

1 Introduction
The unrestrained burning of fossil fuel causes serious environmental issues and negatively affect the humankind; therefore, finding out green energy sources is the main challenge for researchers in the 21st century. Low-temperature fuel cells, which directly convert chemical energy into electricity and produce water as the main side product, have attracted considerable scientific attention as a green power source [1]. Ethanol has emerged as a suitable candidate for low-temperature fuel cell systems because of its much higher energy density than hydrogen [1,2] and lower toxicity and market cost than methanol [3]. Platinum (Pt) is widely used as an effective electrocatalyst to promote oxidation and reduction reactions in fuel cells [6,7] because of its excellent ability to adsorb organic molecules; however, the performance of pure Pt is not very satisfactory [8]. Large Pt loading (1–2 mg/cm2), which is ∼10-fold higher than that of a proton exchange membrane (PEM) fuel cell, is generally used to achieve a satisfactory performance [9,10]; however, the high cost of Pt inhibits its applications as a metal catalyst in direct ethanol fuel cells (DEFCs) [4,5]. Therefore, low Pt loading is an attractive solution for future commercialization of DEFCs.
The complete oxidation of ethanol is associated with the breaking of C–C bonds; thus, the partial oxidation of ethanol generates several intermediates including acetaldehyde and acetic acid and reduces the cell efficiency [3,8,18]. Generally, carbon-supported Pt electrocatalysts are use both as the anode and the cathode. However, carbon supports exhibit poor durability in a harsh environment because of their low graphitic degree and a large amount of defects on the surface and in the amorphous phase, thus significantly reducing the electrochemical surface area (ECSA) of the electrocatalyst [11]. Moreover, the carbonaceous species poisoning in active sites of a Pt nanocatalyst during anodic oxidation [12,13], the sluggish kinetics of anodic oxidation [14], and the extremely weak interaction between Pt and carbon support [15–17] profoundly degrade the long-term performance of a DEFC. To improve the activity and stability of Pt-based electrocatalysts for electrooxidation reactions (EORs), many efforts have been devoted to fabricating catalyst supports with superior corrosion resistance and strong interactions with Pt.
In the present study, a stable and effective Pt/Ti0.8W0.2O2 electrocatalyst toward EORs was prepared by utilizing the attributes of noncarbon catalyst supports and low Pt loading. The 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst was successfully fabricated through a simple and rapid microwave-assisted polyol route (ethylene glycol was used as a reducing agent). The electrocatalytic characterization of the 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst was performed by cyclic voltammetry (CV). The as-prepared 18.5 wt % Pt/Ti0.8W0.2O2 catalyst could be successfully applied to numerous applications including DEFCs, solar cells, biosensors, and photocatalytic water splitting.
2 Experimental procedure
2.1 Chemicals
Titanium (IV) chloride (TiCl4; 99.5%) was obtained from Aladdin. Tungsten hexachloride (WCl6; 99.9%) and hexachloroplatinic acid (H2PtCl6·xH2O) were purchased from Sigma-Aldrich, USA. Absolute ethanol (C2H5OH; 99.9%) and ethylene glycol [(CH2OH)2; 99.5%] were procured from Merck, Belgium.
2.2 Preparation of Ti0.8W0.2O2 nanoparticles
Ti0.8W0.2O2 nanoparticles were synthesized through a one-pot solvothermal route, and WCl6 and TiCl4 were used as the starting precursors for W and Ti, respectively [19]. WCl6 (0.16 g) was first dissolved in 50 mL of absolute ethanol, and 0.176 mL of TiCl4 was then added to the as-obtained solution. The as-prepared solution was then transferred to a Teflon-lined autoclave and placed in an oven at 200 °C for 10 h. The obtained suspension was then naturally cooled to room temperature and washed with acetone and purified water. Finally, the drying step was performed in an oven at 80 °C to collect Ti0.8W0.2O2 nanoparticles for further analysis.
2.3 Preparation of 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst
The 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst was synthesized through a simple and rapid microwave-assisted polyol route, and ethylene glycol was used as a reducing agent. Ti0.8W0.2O2 nanoparticles (110 mg) were first added to 25 mL of ethylene glycol and then magnetically stirred until complete dispersion. The as-obtained suspension was then ultrasonicated for around 30 min, and H2PtCl6 (0.05 M) (2.56 mL) was then mixed with the as-prepared suspension and continuously stirred for 20 min. The pH of the as-obtained mixture was adjusted to 11 by NaOH solution. The reduction reaction was carried out in a microwave oven (800 W, 2450 MHz, Electrolux EMS2047X) at 160 °C and 240 W for 2 min. Finally, the as-obtained product was rinsed several times with acetone and distilled water and then dried at 80 °C overnight for further analysis. The 20 wt % Pt/C electrocatalyst was prepared under the same conditions for comparison.
2.4 Material properties
The structural phases of the as-obtained samples were examined by X-ray diffraction (XRD, D2 PHASER; Bruker, Germany) in the 2θ range of 20°–80°. The surface morphologies and particle sizes of the samples were revealed by transmission electron microscopy (TEM, JEM 1400; JEOL). The elemental compositions of the as-prepared samples were examined by X-ray fluorescence (XRF) analysis. N2 adsorption/desorption isotherms were recorded on a NOVA 1000e instrument at 77 K. For electrical conductivity measurement, the pellets of diameter 10 mm and thickness 1 mm were made from dry samples using a hydraulic press under 300-MPa pressure and then dried at 105 °C overnight.
