

1 Introduction
External stimuli can change the physical properties of materials by driving phase transitions in solid state. For example, temperature, light, pressure, and electric fields have been used to alter magnetic, electrical, and optical properties [1–12]. These studies not only elucidate the unique functionalities of solid-state materials but also are a critical step toward practical applications, as external stimuli can influence physical properties. One class of materials that exhibit important external stimuli–induced switching of physical properties is the family of cyano-bridged metal complexes [1–18], for which charge transfer (CT)–induced phase transition coupled with important structural reorganization is at the origin of large bistability. Indeed, various studies have demonstrated that external stimuli, including temperature, light, pressure, and electric fields, are able to modify their electronic and spin states [19–35]. Among many contributions around the world for developing the field of functional materials, it is fair to emphasize the work of Michel Verdaguer, who has developed a rational approach to new systems, from quantum chemistry to applications and with an intensive use of synchrotron radiation. Indeed, the presence of cyano ligands imparts structural flexibility, allowing for spin state conversion when related to important structural reorganizations. Because of the potential of such complexes, our research has focused on investigating stimulus-induced functionalities in cyano-bridged metal complexes [36–41]. We have studied one Prussian Blue analogue in particular; rubidium manganese hexacyanoferrate, RbxMn [Fe(CN)6](x+2)/3·zH2O, which has diverse functionalities [42]. Herein, we describe its thermally and photoinduced CT–induced structural phase transitions, and we discuss how new ultrafast techniques may allow understanding the basic mechanism behind photoinduced phenomena.
2 Materials and methods
Rubidium manganese hexacyanoferrate (Fig. 1) can be prepared in various forms such as powders and microcrystalline fluid dispersions. Mixing a solution of manganese chloride with a mixed aqueous (aq) solution of rubidium chloride and potassium ferrocyanide yields a light brown powder after filtering and drying the obtained precipitate. The powder comprised 2.1 ± 1.1 μm cubic crystals [43]. On the other hand, mixing a polyethylene glycol monolaurate matrix containing manganese chloride (aq) and rubidium chloride (aq) solutions with polyethylene glycol monolaurate containing potassium ferrocyanide (aq) and rubidium chloride (aq) solutions realizes a fluid dispersion after centrifuging, washing in methanol, and air drying the obtained precipitate. The fluid dispersion is composed of plate-shaped microcrystals measuring 690 ± 250 nm, 250 ± 125 nm, and 94 ± 37 nm in length, width, and height, respectively [44]. Spin-coating a prepared dispersion fluid onto a well-washed glass substrate forms a film with 820 ± 40 nm thick [44]. In the synthesized compound, the valence states of metal ions are mainly RbIxMnII [FeIII(CN)6](x+2)/3·zH2O. A small amount of impurity of RbI2MnII [FeII(CN)6]·zH2O exists depending on x[42,44]. Even if the synthesis is proceeded under similar conditions, the compositions of the samples always differ slightly, ca. ±0.06 difference appears in x. It is likely because of slight differences in temperature, chemical concentration, and stirring speed. The relationship between composition and phase transition phenomenon is an important topic, which was the scope of previous studies [42].
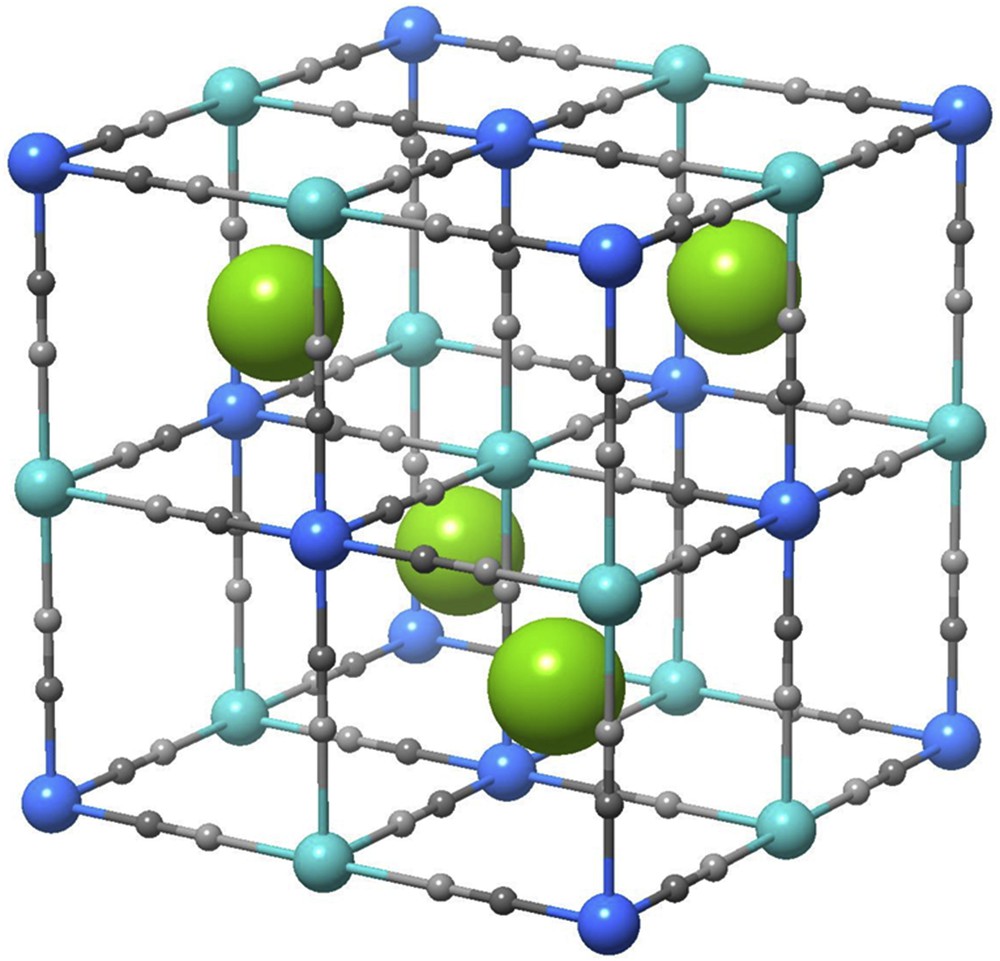
Crystal structure of rubidium manganese hexacyanoferrate. Rb, Mn, Fe, C, and N atoms are denoted as green, aqua, blue, small dark gray, and light gray circles, respectively.
