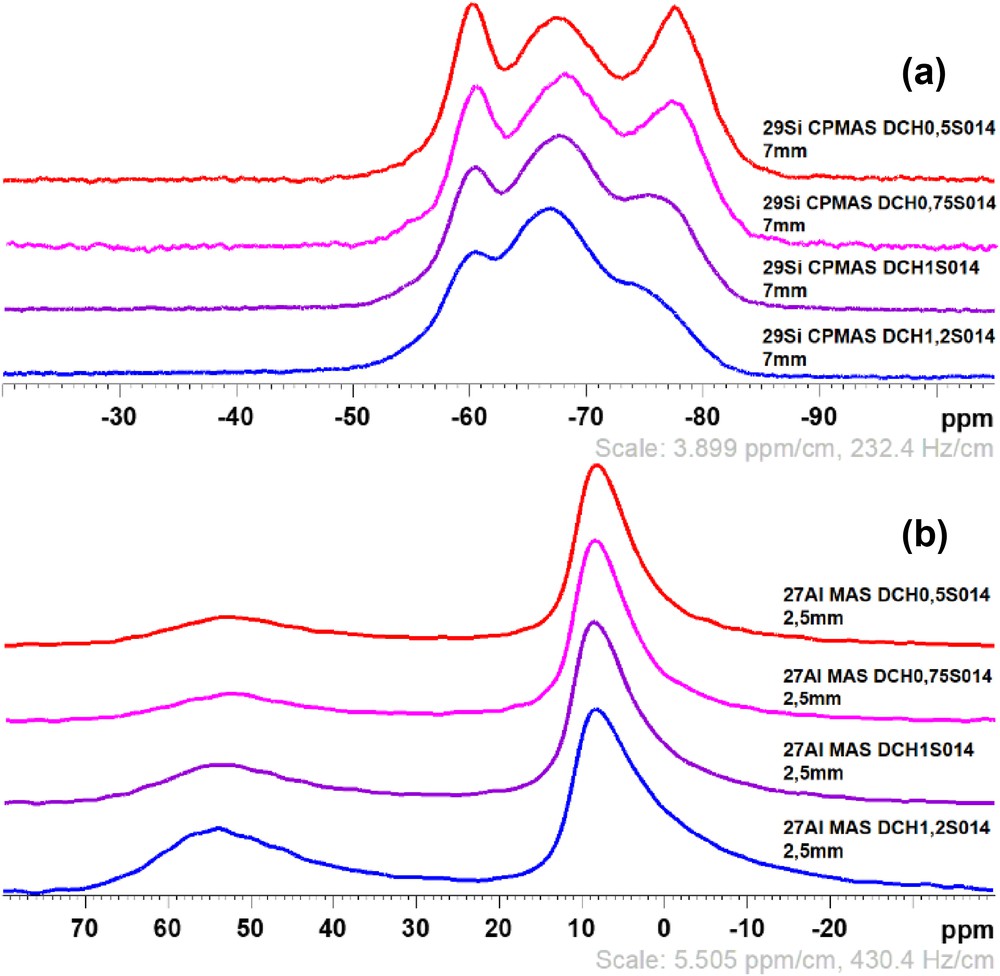
29Si CPMAS NMR (a) and 27Al MAS NMR (b) spectra of the samples S014.
These series of analyses prove the formation of a saponite-like hybrid organic–inorganic material with different aluminum amounts into the structure, as expected in the beginning of this work. To go further in this study, the aluminum precursor has been changed to characterize its impact on the structure of the synthesized materials.
3.2 Syntheses with N-propyltriethoxysilane
Two different magnesium salts, magnesium nitrate and magnesium chloride, have been tested as precursors during this experiment. The composition and calculated molar ratios are reported in Table 2.
Sol–gel synthesis parameters of the samples.