2.5 Electrochemical characterization
The electrochemical properties of the as-synthesized 18.5 wt % Pt/Ti0.8W0.2O2 catalyst were measured using an EC-LAB electrochemistry device equipped with a silver/silver chloride (saturated KCl) reference electrode, a Pt counter electrode, and a glassy carbon working electrode (diameter of 5 mm). To prepare the catalyst ink, the catalyst powder was ultrasonicated for 30 min in a solution consisting of ethanol absolute and 0.5% Nafion. The surface of the glassy carbon disk was polished with 0.5-μm Al2O3 polishing powder (BAS) and then washed with ethanol and purified water. The catalyst electrode was activated for 100 cycles at a sweep rate of 50 mV/s. To examine the ECSA of the as-obtained catalyst, the electrochemical test was carried out in a nitrogen-saturated 0.5 M H2SO4 solution at a sweep rate of 50 mV/s. Furthermore, to investigate the electrocatalytic activity and stability toward EOR of the as-obtained electrocatalysts, the CV and the accelerated durability tests were performed in a nitrogen-saturated 10 v/v% C2H5OH/0.5 M H2SO4 aqueous solution at a sweep rate of 50 mV/s. In the present work, all electrochemical potentials were described against the normal hydrogen electrode (NHE) scale.
3 Results and discussion
3.1 Characterization of Ti0.8W0.2O2 nanoparticles
The structural phases of Ti0.8W0.2O2 and undoped TiO2 nanoparticles were revealed by XRD measurements with respect to the standard spectra of anatase-TiO2 (JCPDS 084–1286) and WO3 (JCPDS 020–1324) (Fig. 1). Ti0.8W0.2O2 nanoparticles possessed the structure of anatase-TiO2 with typical diffraction peaks of the (101), (004), (200), (211), and (204) facets at 25.3°, 37.8°, 48.0°, 55.1°, and 62.7°, respectively. The typical peaks for the (001), (020), and (200) facets of WO3 at 23.08°, 23.70°, and 24.09° were not detected in the XRD profile of Ti0.8W0.2O2 nanoparticles. Moreover, the phase segregation between W and anatase-TiO2 was not observed in the XRD patterns of Ti0.8W0.2O2 nanoparticles, which signifies that W was successfully incorporated into TiO2 lattices. The crystallite sizes of Ti0.8W0.2O2 and undoped TiO2 nanoparticles estimated based on the (101) peak were found to be 7.36 nm and 14.35 nm, respectively (calculated by Scherrer equation [20–22]). The decrease in the crystallite size of Ti0.8W0.2O2 can be ascribed to the incorporation of W into TiO2 lattices [23,24].
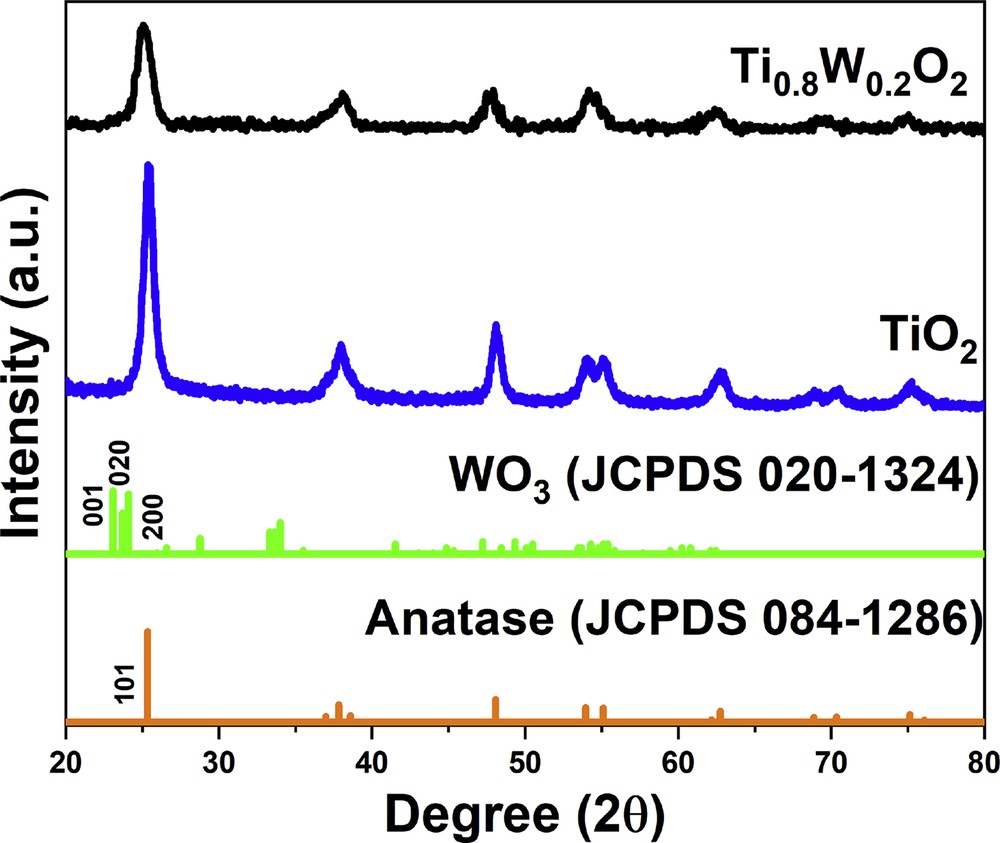
The XRD profile of the as-obtained nanoparticles. XRD, X-ray diffraction.