3 Results and discussion
3.1 Thermally induced phase transition
Rubidium manganese hexacyanoferrates are known for presenting bistability between two states: a low-temperature (LT) tetragonal FeII (S = 0)–CN–MnIII (S = 2) state and a high-temperature (HT) cubic FeIII (S = 1/2)–CN–MnII (S = 5/2) state. As LT and HT states have different entropies, thermal phase transition may occur. In addition, the elastic interactions between metal ions and the lattice mediated by the cyano-bridged ligands are responsible for cooperativity and the appearance of first-order phase transition with broad thermal hysteresis. One example of materials with thermally induced phrase transition is RbxMn [Fe(CN)6](x+2)/3·zH2O (x > 0.61) [42].
Fig. 2a shows the product of the molar magnetic susceptibility (χM) and the temperature (T) versus T plots of a plate-shaped microcrystal of Rb0.97Mn [Fe(CN)6]0.99·0.3H2O [44]. Upon cooling the χMT value in the HT phase decreases around 176 K (= T1/2↓). On the other hand, upon warming the LT phase suddenly increases around 294 K (= T1/2↑). In addition, Rb0.97Mn [Fe(CN)6]0.99·0.3H2O exhibits a very wide thermal hysteresis loop [ΔT (= T1/2↑ − T1/2↓)] of 118 K. Both the change in the magnetic susceptibility and the change in the infrared (IR) spectra shown in Fig. 2b are characteristics of a phase transition from the HT phase comprising FeIII (S = 1/2)–CN–MnII (S = 5/2) to the LT phase composed of FeII (S = 0)–CN–MnIII (S = 2). In addition to the change in electronic state, important structural reorganizations occur during the phase transition, as observed in the X-ray diffraction (XRD) pattern in Fig. 2c. The CT induces, on the one hand, an important variation in the population of the bonding states, and the averaged Fe–C bond distance decreases from ≈2.2 Å (HT) to ≈1.9 Å (LT), which is the main contribution to the global volume contraction from HT to LT phases. On the other hand, the six Mn–N distances, symmetry equivalent in the HT cubic phase, split into four short (≈1.9 Å) and two long (≈2.3 Å along the c axis) distances in the LT phase because of the Jahn–Teller distortion. This structural phase transition is therefore associated with important changes in the lattice parameters from cubic (HT phase; Fm3m, aHT = 10.56065 (10) Å) to tetragonal (LT phase; I4m2, aLT = bLT = 7.0874 (3) Å, cLT = 10.5112 (12) Å at 15 K). Because of the change from F to I cell, . In addition, X-ray photoelectron spectroscopy, synchrotron radiation X-ray powder diffraction, and X-ray absorption near-edge structure (XANES) show that the occurrence of a metal-to-metal CT (MMCT) from FeIII–CN–MnII to FeII–CN–MnIII as temperature is decreased is concomitant with a Jahn–Teller distortion of the produced MnIII (Fig. 3) [45–47].

Rb0.97Mn [Fe(CN)6]0.99 · 0.3H2O microcrystals show a thermally induced phase transition. (a) χMT−T plots under 5000 Oe with cooling (black circles) and warming (white circles) processes. (b) Temperature dependence of the CN stretching mode of the IR spectrum at 300 K (upper) and 20 K (lower). (c) XRD patterns at 300 K (upper) and 15 K (lower), and Rietveld analyses. Observed and calculated intensities of the diffraction patterns are represented. Bars represent the calculated positions of the Bragg reflections in the crystal structure. Panels are modifications of Chem. Mater. 24 (2012) 1324 [44]. Copyright 2012 American Chemical Society.

Reaction scheme between the LT and HT phases. Panels are modifications of Inorg. Chem. 43 (2004) 5231 [40]. Copyright 2004 American Chemical Society.
