

1 Introduction
Continuous emission of carbon dioxide (CO2) from industrial activities has caused serious global warming and climatic challenges [1–5]. Therefore, the conversion of CO2 into useful organic compounds is of great interest and importance [6,7]. CO2 is a stable compound, and the kinetics of its conversion into a radical anion (activated CO2) is very slow [8]. Several methods, such as chemical reduction [9], photochemical reduction [2], biological reduction [10,11], and electrochemical reduction [4], have been reported to activate CO2 and convert it into radical anions. The electrocatalytic reduction of CO2 is one of the easiest ways to activate it in mild conditions (i.e. room temperature and pressure) [12]. However, CO2 reduction at the surface of an ordinary electrode requires high overpotentials. Therefore, a range of electron transfer mediators (catalysts) such as metal complexes [7,13] and organic compounds [3,14] have been used for the electrocatalytic reduction of CO2 at low overpotentials. Most of the proposed catalysts suffer from certain problems, such as difficult and expensive synthesis, deactivation during the electrocatalytic process, and workup complexity in the reduction of CO2 and the use of activated CO2 for the electrosynthesis of organic compounds. In the literature, there are reports on the application of Cu, Ni, Sn, Pt, Zn, Pb, and Ti electrodes, as well as various oxidized forms of these metals, for the electrochemical reduction of CO2 [3,15–24]. Among the transition metals and their oxides, copper and copper oxides are widely used as electrode materials for the electrochemical reduction of CO2. This is owing to the abundance, low cost, and relatively good chemical stability of these materials [3,25]. Despite these advantages, copper electrodes rapidly lose their activity for CO2 reduction [26]. Copper oxides with crystalline structures [20,27] and different morphologies, however, have very good electrocatalytic activity [25,28].
Nickel is applied in micromechanical devices, data storage, magnetic sensors, catalyst manufacturing, and protection against corrosion [29]. For CO2 reduction, Ni electrodes show less electrocatalytic activity than copper electrodes [30]. The electrocatalytic activity of Ni for the reduction of CO2 can be improved by using electrodes made of Ni composites or Ni alloys [21,30–32]. For example, Hori et al. used a Ni-modified copper electrode for the electroreduction of CO2 [27].
There are reports on the use of carbon dioxide to promote the reduction of organic compounds [33,34]. As an example, activated CO2 (CO2•−) reduces pyridine, and isonicotinic acid is finally formed as a major product [29]. Isonicotinic acid synthesis is significant because of its participation in the synthesis of some pharmaceutically important drugs such as isoniazid, terfenadine, and nialamide [35,36]. It also plays important roles as a plant growth regulator (inabenfide), a photosensitive resin stabilizer, an electroplating additive, and an anticorrosion reagent [37].
In the present research, copper oxide nanosheets were anchored on a copper foam and modified by Ni nanorods via a facile and inexpensive electrodeposition method. The copper foam was used as a substrate to provide a large surface area for the growth of copper oxide nanosheets on its 3D frame. Then, the modified electrode (Cu/CuxO/Ni) was used for CO2 reduction. Finally, isonicotinic acid was synthesized by the reaction of pyridine with activated CO2. The results indicate that activated CO2, CO2•−, has two roles in the electrosynthesis of isonicotinic acid. At first, CO2•− transfers one electron to pyridine and forms a pyridine radical anion. Then, this anion reacts with CO2•− to form isonicotinic acid in the presence of air [29].
2 Experimental setup
2.1 Chemicals and instruments
Tetrabutylammonium perchlorate (TBAP) (>98.0%), acetonitrile (ACN), potassium hydroxide (KOH), and pyridine (99.8%) were purchased from Merck Company and used without any further purification. Argon (Ar), carbon dioxide (CO2) with the purity of 99.995%, Cu foam (with the purity of 99.99% and the thickness of 2 mm), and Ni foam (with the purity of 99.99% and the thickness of 1.5 mm) were also used.
Linear sweep voltammetry was carried out using an EG&G PARSTAT 2273 equipped with the Power Suite software in a conventional three-electrode electrochemical cell containing Ni, Cu/CuxO, and Cu/CuxO/Ni electrodes as the working electrodes, Ag/AgCl/KCl (sat'd) as the reference electrode, and a Pt wire as the counter electrode. Controlled potential coulometry (CPC) and constant current coulometry (CCC) were performed using a SAMA 500 electroanalyzer system in an undivided glass cell with a Cu/CuxO/Ni electrode as the cathode, a platinum plate (ca. 6 cm2) as the counter electrode, and Ag/AgCl/KCl (sat'd) as the reference electrode.