Fig. 2 displays TEM images of the as-prepared nanoparticles. It is evident that spherical-shaped Ti0.8W0.2O2 and undoped TiO2 nanoparticles had an average diameter of about 9 nm and 15 nm, respectively. The decrease in the particle size of Ti0.8W0.2O2 can be attributed to the incorporation of W into TiO2 lattices which hinders the grain growth [25–27]. In addition, less Ti0.8W0.2O2 nanoparticle agglomeration was observed in comparison with previous findings [28–30], which can be attributed to the one-pot solvothermal route using inorganic starting precursors without using any surfactant/stabilizer or further heat treatment process. Therefore, smaller particle size and less particle agglomeration imply that the as-obtained Ti0.8W0.2O2 nanoparticles possessed a large surface area and played an important role in the uniform dispersion of the Pt-based catalyst. Moreover, the high-resolution transmission electron microscopy (HR-TEM) image in Fig. 2 (b) (inset) displays well-defined fringes of 3.4-Å size corresponding to the (101) crystal plane of anatase-TiO2, which closely agrees with XRD patterns (Fig. 1).

(a,b) TEM images and (b, inset) HR-TEM image of the Ti0.8W0.2O2 nanoparticles; (c, d) TEM images of the undoped TiO2. TEM, transmission electron microscopy; HR-TEM, high-resolution transmission electron microscopy.
Fig. 3 (a) depicts the results of XRF spectroscopy for Ti0.8W0.2O2 nanoparticles, and the proportions of Ti and W were found to be 79.40 and 20.60, respectively; hence, it signifies that the elemental compositions of Ti0.8W0.2O2 nanoparticles were effortlessly controlled by adjusting the proportions of starting precursors. Furthermore, a relatively uniform distribution of compositional elements in Ti0.8W0.2O2 nanoparticles was observed by elemental mapping (Fig. 3 (b–d)).
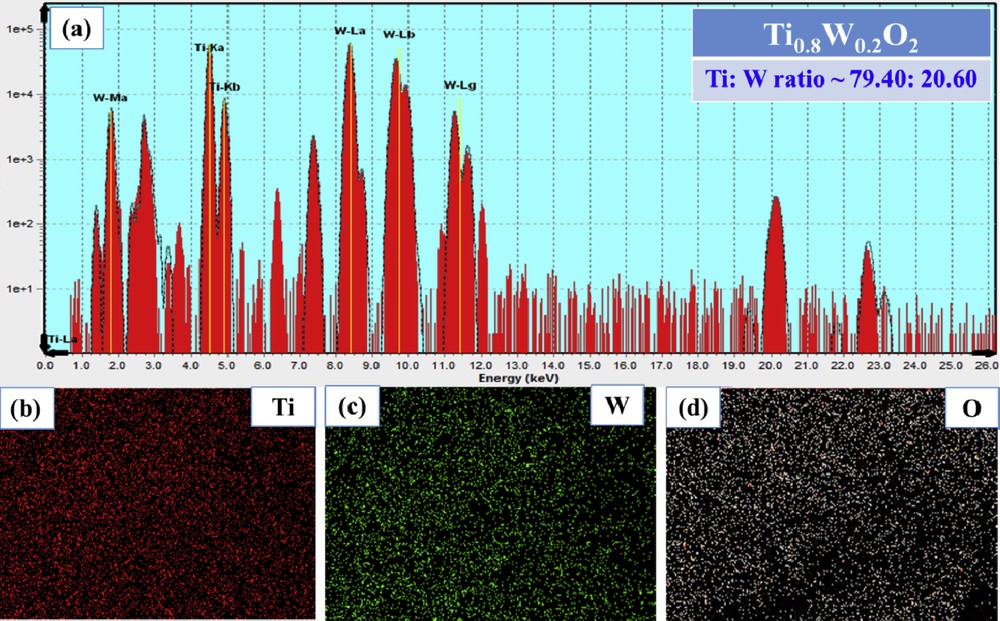
(a) XRF spectroscopy; (b–d) elemental mapping of the as-obtained Ti0.8W0.2O2. XRF, X-ray fluorescence.
The N2 adsorption/desorption isotherms of Ti0.8W0.2O2 and undoped TiO2 nanoparticles exhibited hysteresis loops of type IV curves (Fig. 4). It indicates that the as-obtained nanoparticles possessed a porous structure. The surface areas of Ti0.8W0.2O2 and undoped TiO2 nanoparticles were calculated as 152.32 m2/g and 125.51 m2/g, respectively (Fig. 4 (a, c)); hence, the increase in the surface area of Ti0.8W0.2O2 nanoparticles could be ascribed to its smaller particle size in comparison to undoped TiO2 (Fig. 2 (a–d)). Furthermore, the pore sizes of Ti0.8W0.2O2 and undoped TiO2 were measured to be ∼2.46 nm and ∼2.68 nm, respectively, by the Barrett–Joyner–Halenda (BJH) analysis (Fig. 4 (b, d)). This suggests that the as-synthesized Ti0.8W0.2O2 and undoped TiO2 were mesoporous materials, thus facilitating the uniform distribution of Pt and the electron transfer during electrochemical reactions [7,31]. Therefore, the as-obtained Ti0.8W0.2O2 nanoparticles possessing a larger surface area than noncarbon supports [32–38] (Fig. 5 (a)) were promising supports for the Pt nanocatalysts.