As a class II mixed-valence compound, the CT–induced phase transition in rubidium manganese hexacyanoferrates can be represented by two parabolic potential–energy curves in the nuclear coordinates of the coupled vibrational modes [48,49]. Because of the valence isomers, the ground state surface exhibits two minima in the vibrational coordinates, where these two vibronic states interact. Fig. 4a shows the free energy surface of this mixed-valence system [43], at T = 0 K where the ground state is the FeII–MnIII vibronic state (Fig. 4a, black line). An elongation-type Jahn–Teller distortion because of MnIII results in two energy minima for FeII–MnIII (Fig. 4a, gray curve). Increasing the temperature changes the free energy surfaces due to the gain in entropy, which stabilizes the higher entropy FeIII–MnII state (Fig. 4a,c) above the phase transition temperature Tp.
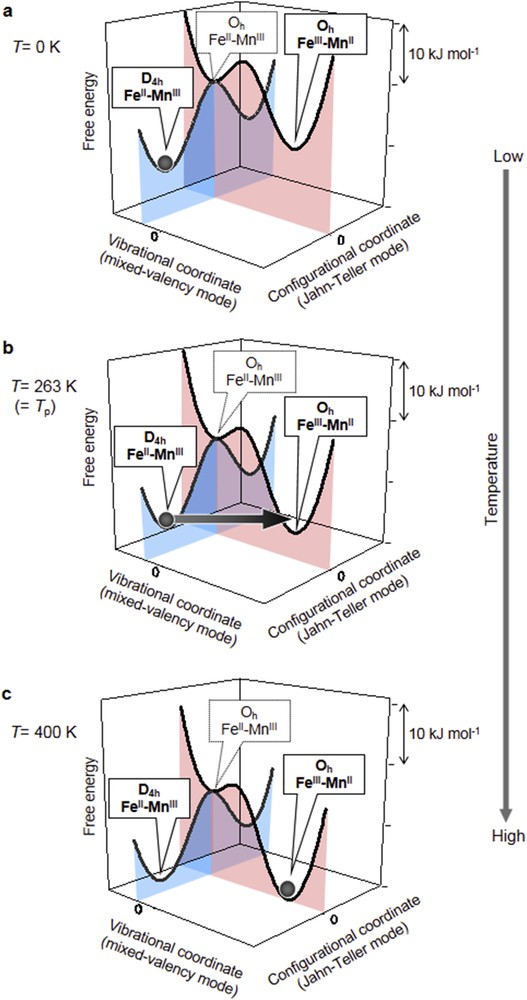
Free energy surfaces of this system in mixed-valence (black curve) and Jahn–Teller (gray curve) modes. FeII–MnIII (FeIII–MnII) is a ground (metastable) state at (a) T = 0 K, (b) T = 263 K (= Tp). (c) FeIII–MnII (FeII–MnIII) is a ground (metastable) state at T = 400 K. Gray spheres indicate population. Panels are modifications of Inorg. Chem. 43 (2004) 5231 [43]. Copyright 2004 American Chemical Society.
The thermally induced CT phase transition has a mechanism that is related to the structural flexibility of rubidium manganese hexacyanoferrate. First principles calculations confirm that the IR spectrum (Fig. 2) of the LT phase displays a CN stretching mode ν(C≡N) of FeII–CN–MnIII around 2100 cm−1, whereas the far-IR spectrum reveals transverse modes δ(Fe–CˆN–Mn): a transverse translational mode at 304 cm−1 and a transverse librational modes at 253 and 503 cm−1 [50,51]. Of particular interest is the effect of the transverse modes δ(Fe–CˆN–Mn) on electronic states. Fig. 5 shows two types of Fe–CN–Mn structural configurations for which the molecular orbitals were calculated using the discrete-variable Xα method [44]. One is the Fe–CN–Mn linear configuration (Fig. 5a), and the other one is a distorted configuration (Fig. 5b), which mimics the effect of the transverse translational mode. The linear configuration has a metal ion charge distribution corresponding to a Fe3dzx−C2px≡N2px−Mn3dzx molecular orbital, which is similar to the LT phase for Fe2+−C≡N–Mn3+ in the classical valence states. The calculated distribution is 49% (Fe) and 26% (Mn). On the other hand, the charge distributions for the distorted configuration are 22% for Fe and 58% for Mn, indicating that the valence state is close to Fe3+−C≡N–Mn2+ (the HT phase). Hence, the distortion due to transverse translational mode causes interchanging of the valence states of the metal ions. These results suggest that a thermal excitation of the transverse translational mode can play an important role for driving the CT phase transition.

Fe3dzx−C2px≡N2px−Mn3dzx magnetic orbitals for the (a) linear configuration with ∠Fe–C≡N–Mn angle of 180° and (b) distorted configuration due to transverse translational mode with ∠Fe–C≡N = ∠Mn–N≡C = 168° calculated by the discrete-variable Xα method. Panels are modifications of Chem. Mater. 24 (2012) 1324 [44]. Copyright 2012 American Chemical Society.
3.2 Photoinduced phase transition
The strong cooperativity of the CT process in rubidium manganese hexacyanoferrates and the coupled important structural reorganizations are responsible for the stabilization of photoinduced CT state, which alters the electronic spin state. This opens the way for controlling optically the magnetization of this family of systems. Indeed, the photodemagnetization effect, induced by a single laser pulse light, and the reversible photomagnetic switching between ferromagnetism and antiferromagnetism, induced by alternating green and blue light irradiation, were observed in a powder-type sample of rubidium manganese hexacyanoferrate and are described hereafter [52,53].