| Sample | Mg source | Mg amount (g) | Al amount (g) | Si amount (g) | Si/Mg molar ratio | Si/Al molar ratio | Mg/Al molar ratio | pH | Mass obtained (g) |
| DCHT006 | Mg(NO3)2·6H2O | 1.9 | – | 1.9 | 1.3 | – | – | 10.5 | 0.6 |
| DCH0.5S009 | MgCl2·6H2O | 1.7 | 0.5 | 2.0 | 1.2 | 7.0 | 6.0 | 11 | 0.5 |
| DCH0.5S010 | Mg(NO3)2·6H2O | 2.1 | 0.5 | 2.0 | 1.2 | 6.9 | 6.0 | 10.5 | 0.6 |
| DCH0.75S009 | MgCl2·6H2O | 1.8 | 0.7 | 1.9 | 1.1 | 4.3 | 4.0 | 10.5 | 0.7 |
| DCH0.75S010 | Mg(NO3)2·6H2O | 2.3 | 0.7 | 2.0 | 1.1 | 4.3 | 4.0 | 10 | 0.6 |
| DCH1S009 | MgCl2·6H2O | 1.9 | 1.1 | 2.0 | 1.0 | 3.0 | 3.0 | 11 | 0.6 |
| DCH1S010 | Mg(NO3)2·6H2O | 2.5 | 1.1 | 2.0 | 1.0 | 3.0 | 3.0 | 10 | 0.7 |
| DCH1.2S009 | MgCl2·6H2O | 2.1 | 1.4 | 1.9 | 0.9 | 2.3 | 2.5 | 10.5 | 0.7 |
| DCH1.2S010 | Mg(NO3)2·6H2O | 2.7 | 1.3 | 2.0 | 0.9 | 2.3 | 2.5 | 11 | 0.8 |
X-Ray diffractograms of these samples are shown in Fig. 5. Six main reflections are observed: 11.4°, 22.8°, 34.6°, 38.6°, 46°, and 60.8°–61.8° 2θ. Even if these samples are poorly crystallized according to the broadness of their peaks, their position does not correspond to hybrid organic–inorganic saponite-like materials [12–18]. According to the general indexation of hydrotalcite diffraction patterns in the literature, these peaks are assigned to (003), (006), (012), (015), (018), and (110) reflections, respectively [19–23]. As these patterns show no (001) reflection, this parameter can be estimated by multiplying the distance d003 by 3, which corresponds to the distance d001 according to the periodicity observed in the diffraction patterns. According to the Bragg law 2dhkl sin(θ) = nλ, the first peak indicates a distance d003 of about 7.9–7.7 Å, which corresponds to a c-cell parameter of about 2.57 Å. As a comparison, saponite-like materials have a basal distance of about two times the thickness of tetrahedral sheets plus the octahedral sheet and the interlayer distance, which corresponds to a final d001 value of about 11–15 Å under a relative humidity of 80%, sometimes greater depending on the size of the counterions and the structure content [7,8,17,18]. The distance observed in these diffraction patterns is much lower than the basal distance of saponite-like materials (around 10 Å plus the length of the organic chain), indicating that the structure of the synthesized materials cannot correspond to hybrid organic–inorganic saponite-like structures but to hydrotalcite-like compounds. Moreover, an interesting feature is generally observed in the low angle region: lamellar materials exhibit a peak around 60° 2θ corresponding to the (060;330) reflection. The position of this peak depends on the occupancy of the octahedral sheet: high angle values indicate small distances, typically dioctahedral nature (e.g., around 1.49 Å, Al3+ or Fe3+ with a vacant site), and the contrary indicates a trioctahedral nature (e.g., around 1.52 Å, Mg2+ or Fe2+). In this case, the width of this peak indicates the presence of two different features: one for the trioctahedral character around 1.51 Å, and the second peak around 1.54 Å (quite visible in the sample DCH0.5S010) for the dioctahedral nature of the synthesized materials. This indicates the presence of both divalent and trivalent cations into the octahedral sheet, respectively, Mg2+ and Al3+ considering the synthesis reactants in our case. Another interesting point is that no other phases are observed, impurities being generally represented by thin peaks in X-Ray diffractograms. Moreover, the pattern in Fig. 5 looks quite similar whatever the Si/Al substitution rate (between 0.5 and 1.2 in the structural formula of hybrid organic–inorganic saponite-like materials), whereas a shift in the main peaks (001) and (060) is generally observed in diffractograms of saponite-like materials related to changes in the structure of these lamellar compounds [12–18].