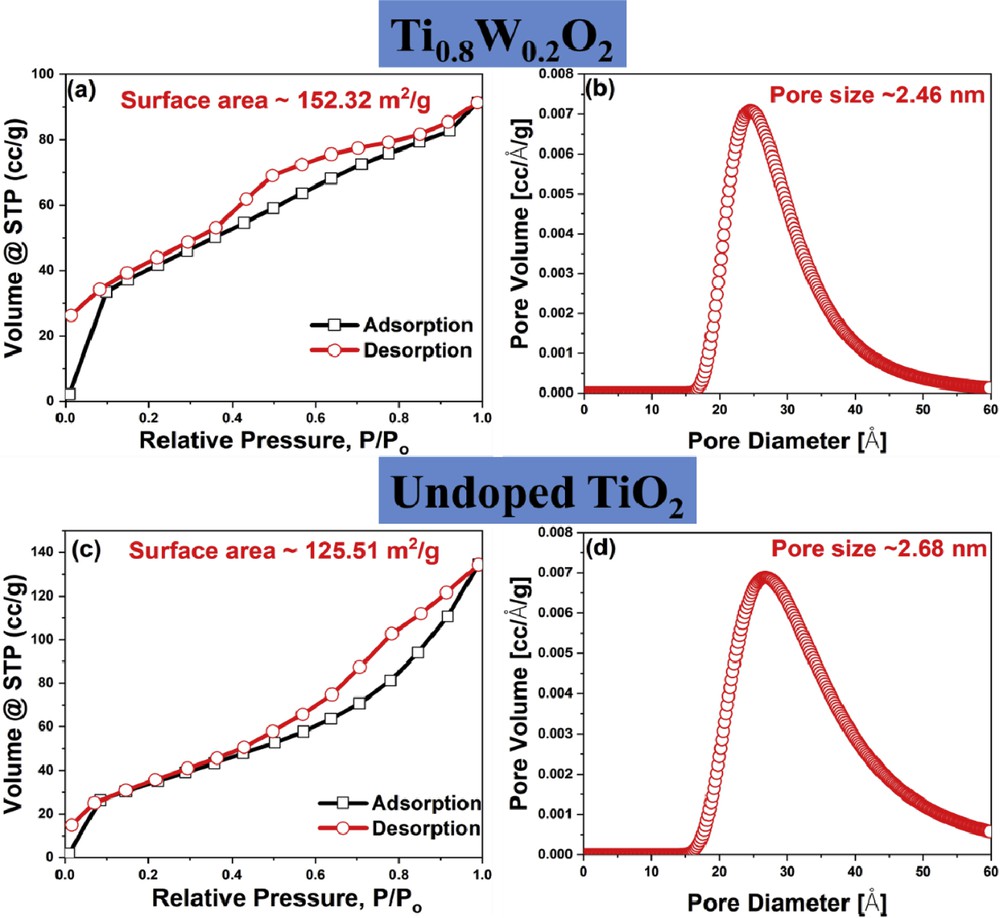
N2 adsorption/desorption isotherms of (a) Ti0.8W0.2O2 and (c) undoped TiO2; (b, d) pore size distribution of Ti0.8W0.2O2 and undoped TiO2, respectively.
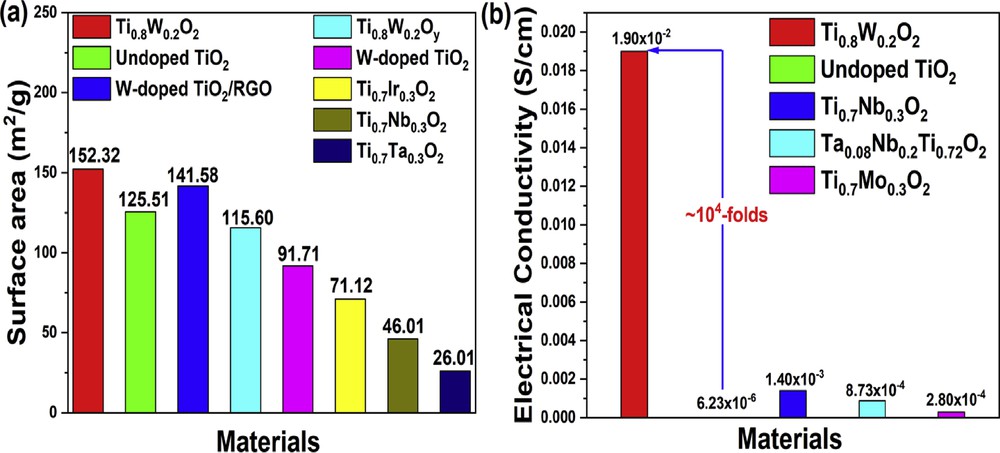
Comparison of (a) the surface area and (b) the electrical conductivity between the as-synthesized Ti0.8W0.2O2 and noncarbon support in the previous studies.
In a fuel cell, one of the most important requirements for catalyst support is electrical conductivity, which facilitates electron transmission during anodic oxidation. The as-prepared Ti0.8W0.2O2 nanoparticles yielded an electrical conductivity of 1.90 × 10−2 S/cm, which is about 104-fold higher than that of undoped TiO2 nanoparticles (6.23 × 10−6 S/cm2) (Fig. 5 (b)). This suggests the successful incorporation of W into anatase-TiO2 and the formation of the “aliovalent ions” effect [39,40]. Furthermore, the as-synthesized Ti0.8W0.2O2 exhibited much higher electrical conductivity than previously reported noncarbon supports [35,37,41], implying that the Ti0.8W0.2O2 nanoparticles were promising supports for the Pt-based electrocatalyst.