3.2.1 Photodemagnetization phenomenon
A spontaneous magnetization appears in the LT phase at less than 12 K, with a saturated magnetization (Ms) of 3.6 μB at 2 K and a coercive field (Hc) of 1800 Oe. The ferromagnetic spin ordering of the MnIII (S = 2) ions can explain the Ms value, and this ferromagnetism may be explained by a mixed-valence mechanism [54,55].
The LT phase has a broad optical absorption band around 400–600 nm, which is because of the MMCT band [56]. Thus, we investigated the photomagnetic effect of the LT phase using a pulsed laser with wavelength of 532 nm (pulse width, 6 ns). As shown in Fig. 6a and b, irradiating with a pulsed laser (power density of P = 130 mJ·cm−2·pulse−1·at 3 K) leads to demagnetization. The inset of Fig. 6a shows the dependence of the photodemagnetization with the laser power density (P) on the one-shot-pulsed-laser irradiation at 3 K. The magnetization value decreases for P > 9.3 mJ·cm−2·pulse−1 (= Pth; laser power density threshold). On the other hand, for P < Pth, irradiation does not affect magnetization even after 1000 shots. A P value greater than Pth enhances the photoconversion. The LT phase is recovered upon annealing at 150 K. The IR spectra before and after the one-shot-laser-pulse irradiation indicate that a photoinduced CT phase transition from the LT to HT phases is responsible for the observed photodemagnetization. Fig. 7, which plots the photoinduced phase transition of the adiabatic potential energy as a function of the order parameter [57], indicates that irradiation excites the ground state to the Franck–Condon state. The Franck–Condon state either undergoes a structural trapping toward a hidden substable state or relaxes to the ground state. The behavior of the excited state depends on P. It proceeds to the photoproduced HT phase for P > Pth, but relaxes to the ground state for P < Pth. Because the thermal energy barrier (ΔG) separates the photoinduced HT and LT phases, the photoinduced HT phase is then stabilized in a low-temperature range.

Effect of the one-shot-pulsed laser irradiation. (a) Magnetization versus time plots under 200 Oe before (white circles) and after one-shot-pulsed-laser irradiation (130 mJ·cm−2·pulse−1) (black circles). Inset shows the laser power density (P) dependences of the photodemagnetization with one-shot-pulsed-laser irradiation under 200 Oe. (b) Magnetization versus temperature plots under 200 Oe before (white circles), after the one-shot-pulsed-laser irradiation (black circles), and after thermal treatment (white squares). Panels are modifications of Appl. Phys. Lett. 82 (2003) 1245 [52]. Copyright 2003 American Institute of Physics.
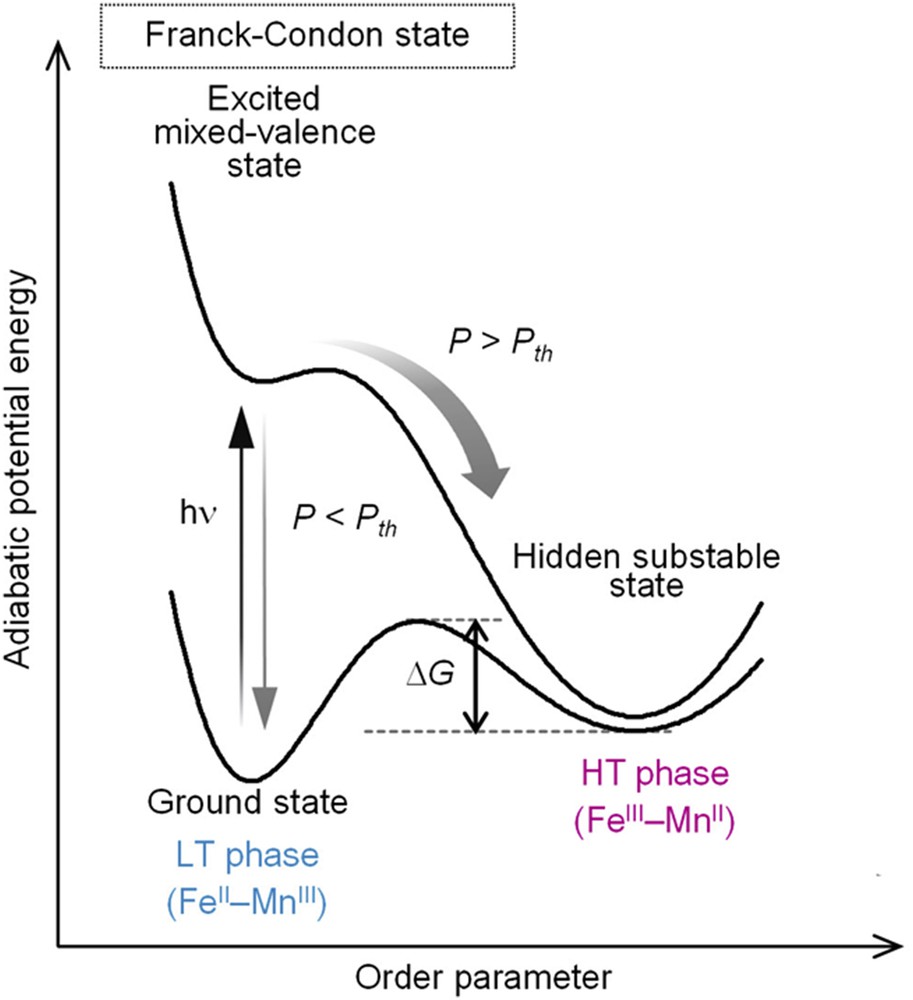
Schematic of the one-shot-pulsed-laser–induced phase transition from the stable FeII–MnIII phase to the hidden substable FeIII–MnII phase. Pth indicates the laser power density threshold. Panel is a modification of Appl. Phys. Lett. 82 (2003) 1245 [52]. Copyright 2003 American Institute of Physics.