X-ray powder diffraction (XRD) patterns were taken in the reflection mode of Cu Kα (λ = 1.5406 Å) radiation in a 2θ range from 30° to 80° on a senware AW-DX300 X-ray diffractometer. The surface morphologies of the coatings were studied by scanning electron microscopy (SEM) (TESCAN, Vega3, and Czech).
2.2 Preparation of a Cu/CuxO nanosheets electrode
CuO–Cu2O (CuxO) nanosheets were coated on a copper foam (0.25 cm2) by an electrochemical anodization method in an alkaline solution [3]. Briefly, a piece of Cu foam was first cleaned in a 1.0 M HCl solution to remove any oxide layer on it. Then, the electrode was rinsed with water and dried up under Ar steam. The copper foam, as the working electrode, was immersed into a 3.0 M KOH aqueous solution at 40 °C, and the anodization process was carried out at a current density of 30 mA cm−2 for 20 min. In addition, a platinum (Pt) plate was used as the counter electrode. Finally, the dark black film fabricated on the copper foam was rinsed with deionized water and ethanol and dried in an electrical oven at 60 °C for 2 h.
2.3 Preparation of a Cu/CuxO/Ni nanorods electrode
Ni was coated on a Cu/CuxO foam substrate using a Watts-type bath [38,39]. The bath contained nickel sulfate (NiSO4·6H2O, 0.10 M), nickel chloride (NiCl4·6H2O, 0.02 M), a critical micelle concentration (CMC) of sodium dodecyl sulfate (SDS, 0.06 g L−1) as a surfactant, and the complexing agent of trisodium citrate (Na3C6H5O7, 0.50 M). Citric acid was added to the electrodeposition bath solution, and pH was adjusted to 4.5. The Cu/CuxO electrode, as a cathode, and the platinum plate, as an anode, were immersed into the bath for Ni nanorod electrodeposition. The solution was stirred moderately using a magnetic stirrer during the electrodeposition process. The CCC method was used to electrodeposit the Ni nanorods on the Cu/CuxO electrode surface.
2.4 Electrolysis of pyridine in the presence of CO2 at the Cu/CuxO/Ni electrode surface
The CPC method was used to run the process of pyridine electrolysis in 50.0 mL of ACN containing 0.1 M TBAP and 1.0 mmol of pyridine. Before every experiment, the solution was bubbled with Ar gas for 10 min. CO2 was also bubbled into the solution during the electrolysis, and a constant potential of −1.5 V was applied to the Cu/CuxO/Ni working electrode. The results showed that pyridine conversion was about 100% after 3.0 F mol−1 of the starting compound (pyridine) was spent at room temperature. After the electrolysis, the solvent was evaporated completely, and the residue was dissolved in diethyl ether and filtered (5 × 20 mL). As the ether evaporated, the product was identified by Fourier-transform infrared (FTIR), 1H, and 13C nuclear magnetic resonance (NMR) spectroscopies. The spectral characteristics of the products were determined as follows (see Figs. S1–S6 of the Supplementary Information): isonicotinic acid: 1H NMR (CDCl3, 400 MHz): δ 7.47 (d.d, 2H, j = 3.2), δ 7.66 (d.d, 2H, j = 3.2), δ 7.95 (s, 1H, CO2H),13C NMR (CDCl3, 100.6 MHz): δ 128.72, 130.83, 132.41, 167.63 ppm, IR: 3379 cm−1 (O–H), 1714, 1665 (C=O), 1590 (C=C), 1095 (C-O),; 4,4′- bipyridine: 1H NMR (CDCl3, 400 MHz): 1H NMR: 7.48 (d, 4H, j = 6), 8.68 (d, 4H, j = 5), 13C NMR (CDCl3, 100.6 MHz): δ 121.44, 145.57, 150.67 ppm; FT-IR: 1696, 1653 cm−1 (C=N), 1578, 1555 cm−1 (C=C), 1453, 1248, 1085, and 756 cm−1.
3 Results and discussion
3.1 Characterization
Fig. 1 displays the XRD pattern of the (A) Cu/CuxO and (B) Cu/CuxO/Ni electrodes. As the figure suggests, the crystallographic parameters obtained from the XRD data of the Cu/CuxO and Cu/CuxO/Ni electrodes are in accordance with the crystallographic planes of Cu (JCPDS No. 00-004-0836), CuO (JCPDS No. 00-045-0937), and Cu2O (JCPDS No. 01-074-1230). In addition, as it can be seen in the XRD pattern, there exist three broad peaks (signed as *) for the Cu/CuxO/Ni electrode in a position between the peaks of pure Cu (JCPDS No. 00-004-0366) and Ni (JCPDS No. 00-001-1258). These broad peaks have more prominence and less shift than the copper peaks. The explanation for this result is that the Cu–Ni alloy could possibly be formed by the substitution of Cu with Ni [40], and little shifts occurred in the diffraction peaks. In addition, the reduction of the oxidized forms of copper along with Ni ions must have increased the peak intensity of Cu. The diffraction peak at the 2θ angle of 37.6° is probably related to the nickel oxide (JCPDS No. 00-047-1049).