3.2 Properties of 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst
Fig. 6 compares the XRD pattern of the as-obtained Ti0.8W0.2O2-supported Pt electrocatalyst with those of anatase-TiO2 (JCPDS 084–1286) and face-centered cubic (fcc) Pt (JCPDS 04–0802). The as-prepared Ti0.8W0.2O2-supported Pt electrocatalysts exhibited the typical Pt peaks corresponding to (111), (200), and (220) crystal planes observed at 2θ positions of 39.76°, 46.24°, and 67.45°, respectively. It is noteworthy that the phase separation between W and anatase-TiO2 was not detected, which implies Ti0.8W0.2O2 nanoparticles had a stable structure.
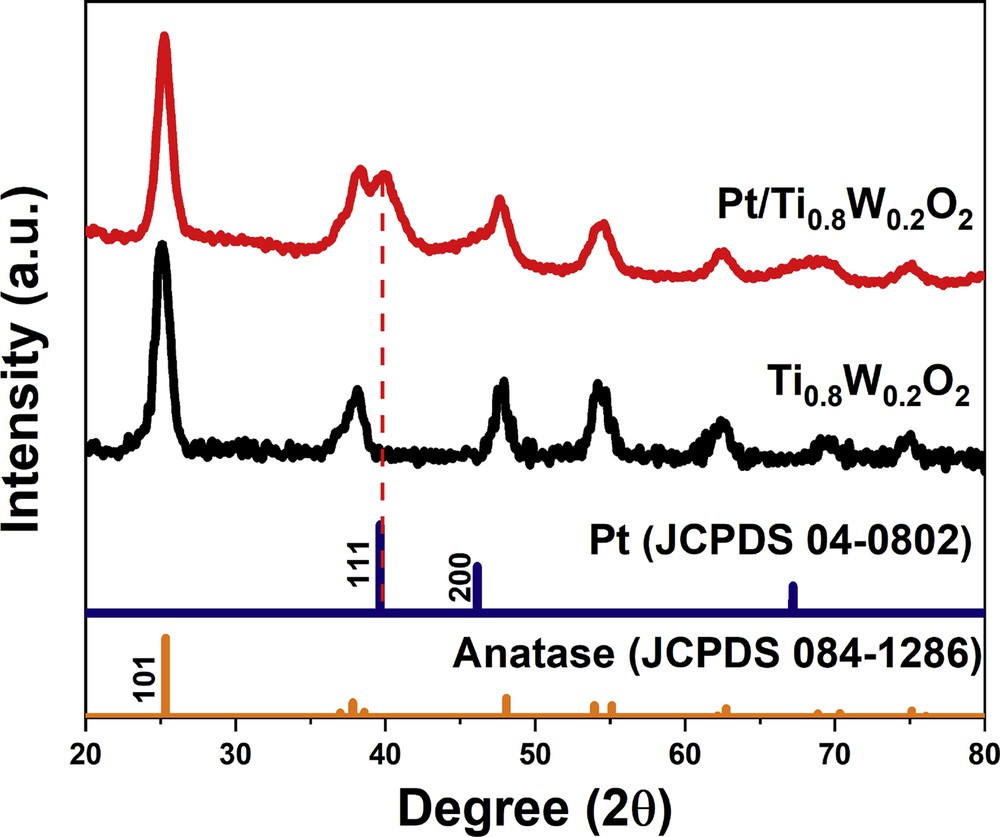
The XRD pattern of the Ti0.8W0.2O2-supported Pt electrocatalyst. XRD, X-ray diffraction.
The morphology and particle size of the anchored Pt nanoform over the Ti0.8W0.2O2 support were examined by TEM. TEM images of the Pt/Ti0.8W0.2O2 electrocatalyst at different scale bars are exhibited in Fig. 7 (a–c). Pt nanoforms anchored on Ti0.8W0.2O2 nanoparticles exhibited a spherical-shaped morphology with an average diameter of several nanometers. Furthermore, relatively uniform distribution and less agglomeration of Pt nanoforms over Ti0.8W0.2O2 nanoparticles were also observed, which can be attributed to the microwave-assisted polyol route that incorporates rapid reduction and fast formation of metal nuclei, thus restricting further particle growth [42,43]. Moreover, the H-TEM image of the 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst exhibited a well-defined fringe of 2.3-Å size corresponding to the (111) crystal plane of fcc platinum. The formation of Pt nanoforms along the (111) facet improved the catalytic activity of 18.5 wt % Pt/Ti0.8W0.2O2 toward EOR because of thier low poisoning rate [44].