3.2.2 Reversible switching of ferromagnetism and antiferromagnetism
As mentioned previously, the LT phase of rubidium manganese hexacyanoferrate displays a MMCT band at 400–600 nm [56]. Hence, green laser with wavelength of 532 nm is a suitable light irradiation source. Fig. 8a shows the IR spectra with the light irradiation of a continuous wave diode green laser (wavelength, 532 nm; power, 30 mW·cm−2). The broad peak around 2100 cm−1 in the LT phase before irradiation is due to FeII–CN–MnIII. A sharp peak appears at 2153 cm−1 and the FeII–CN–MnIII peak decreases upon irradiation with 532 nm light. As the HT phase also has a peak at 2153 cm−1, the sharp peak is assigned to MnII–NC–FeIII. Next, we investigated the photoreversibility. Because our results indicate similar valence states for the photoinduced and HT phases, and the HT phase has a ligand-to-metal CT band around 410 nm [56], we irradiated the photoinduced phase with blue light from a filtered Xe lamp (wavelength, 410 ± 30 nm; power, 13 mW·cm−2). Fig. 8a (right), which plots the peak intensities of FeII–CN–MnIII as a function of irradiation time, shows that the FeIII–CN–MnII peak decreases and the FeII–CN–MnIII peak increases. This switching is repeatedly observed. Fig. 8b (left) plots the magnetization value while alternately irradiating with 532 and 410 nm light. The magnetization value decreases upon irradiating the LT phase with 532 nm light at 3 K (black circles). However, the magnetization value increases upon successively irradiating the photoinduced phase with 410 nm light (white circles). This behavior is reversible and cycling is shown in Fig. 8b (middle). Fig. 8b (right) shows the reverse process from the photoinduced phase to the LT phase. The magnetization increases up to a plateau-M425nm upon initial irradiation with 425 nm light, but the magnetization increases up to a plateau-M410nm upon subsequent irradiations with 410 nm light because of the photostationary state between the photodemagnetization (LT → photoinduced phase) and the photoinduced magnetization (photoinduced → LT phase).

Visible-light reversible photomagnetic effect. (a) IR spectrum shows a visible-light reversible change. Irradiation-time dependence at 3 K upon irradiation with 532 and 410 nm light (left). Peak intensity at 2108 cm−1 as a function of irradiation with 532 nm light (black circles) and 410 nm light (white circles) (right). (b) Magnetization versus temperature curves under 200 Oe (left) before irradiation (white squares), after irradiation with 532 nm light for 100 min (black circles), after irradiation with 410 nm light for 80 min (white circles), and after the thermal annealing treatment of 180 K (black squares). Magnetization versus irradiation-time plot at 3 K by alternating between 532 nm (black circles) and 410 nm (white circles) light irradiation, and the magnetization value after thermal treatment (Δ) at 180 K (black square) (middle). Photostationary state between photodemagnetization and photoinduced magnetization. Magnetization versus irradiation-time plot at 3 K before irradiation (black square), and upon irradiation with 425 nm light (black circles) and 410 nm light (white circles) (right). Panels are modifications of Chem. Mater. 20 (2008) 423 [53]. Copyright 2008 American Chemical Society.
The mechanism for the reversible photomagnetism is shown in Fig. 9. The MMCT (FeII → MnIII) band in the LT phase is excited to photoexcited state I upon irradiation with 532 nm light. Photoexcited state I, which has the same valence state as the HT state, is a metastable phase because of a sufficient ΔG. Exciting the ligand-to-metal CT (CN– → FeIII) band of [Fe(CN)6]3− in the photoinduced phase by 410 nm light excites the state to photoexcited state II. This state subsequently proceeds to the LT phase. The photoinduced phase is an antiferromagnet [53], and ferromagnetic coupling between the MnIII (S = 2) sites results in a LT ferromagnetic phase [56]. Consequently, the magnetization value is changed by optically switching between the LT phase and the photoinduced phase.