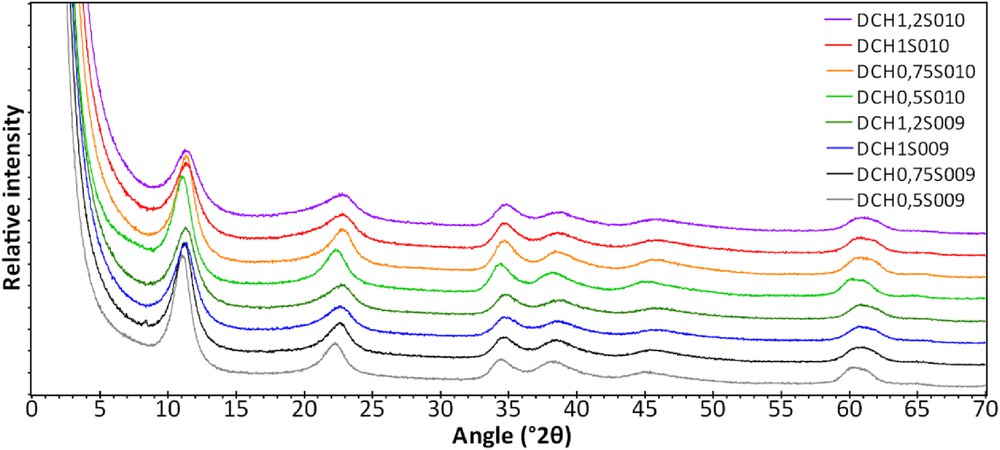
XRD patterns of the samples S009 (down) and S010 (up) synthesized by sol–gel method with rising Si/Al substitution rates from 0.5 to 1.2 for each series, washed three times with ethanol.
Considering these observations, FT-IR analyses have been performed on the synthesized samples using potassium bromide pellets. As seen from Fig. 6, the spectra obtained show several characteristic bands: the broad and strong absorption band in the range of 3700–3300 cm−1 corresponding to the OH stretching vibration of the surface and interlayer water molecules, as the OH bending vibration centered at 1660 cm−1. The three bands observed in the range of 2950–2900 cm−1 are related to the CH stretching vibration, and the presence of the stretching SiC vibration band around 1130 cm−1 proves the integrity of silicon and its organic part into the sample. Additional bands in the range of 1300–1200 cm−1 show the presence of magnesium in the materials. It is worthy to note that the positions of the vibrational bands do not change whatever the Si/Al substitution rate into the samples, and stretching SiAl bands are not observed in the 3500–3800 cm−1 range, similar to MgAl layered double hydroxide (LDH) samples.
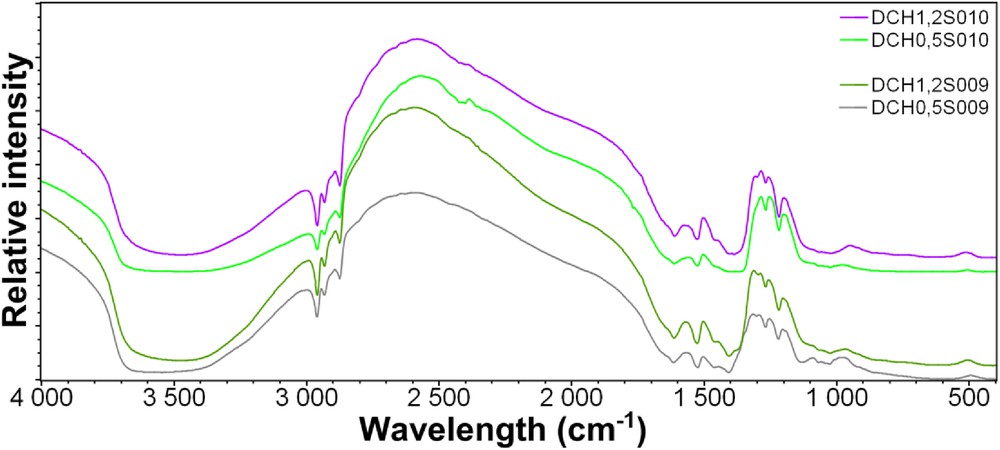
FT-IR spectra of the series of samples S009 and S010 recorded from 400 to 4000 cm−1.
The samples DCH0.5S009 and DCH1.2S009 have been analyzed using 27Al and 29Si solid-state NMR, as shown in Fig. 7. A third sample has been analyzed by 29Si CPMAS NMR, DCHT005, a talc-like hybrid sample synthesized without aluminum to compare the T-type environments in these materials. Fig. 7(a) shows three separate peaks, −67.9, −56.9, and −48.3 ppm, which correspond, respectively to T3, T2, and T1 environments, but in a very low amount according to the single pulse MAS analyses. This means that only very small amounts of silicon atoms are located into the structure of the synthesized materials. However, T3 environments seem to be more important than T2 and T1 ones, indicating a high condensation of silicon species into the tetrahedral sheets except in the sample DCH1.2S009, which is supposed to have the higher substitution rate and a lower Si/Al molar ratio. For 27Al NMR spectra, Fig. 7(b), a sample has been prepared without silicon precursor (DCH AlMg01), corresponding to the chemistry of a pure hydrotalcite phase. All of the samples exhibit one strong peak around 9.1 ppm, and a very small one at 61.3 ppm is observed in the sample DCH1.2S009. The position of the first peak indicates the presence of aluminum in octahedral positions (Al VI), as proved by the sample without silicon source, whereas the second shows the same element in tetrahedral environment (Al IV), which is not possible in pure hydrotalcite phases. According to the results obtained by 27Al and 29Si NMR, it can be assumed that the synthesized materials are made of two different compounds: the first is constituted of traces of saponite-like structure, with silicon and aluminum in tetrahedral sheets, and the second part is essentially made of aluminum in an octahedral environment.