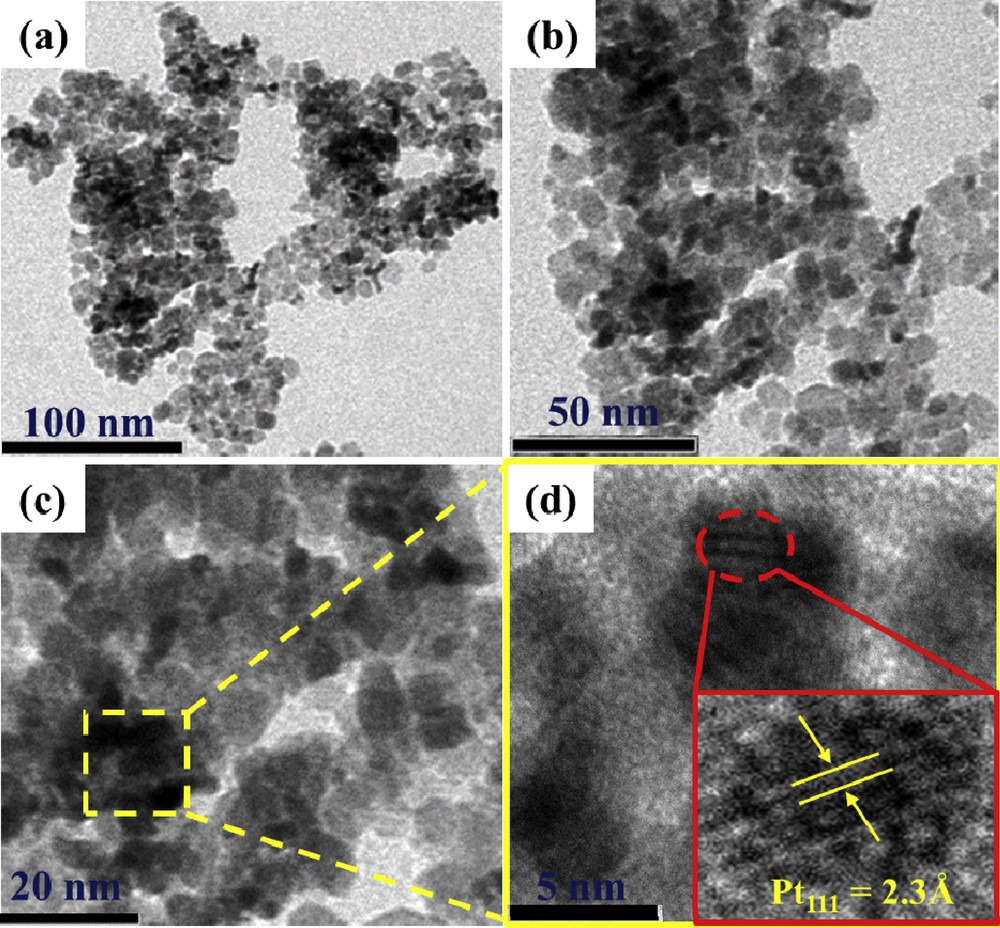
(a–c) TEM images and (d) HR-TEM image of the as-synthesized Ti0.8W0.2O2-supported Pt electrocatalyst. TEM, transmission electron microscopy; HR-TEM, high-resolution transmission electron microscopy.
To investigate the distribution of Pt over Ti0.8W0.2O2 nanoparticles, the scanning electron microscope (SEM)/energy-dispersive X-ray spectroscopy (EDX) mapping measurement was used. The amount of Pt loading over Ti0.8W0.2O2 nanoparticle surfaces was measured as 18.45 wt %, which is approximately equal to the theoretical Pt loading (18.5 wt %) (Fig. 8 (b)). Moreover, elemental mapping in Fig. 8 (c–e) demonstrates the relatively uniform dispersion of Pt on Ti0.8W0.2O2 nanoparticles. These results suggest that the adopted simple and rapid microwave-assisted polyol approach was a suitable process to anchor Pt on Ti0.8W0.2O2 nanoparticles. Moreover, the dispersion of Ti and W before and after Pt deposition remained almost unchanged (Fig. 8 (c, d)), which implies that the as-synthesized Ti0.8W0.2O2 nanoparticles possessed a stable structure in the reduction environment.
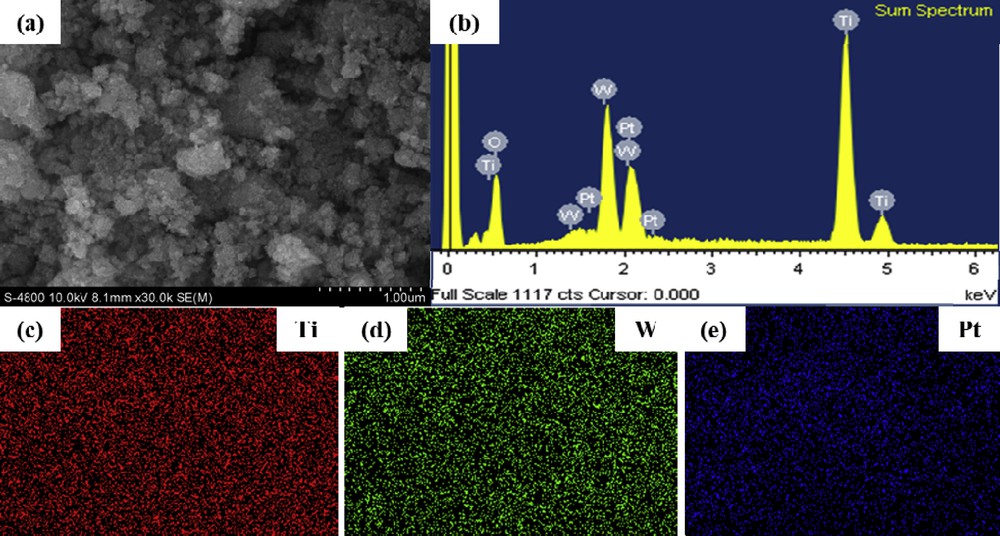
SEM/EDX mapping of the Ti0.8W0.2O2-supported Pt electrocatalyst (a) SEM image; (b) EDX spectroscopy and (c-e) elemental mapping of the Ti0.8W0.2O2-supported Pt electrocatalyst.