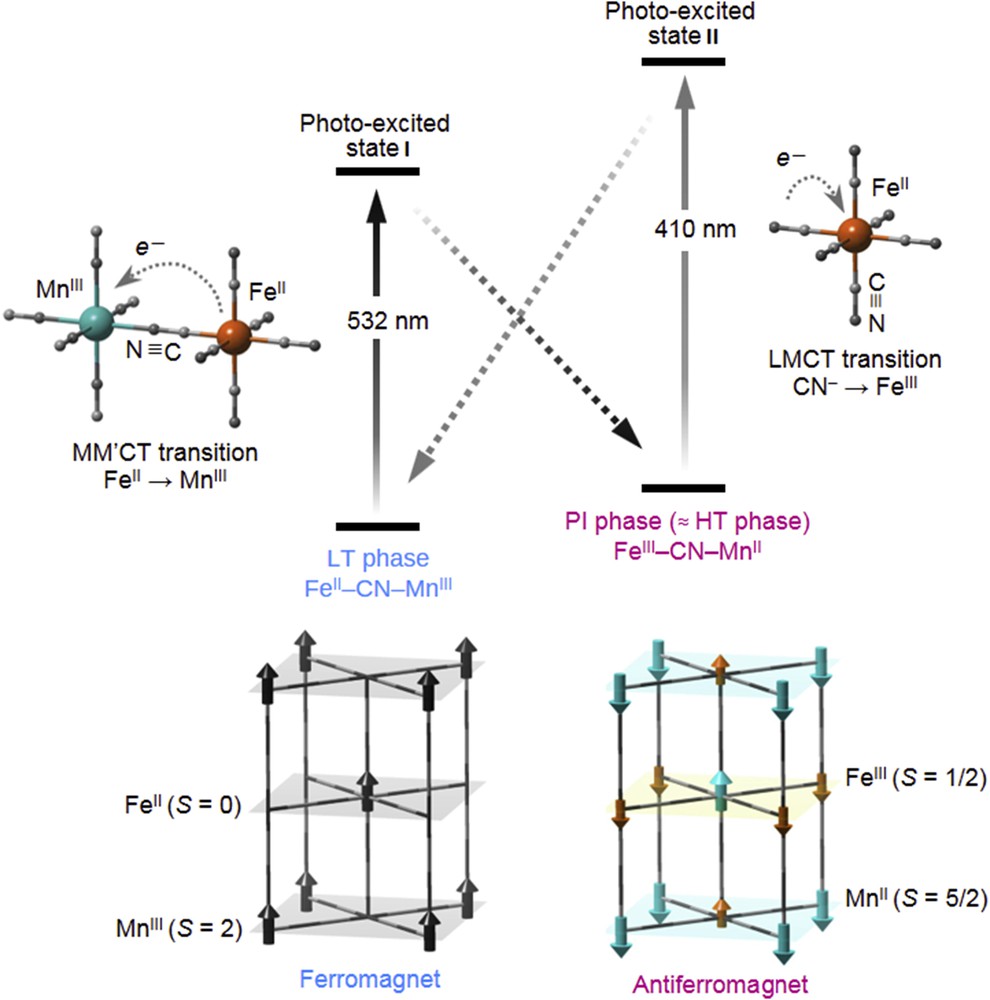
Schematic of the visible-light reversible photomagnetic switching effect. Reversible CT between FeII–CN–MnIII and FeIII–CN–MnII states (upper), and spin ordering for the LT and HT phases (lower). LT phase is ferromagnetic because of the coupling between the MnIII sites. HT phase is antiferromagnetic. Black arrows in the LT phase represent the spins of MnIII. Aqua and orange arrows in the HT phase indicate the spins of MnII and FeIII, respectively. Panel is a modification of Chem. Mater. 20 (2008) 423 [53]. Copyright 2008 American Chemical Society.
A visible light-induced reversible photomagnetism occurs between the ferromagnetic and antiferromagnetic phases by alternating between 532 and 410 nm light irradiation. Although photomagnetism has been reported in various compounds, rubidium manganese hexacyanoferrate is the first photomagnetic material to exhibit a reversible optical switching between a ferromagnet and an antiferromagnet.
3.2.3 Photoswitching inside the thermal hysteresis
Single-shot laser switching was also observed in the hysteresis domain of bistability: a nanosecond (ns) laser pulse (532 nm) can convert the LT phase to the HT phase above a threshold excitation density, and full conversion can be achieved, as observed by IR spectroscopy [58]. The IR probe is very sensitive to the electronic and structural reorganizations accompanying the change in the electronic state, through the frequency shift and splitting of the CN stretching mode. However, this local probe cannot discriminate the nature of the macroscopic conversion mechanism being either a homogeneous process or a phase separation process. An XRD study of the photoinduced phase transition of the Rb0.94Mn [Fe(CN)6]0.98·2.5H2O system induced by a single laser shot inside the thermal hysteresis was reported recently [40]. It shows that above a threshold excitation density (Fig. 10), the ns laser pulse induces a stable long-range ordered cubic phase when conversion is partial or complete.