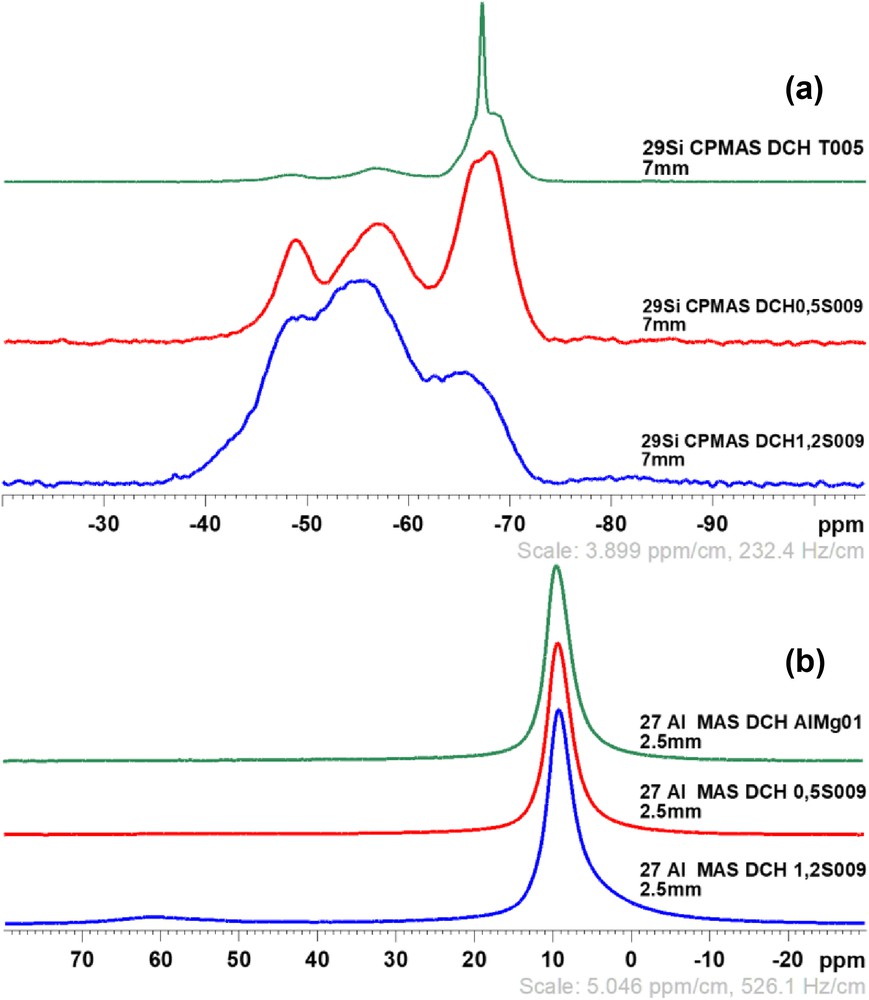
29Si CPMAS NMR spectra (a) of the samples DCH0.5S009 (red) and DCH1.2S009 (blue) as compared with a synthesized talc-like hybrid (green), and 27Al MAS NMR spectra (b) of the samples S009 as compared with a sample synthesized only with aluminum and magnesium (green).
The thermal stability of the samples has been tested using TGA from 25 to 600 °C with a slope of 5 °C min−1, and is shown in Fig. 8. In all cases, the curves can be divided into two main regions: the first one, ranging from 25 to 250 °C, is related to the dehydration (loss of the surface water) of the samples. It corresponds to a mass loss of approximately 16–18% in the samples. The second region is located between 250 and 600 °C and is related to the dehydroxylation and decomposition of the organic part, with a final mass loss of almost half of the starting mass (around 43–47%). Moreover, the sample DCHT006, a talc-like hybrid material synthesized the same way without aluminum, presents higher thermal stability than other materials, which can be explained by the insertion of aluminum into the structure of these lamellar materials inducing structural defects and cavities.