The electrochemical characterization of the Ti0.8W0.2O2-supported Pt electrocatalyst and the conventional Pt/C electrocatalyst was performed by CV (Fig. 9). The CV curves of different electrocatalysts revealed the presence of multiple peaks in hydrogen adsorption/desorption regions, thus exposing different Pt crystallographic planes and the high crystallinity of the electrocatalyst [3,45]. The ECSA values of 18.5 wt % Pt/Ti0.8W0.2O2 and 20 wt % Pt/C were measured as 42.72 m2/gPt and 51.79 m2/gPt, respectively; therefore, the decrease in ECSA value of the 18.5 wt % Pt/Ti0.8W0.2O2 catalyst can be attributed to its low Pt loading. The CV curve of the Pt/Ti0.8W0.2O2 electrocatalyst exhibited the positively shifted onset potential of oxidation and the significantly suppressed intensity relative to the Pt/C catalyst, thus implying the weaker binding between OH species and the 18.5 wt % Pt/Ti0.8W0.2O2 catalyst which was evidenced as beneficial effect toward oxygen reduction reaction (ORR) [46,47].
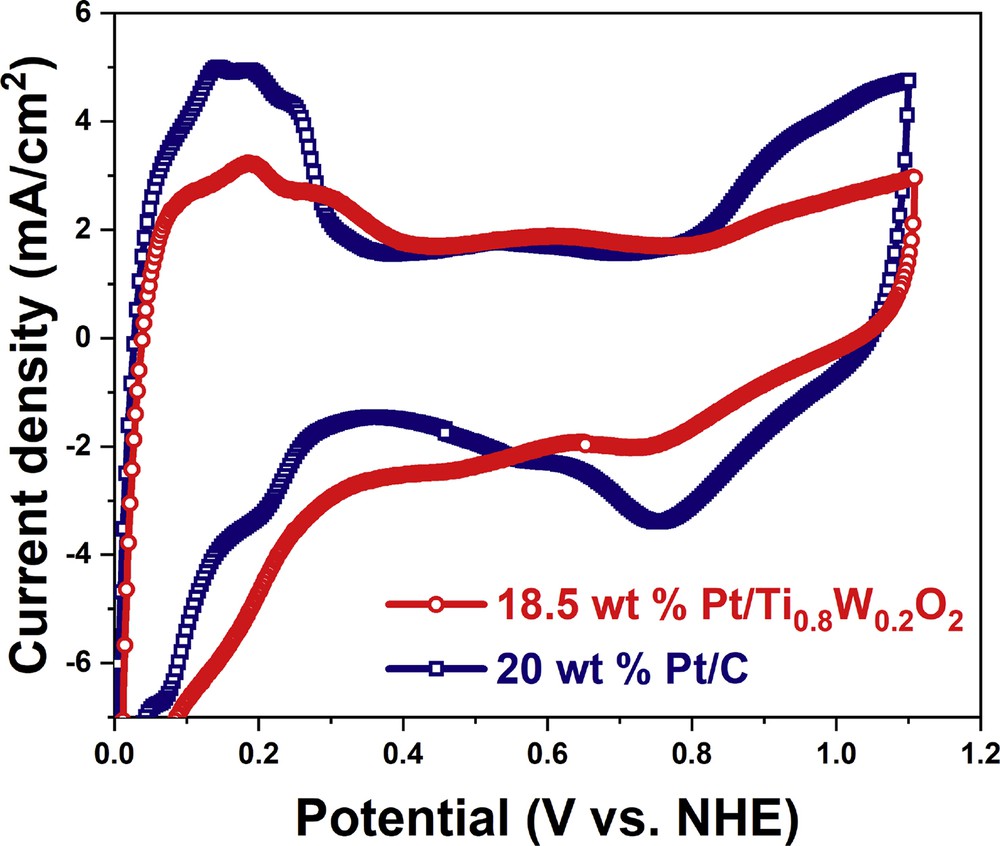
CV curves of the different catalyst in acidic media. CV, cyclic voltammetry.
The electrocatalytic activity of the Ti0.8W0.2O2-supported Pt electrocatalyst and the conventional Pt/C electrocatalyst toward EOR was examined by CV measurement (Fig. 10). The ethanol electrooxidation peaks for 18.5 wt % Pt/Ti0.8W0.2O2 and 20 wt % Pt/C were observed at 0.90 V and 1.28 V, respectively, versus NHE (Fig. 10 (a)). Therefore, the 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst exhibited the facile removal of adsorbed carbonaceous intermediates and manifested a better CO tolerance than the carbon-supported Pt electrocatalyst [48,49]. The 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst displayed a current density of 20.90 mA/cm2, which was comparable with that of the conventional 20 wt % Pt/C electrocatalyst (25.78 mA/cm2). Moreover, the onset potential of the 18.5 wt % Pt/Ti0.8W0.2O2 catalyst was found as to be 0.70 V versus NHE, which was about 1.23-fold lower than that of the 20 wt % Pt/C electrocatalyst (0.80 V vs NHE) (Fig. 10 (b)); hence, it indicates that the 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst exhibited better ethanol electrooxidation than the conventional 20 wt % Pt/C electrocatalyst. The moderate activity of 18.5 wt % Pt/Ti0.8W0.2O2 toward EOR can be attributed to the strong interactions between Pt nanoparticles and the TiO2-based support and manifested beneficial effects on catalytic activity/selectivity [6,50]. Furthermore, the If/Ib value (the ratio between positive-going scan [If] and negative-going sweep [Ib]) was measured to examine the poisoning resistances of the as-obtained electrocatalysts during the anodic EOR. The 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst resulted in an If/Ib value (1.49) about 2.12 times higher than the conventional 20 wt % Pt/C electrocatalyst (0.70).