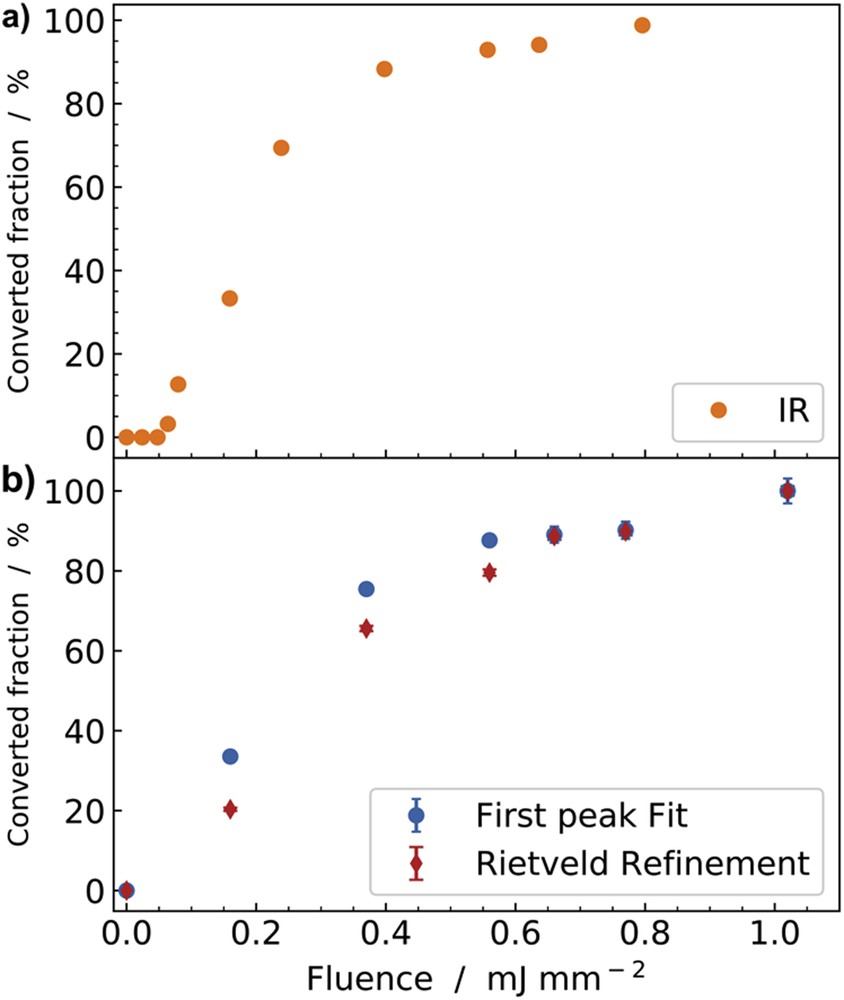
Fraction converted from LT to HT after a single shot as a function of the power density inside the thermal hysteresis. (a) Data shown in orange were obtained by IR spectroscopy after ns laser excitation at 295 K. (b) Data show X-ray study performed at 240 K and the fractions were obtained by using pseudo-Voigt fitting of the intensities of (111)HT and (101)LT peaks (blue) and by Rietveld refinement of the diffraction pattern (red). Adapted with permission from J. Appl. Phys. 97 (2005) 10M508 [58] and Eur. J. Inorg. Chem. (2019) doi.org/10.1002/ejic.201801478 [40]. Copyright 2005 American Institute of Physics and Copyright 2019 Wiley-VCH.
3.2.4 Ultrafast photoswitching dynamics
To control the light-induced properties, and especially for photoactive cyano-bridged metal complexes, it is compulsory to understand the elementary mechanisms allowing structural trapping of new electronic states. However, this requires watching at materials on the timescale of elementary electronic and structural dynamics, that is, on timescale shorter than typical atomic vibrations, which fall in the 10–100 fs regime. Ultrafast studies were performed in cyanide-bridged bimetallic assemblies, using time-resolved optical [59], Raman [60], XRD [61], and X-ray spectroscopy [62]. These experiments with limited time resolution discussed the out-of-equilibrium dynamics and have shown, for example, that the lifetime of the CT states is in the 1–10 μs range in the 200–300 K range. We performed time-resolved XANES measurements on RbMnFe powder in the low temperature phase and monitored the transformation induced by a laser flash at 530 nm, by probing the time evolution of XANES changes ΔI(t) at 6557 eV. The signal decrease in the time trace reported in Fig. 11 is characteristic of the MnIIIFeII → MnIIFeIII electron transfer, and it indicates that the electron transfer occurs within the ≈100 ps time resolution of the experiment, which is responsible for the apparent change before dt = 0. These results open new possibilities for extending XANES and XRD studies at synchrotron or at X-ray free electron laser (X-FEL), and so for tracking the intrinsic dynamics at sub-picosecond timescale. Indeed, X-FELs represent a new generation of incredibly short and ultra-bright X-ray source, allowing for developing ultrafast studies of photoinduced CT processes, as X-FELs provide probes sensitive to both electronic and structural reorganizations. We are now able to monitor transformations on the femtosecond timescale (1 fs = 10−15 s), that is, beyond the Born–Oppenheimer approximation [63,64]. For illustrating these capabilities, we can mention the recent work on the photoactive [FeII(bpy)3]2+ system, showing bistability between a ground low-spin (LS) state (S = 0) with an electronic distribution t2g6eg0L0, where L refers to unoccupied ligand orbitals and a high-spin (HS) state (S = 2, t2g4eg2L0). Because the eg orbitals are antibonding, an important elongation of the metal–ligand bond occurs in the HS state and this structural reorganization is at the origin of light-induced excited spin-state trapping. Zhang et al.[65] performed an fs X-ray emission spectroscopic study at the Linac Coherent Light Source (LCLS) X-ray Free-electron Laser (X-FEL) [65] which evidenced a short passage via the 3T state from the metal-to ligand CT (t2g5eg0L1) to the HS state. Lemke et al. [66] used XANES at the LCLS X-FEL for decoupling the time evolution of the electronic state from the structural dynamics. It was shown that the initial photoinduced metal-to ligand CT state decays within 120 fs toward the HS state, with a 70 fs passage through the 3T. In addition, the structural trapping of this HS state was shown to result from the activation and damping of the molecular breathing mode, moving the system from the LS to the HS potentials toward longer Fe–N bonds. Our goal now is to apply such ultrafast techniques [67–69] for understanding the photoswitching process in Prussian Blue analogues. Indeed, different optical excitation processes (d–d vs CT) may open different switching pathways. By tracking XANES changes at the Mn and Fe edges one may dream of tracking how CT dynamics and structural trapping occur and couple in real time.