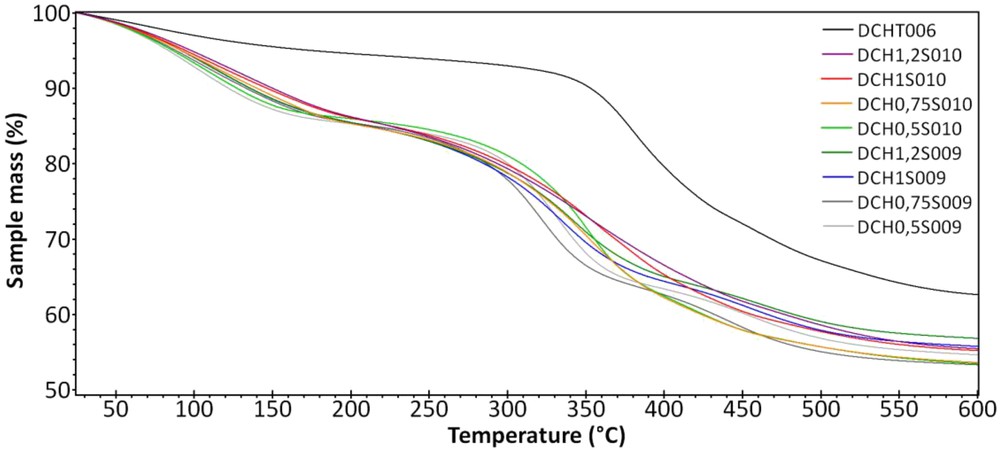
TGAs of the samples S009 and S010, from 25 to 600 °C. DCHT006 is a talc-like hybrid synthesized the same way, without aluminum.
By using this synthesis method in our study, MgAl LDH has, however, been surprisingly formed during the synthesis of hybrid organic–inorganic saponite-like materials. Hydrotalcite belongs to the LDH family with a structure based on brucite (Mg(OH2))-like layers (similar to octahedral sheets in natural clays), in which a part of the divalent cations are substituted by trivalent ones, for example, a part of Mg2+ ions are replaced by Al3+ ions [19–31]. These structural substitutions generate positive charges within the stacked layers, which are kept electronically neutral by the intercalation of anions and water among the interlayer spaces, as seen in Fig. 9. The general structural formula of LDHs can be denoted as
| [MxIIMyIII(OH)(2x + 2y)](An−)(y/2)·mH2O |
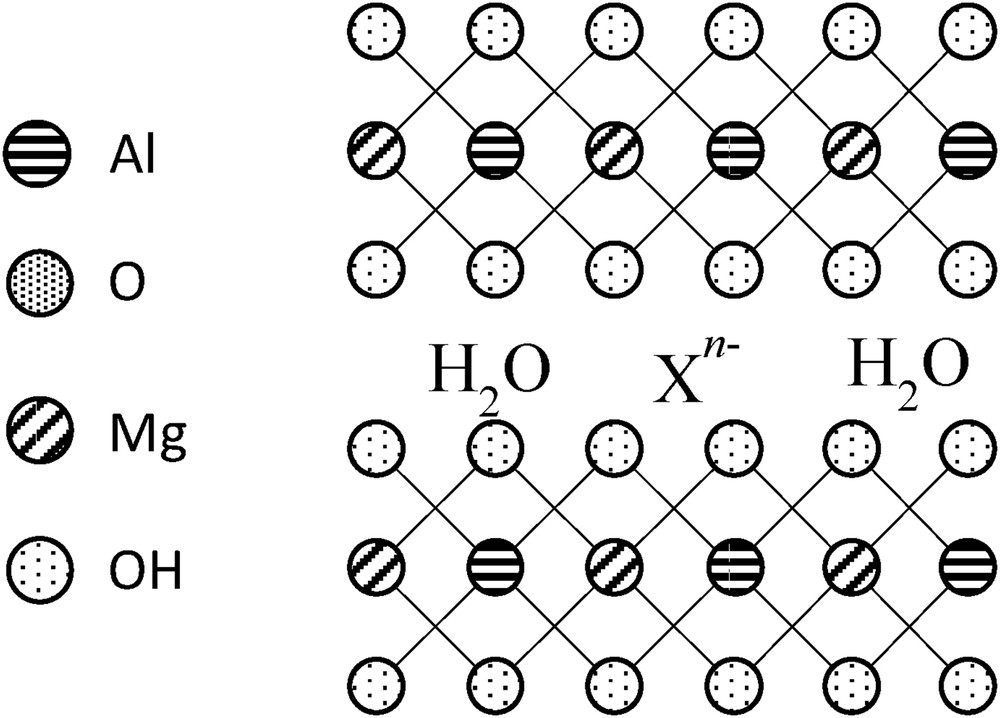
Schematic representation of an AlMg LDH (also called hydrotalcite).