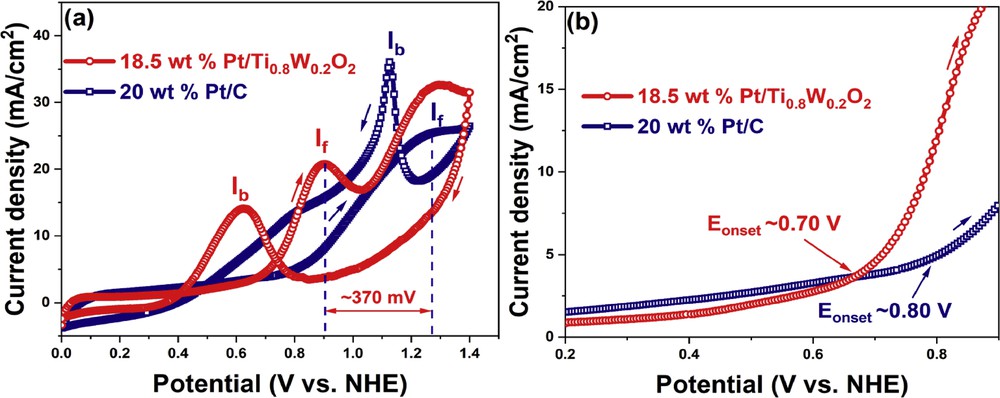
(a) CV curves and (b) onset potential of the different electrocatalyst in ethanol acidic media at a sweep rate of 50 mV/s. CV, cyclic voltammetry.
The electrocatalytic stability of the 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst and the conventional 20 wt % Pt/C electrocatalyst was investigated based on 2000 cycling tests in an acidic ethanol medium (Fig. 11). The Ti0.8W0.2O2-supported Pt electrocatalyst exhibited superior electrocatalytic stability toward EOR in comparison with the conventional carbon-supported Pt electrocatalyst. After 2000 cycling tests, the current density of the 18.5 wt % Pt/Ti0.8W0.2O2 electrocatalyst dropped from 20.90 mA/cm2 to 18.23 mA/cm2 (a degradation of 12.77%). However, the current density of the conventional 20 wt % Pt/C electrocatalyst dropped from 25.478 mA/cm2 to 19.29 mA/cm2 (a decay of 25.17%). Therefore, the superior chemical stability of the Ti0.8W0.2O2-supported Pt electrocatalyst can be ascribed to the initial structural and chemical durability of the TiO2-based support in the acidic media [41]. Moreover, the incorporation of W into the anodic electrocatalyst caused several beneficial effects: (i) It provided necessary OHad species at less positive potential than Pt (bifunctional mechanism) and facilitated the oxidation of carbonaceous intermediate species on the electrocatalyst surface; (ii) it modified the electronic interplays between Pt and the CO adsorbate (electronic effect); and (iii) it increased the catalytic activity through the “hydrogen spillover” effect [13,51–53].
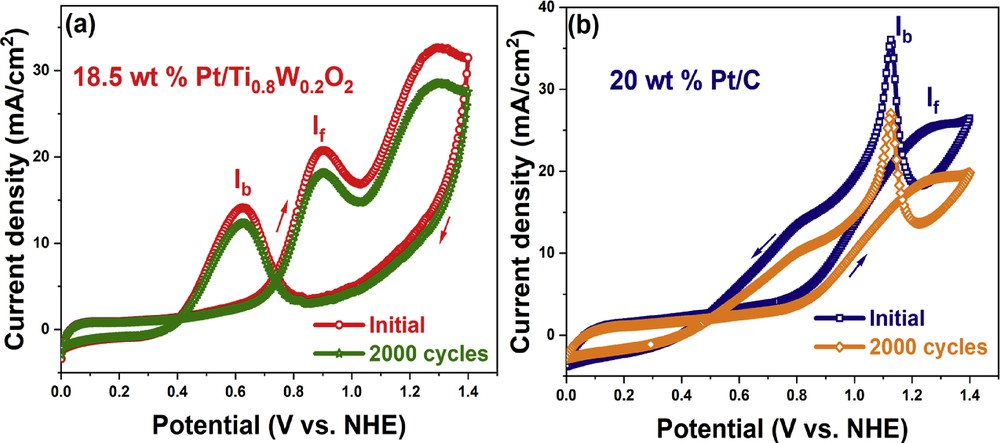
CV curves after 2000 cycling test of the different electrocatalyst in ethanol acidic media. CV, cyclic voltammetry.
4 Conclusion
A stable and effective Pt/Ti0.8W0.2O2 electrocatalyst toward EORs was prepared by utilizing the attributes of noncarbon catalyst supports and low Pt loading. The Ti0.8W0.2O2-supported Pt electrocatalyst was successfully fabricated through a simple and rapid microwave-assisted polyol route (ethylene glycol was used as a reducing agent). The electrocatalytic characterization of the Ti0.8W0.2O2-supported Pt electrocatalyst was carried out by CV. The Ti0.8W0.2O2-supported Pt electrocatalyst was found to be a promising anodic electrocatalyst and possessed the ability to replace the conventional 20 wt % carbon-supported Pt electrocatalyst, albeit its lower Pt loading (18.5 wt %). The Ti0.8W0.2O2-supported 18.5 wt % Pt electrocatalyst resulted in much lower onset potential (0.70 V vs NHE), higher If/Ib value (1.49), and superior electrocatalytic stability (12.77% of initial current density) than the conventional carbon-supported Pt electrocatalyst (0.80 V, 0.70, and 25.17%, respectively).
Acknowledgments
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.03-2018.367