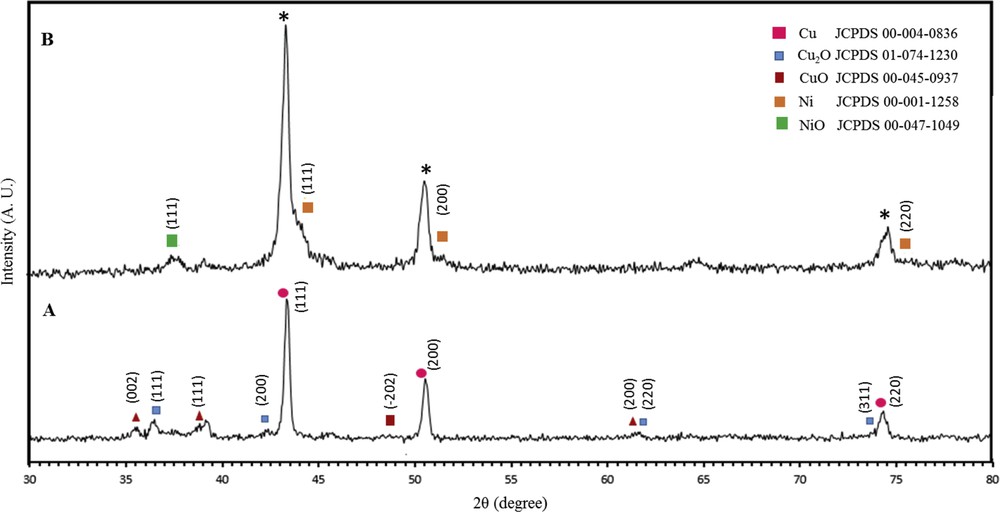
XRD pattern of the (A) Cu/CuO–Cu2O (Cu/CuxO) and (B) Cu/CuO–Cu2O/Ni (Cu/CuxO/Ni) foam electrodes. XRD, X-ray diffraction.
To study the surface morphology of the Cu foam, Cu/CuxO, and Cu/CuxO/Ni, SEM micrographs were taken. As Fig. 2 shows, the Cu foam has a three-dimensional (3D) interconnected microporous structure with a smooth surface. Fig. 3A–D show the SEM micrographs with different magnifications of the CuO–Cu2O nanosheets obtained by anodization. As it can be seen, after anodization in air, the surface of the Cu framework was covered by a dense array of uniform, straight, and long nanosheets. In addition, the electrode surface morphology showed that the nanosheet clusters on the Cu foam skeleton formed flower-like nanostructures (Fig. 3D). The SEM micrographs (with different magnifications) of the Ni nanorods electrodeposited at the CuxO nanosheets surface are presented in Fig. 4A–D. It can be seen that the Ni nanorods have grown in a random orientation.

The SEM image of the framework of Cu foam electrode. SEM, scanning electron microscopy.

(A)–(D) The SEM images of Cu/CuO–Cu2O nanosheets with different magnification. SEM, scanning electron microscopy.

(A)–(D) The SEM images of Ni nanorods electrodeposited at the CuxO nanosheets surface (Cu/CuO–Cu2O/Ni) with different magnification. SEM, scanning electron microscopy.
3.2 Electrochemical activation of CO2 by the Cu/CuxO/Ni electrode and its application in the electrosynthesis of isonicotinic acid
The electrocatalytic activity of the Ni, Cu/CuxO, and Cu/CuxO/Ni electrodes for CO2 reduction was evaluated. Fig. 5 shows the linear sweep voltammograms of these electrodes in an ACN solution (0.10 M TBAP) in a potential range from −0.46 to −2.0 V. Voltammograms (a) and (b) of Fig. 5 show the voltammetric responses of the Ni and Cu/CuxO electrodes in an ACN solution after being bubbled with Ar gas for 10 min, respectively. Voltammograms (c) and (d) show the voltammetric responses of the same electrodes in a solution containing CO2. A comparison of the voltammograms in the absence (voltammograms (a) and (b)) and the presence of CO2 (voltammograms (c) and (d)) showed that the cathodic currents increased in the presence of CO2. This result indicates that both electrodes are electrocatalytically active for the reduction of CO2 to CO2•− based on an EC′ mechanism [41]. To investigate the simultaneous effect of the electrocatalytic activity of Ni and Cu/CuxO, linear sweep voltammetry (voltammogram e) was carried out to check the CO2 electrocatalytic reduction at the surface of the Ni nanorods–modified Cu/CuxO (Cu/CuxO/Ni) electrode. The results indicate that a Cu/CuxO/Ni electrode has better electrocatalytic activity to reduce CO2 than a Ni electrode or a Cu/CuxO electrode alone. The high electrocatalytic performance of the Cu/CuxO/Ni electrode is probably due to the synergistic effect of the adjacent CuxO and the Ni sites of this proposed modified electrode [42]. Voltammograms (e) to (h) correspond to the Cu/CuxO/Ni electrodes after the deposition of Ni nanorods on a Cu/CuxO electrode for different times of 600, 1200, 1500, and 1800 s in the presence of CO2. As it can be seen, an increase in the time of Ni nanorods deposition on the Cu/CuxO electrode surface to 1200 s improved the electrocatalytic activity of the modified electrode for the reduction of CO2. The decrease in the catalytic current at a higher Ni deposition time to 1800 s was probably due to the increase in the thickness of the Ni layer on the Cu/CuxO surface. In other words, the higher the deposition time, the fewer the CuxO sites exist adjacent to Ni sites, and the weaker the synergistic effect of these sites.