To go further in the formation of this material, a different organoalcoxysilane has been tested in the same conditions.
3.3 Syntheses with trimethoxyphenylsilane
Synthesis carried out with trimethoxyphenylsilane was performed using magnesium chloride hexahydrate salt as a magnesium source. The composition and calculated molar ratios are reported in Table 3.
Sol–gel synthesis parameters of the samples synthesized with trimethoxyphenylsilane.
| Sample | Mg amount (g) | Al amount (g) | Si amount (g) | Si/Mg molar ratio | Si/Al molar ratio | Mg/Al molar ratio | pH | Mass obtained (g) |
| DCH0.5S013 | 1.8 | 0.5 | 2.0 | 1.2 | 7.0 | 6.0 | 11 | 1.4 |
| DCH0.5S013b | 1.8 | 0.5 | 2.0 | 1.2 | 7.0 | 6.0 | 10.5 | 0.9 |
| DCH0.75S013 | 1.9 | 0.8 | 2.0 | 1.1 | 4.3 | 4.0 | 11 | 1.2 |
| DCH0.75S013b | 1.9 | 0.8 | 2.0 | 1.1 | 4.3 | 4.0 | 11 | 0.9 |
| DCH1S013 | 2.1 | 1.1 | 2.0 | 1.0 | 3.0 | 3.0 | 10 | 0.9 |
| DCH1S013b | 2.1 | 1.1 | 2.0 | 1.0 | 3.0 | 3.0 | 10.5 | 1.4 |
| DCH1.2S013 | 2.2 | 1.4 | 2.0 | 0.9 | 2.3 | 2.5 | 10.5 | 1.1 |
| DCH1.2S013b | 2.2 | 1.4 | 2.0 | 0.9 | 2.3 | 2.5 | 11 | 2.2 |
XRRD patterns of the samples synthesized with trimethoxyphenylsilane are shown in Fig. 10. Even if some impurities are visible, six main reflections are observed: 5.7°–7°, 11.4°, 22.8°, 34.8°, 38.5°, and 60.7° 2θ, which are quite similar to the ones observed in the previous samples. Moreover, this first diffraction peak at small angle tends to shift to lower angle values when the substitution rate of silicon by aluminum increases. According to the Bragg law and the general indexation of hybrid lamellar materials in the literature, this first reflection corresponds to basal reflection values (001) between 13 (sample DCH0.5S013) and 15.5 Å. Even if the samples exhibit hydrotalcite-like features, the presence of this basal distance (001) plus the (060) diffraction peak at 1.52 Å also indicates the formation of a saponite-like material, with octahedral sheets containing divalent cations (magnesium according to our precursors).
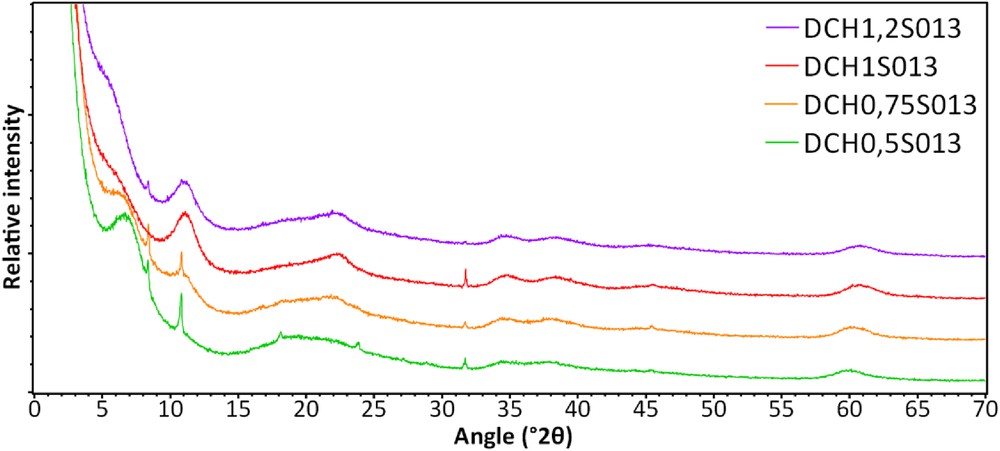
XRD patterns of the samples synthesized with trimethoxyphenylsilane by sol–gel method, washed three times with distilled water.