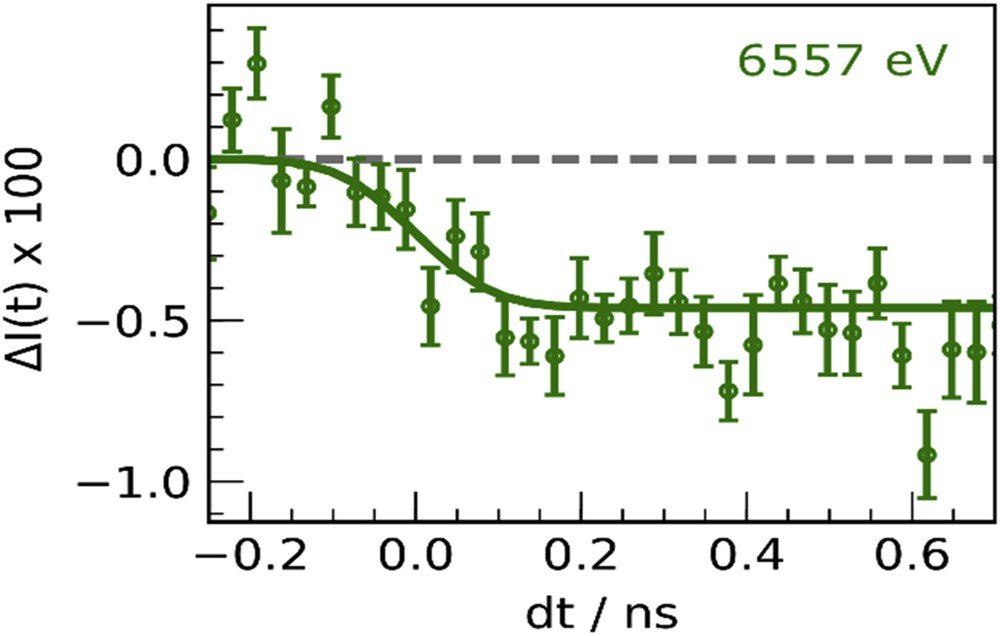
Time scans of relative absorption change 6557 eV for RbMnFe, after fs laser excitation at 530 nm. Adapted with permission from Eur. J. Inorg. Chem. (2018) 272 [39]. Copyright 2018 Wiley-VCH.
4 Conclusions and outlook
Rubidium manganese hexacyanoferrate exhibits various functionalities, and this article is an overview of its basic properties. However, these materials display more exotic functionalities, including photoinduced phase collapse [70], second harmonic generation [71], ferroelectric-ferromagnetism [32], electric field–induced CT structural phase transition [34], zero-thermal expansion behavior [44], and so forth. Investigations on such switchable complexes are critical not only to enhance basic knowledge of functional metal complexes, but also to realize practical applications of innovative functional materials. It is also important to underline that in addition to the development of materials, new techniques emerge for controlling properties on demand [72]. As indicated previously, ultrafast laser-based pump-probe techniques are particularly promising, in which a pump pulse prepares the system in a nonequilibrium state whose temporal evolution is monitored by a delayed probe pulse. However, ultrafast optical excitation often exhibits a limited control, because most of the energy deposited in the electronic subsystem is incoherently redistributed in the material, effectively yielding an ultrafast temperature increase in the out-of-equilibrium dynamics. New alternatives appear for controlling materials on ultrafast timescale, consisting in using THz or IR pulses [72]. This more selective energy deposition allows for exciting directly a coherent structural mode able to drive transformation. Given the coupled structural and electronic reorganizations responsible for the emergence of function in Prussian Blue analogues, this is a promising route for new developments. We therefore strive to contribute to this field through research on rubidium manganese hexacyanoferrate, both by developing materials and new ways to control them.
Acknowledgments
The present research was supported partly by a JSPS Grant-in-Aid for specially promoted Research (Grant Number 15H05697), JSPS KAKENHI (Grant Number JP16H06521) Coordination Asymmetry, and JSPS Grants-in-Aid for Scientific Research on Innovative Areas Soft Crystals (area no. 2903, 17H06367). We also recognize the Cryogenic Research Center, the University of Tokyo, and Nanotechnology Platform, which are supported by the MEXT. The authors also gratefully acknowledge the Agence Nationale de la Recherche for financial support under grant ANR-16-CE30-0018. Parts of this research were carried out in the frame of the IM-LED LIA (CNRS).