Solid-state NMR experiments have also been performed on these samples to probe the local environment of silicon and to study the position of aluminum (whether aluminum is present in tetrahedral sheets (Al IV) and/or in octahedral sheets (Al VI) in the structure). Fig. 11(a) corresponds to the 29Si CPMAS analyses of the samples DCH0.5S013 and DCH1.2S013, the two extreme samples of the series according to the Si/Al substitution rates. These two samples exhibit three main local environments, −75.5, −68, and −60.7 ppm for T3, T2, and T1 environments, respectively. These values are slightly greater than the ones observed in the previous samples but are still in the range of T-types environments. However, T3 environments seem to be of greater importance in the sample DCH0.5S013 with the lowest Si/Al substitution rate, whereas T2 and T1 environments are dominant in the sample DCH1.2S013, indicating a lower condensation of silicon in the structure, and more structural defects than the first sample. Regarding the 27Al MAS NMR on the four samples, as shown in Fig. 11(b), the four spectra display two resonances, a first one around 9 ppm, and a smaller one at 54 ppm, which indicates the presence of aluminum in both octahedral (Al VI) and tetrahedral (Al IV) positions, respectively.
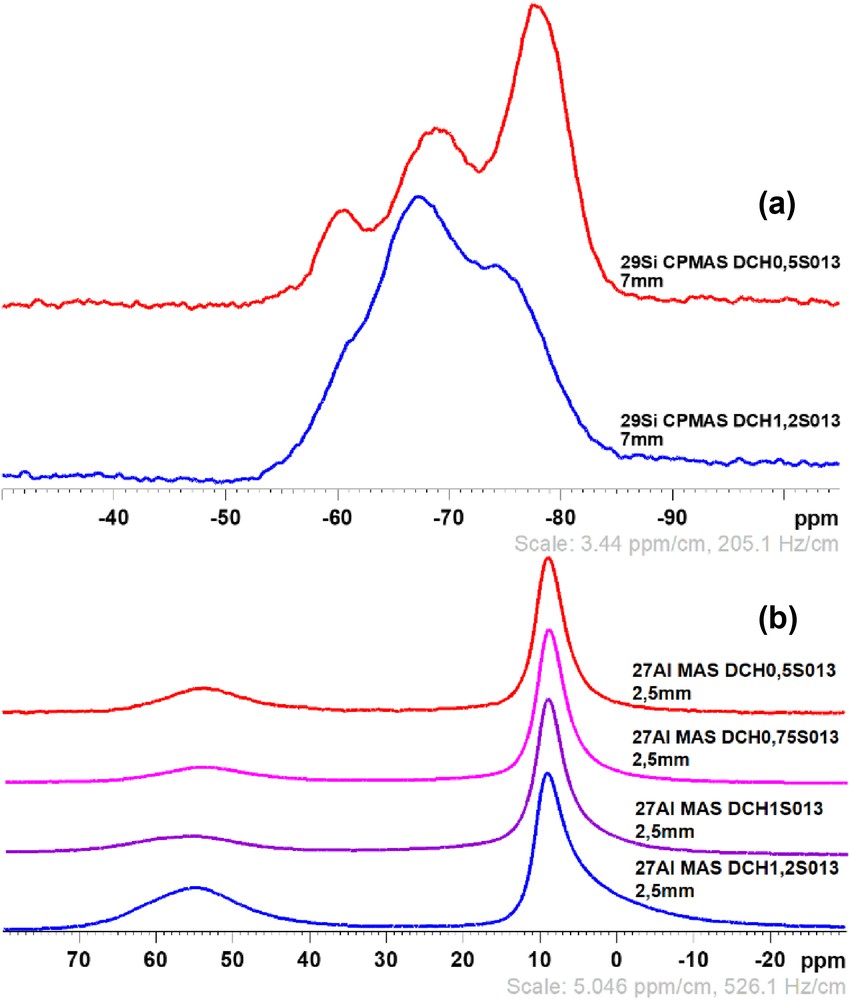
29Si CPMAS NMR spectra (a) of the samples DCH0.5S013 (red) and DCH1.2S013 (blue), and 27Al MAS NMR spectra (b) of the samples S013.
The results obtained with these materials synthesized using trimethoxyphenylsilane confirm our previous observations with the N-propyltriethoxysilane: XRRD data prove the formation of two different lamellar materials, and 27Al NMR analyses show the presence of aluminum in both octahedral and tetrahedral sheets, which is, respectively, observed in hydrotalcite and saponite-like materials. Moreover, it seems that the increase in the aluminum content in the samples induce more defects in the structures and a greater ratio of LDH formed during the sol–gel synthesis.
The most plausible hypothesis to the formation of such a material instead of the expected hybrid organic–inorganic saponite-like product is the nonformation of tetrahedral SiAl sheets, which may be related to the precursors. According to the results mentioned previously, NMR data show the synthesized materials are mainly made of aluminum in octahedral positions, even if some traces of tetrahedral silicon are detected. The XRD patterns of the synthesized materials are similar to MgAl LDHs called hydrotalcite, and the remaining silicon into the samples can be related to the presence of some layers of lamellar hybrid organic–inorganic, as expected at the beginning of this study.
4 Conclusions
As hybrid organic–inorganic saponite-like materials were expected by the use of the sol–gel synthesis process, the experiments have been performed according to Jaber and Miehé-Brendlé [6]. Depending on the precursors, XRRD and solid-state NMR results show the formation of another type of lamellar material made of magnesium and aluminum, even if silicon has been used during the synthesis process. The presence of small amounts of silicon into the samples shown by 29Si solid-state NMR proves the formation of some saponite-like layers with silicon and aluminum in tetrahedral positions, but the samples are mainly made of a stacking of brucite layers with magnesium partly substituted by aluminum, also called MgAl hydrotalcite or LDH. Aluminum is present in octahedral sheets (Al VI), and the basal distance between the layers is estimated at 7.7 Å according to XRRD analyses, which is much lower than the values expected with saponite-like materials plus the length of the organic chain. Aluminum acetylacetonate is known in the synthesis of lamellar materials, but is not always the best choice depending on the type of material expected.
By changing the different precursors, especially the aluminum source, the analyses showed the formation of (1) almost pure MgAl LDHs, also called hydrotalcites, (2) a mixed phase between hydrotalcite and saponite-like structure, or finally (3) pure saponite-like materials by changing the aluminum salt, as outlined in Fig. 12. The changes in the formation of these lamellar materials may be related to the affinity of the aluminum precursors over the organoalcoxysilane: aluminum acetylacetonate might have a lower affinity to N-propyltriethoxysilane and trimethoxyphenylsilane than aluminum chloride, and this interaction reduces hydrolysis and condensation rates during the addition of sodium chloride, as shown by Danks et al. [32]. This is the reason why the choice of the appropriate precursors for the sol–gel synthesis of lamellar materials is an important parameter to take into account. Further studies concerning the formation mechanisms of these materials instead of saponite-like hybrids are under progress.
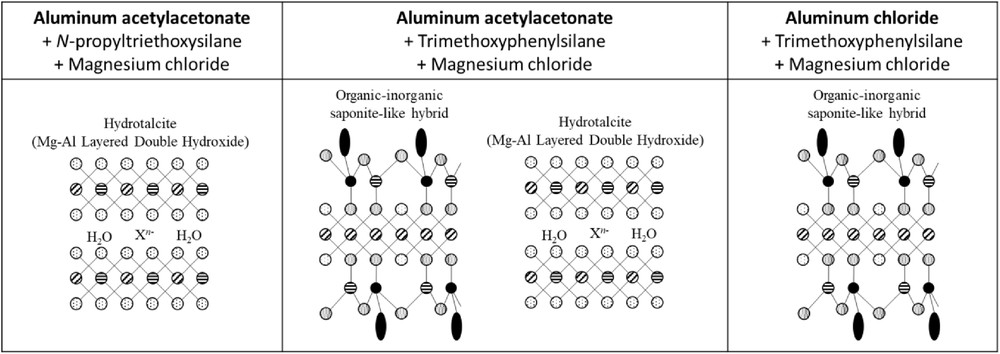
Summary of the sol–gel synthesis conditions and impact of the different precursors.
Acknowledgments
This work was supported by Université de Haute Alsace and École Doctorale Physique et Chimie-Physique (ED 182).
XRD, FT-IR, TGA, and NMR measurements were performed on the technical platforms of IS2M. The authors are very grateful to L. Michelin, H. Nouali, and S. Rigolet for their contributions.