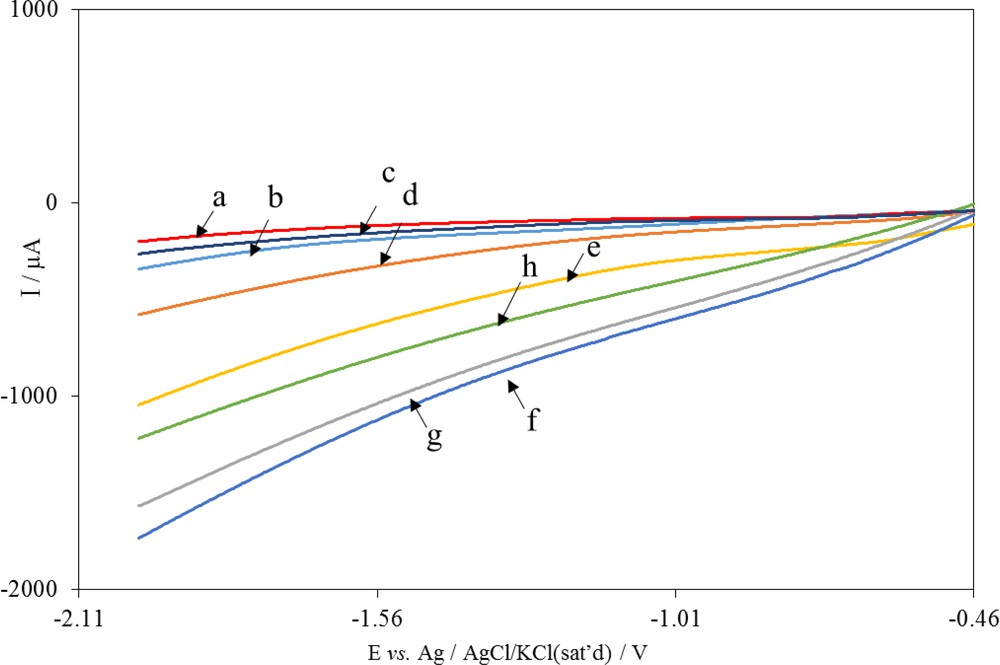
Linear sweep voltammograms of the (a) Ni electrode and (b) Cu/CuxO electrode in ACN (0.1 M TBAP) solution after removing O2, (c) as (a) after addition of CO2, (d) as (b) after addition of CO2, (e) to (h) as (d) after deposition of Ni nanorods at the Cu/CuxO electrode surface for different times of 600, 1200, 1500 and 1800 s, respectively. Scan rate: 0.1 V s−1. ACN, acetonitrile; TBAP, tetrabutylammonium perchlorate.
To continue, the use of the CO2 product obtained through electrocatalytic reduction (i.e. activated CO2, or CO2•−) was investigated for the electrocarboxylation of pyridine in mild conditions. Fig. 6 shows the voltammetric responses of the Cu/CuxO/Ni electrode in a solution after bubbling Ar in it (voltammogram (a)). The solution contained pyridine (voltammogram (b)) and CO2 (voltammogram (c)) in a potential range from −0.46 to −2.0 V. As shown, the voltammogram of the electrode remained unchanged after the addition of pyridine, which indicates that the reduction of pyridine would not take place even by applying the potential of −2.0 V. It is also indicated that there was no interaction between pyridine and the modified electrode. The direct reduction of CO2 on bare glassy carbon electrode occurs at potentials more negative than −2.0 V vs saturated calomel electrode (SCE) in most solvents [43]. As already mentioned, the increase in the current response of voltammogram (c), compared with voltammogram (a), was due to the electrocatalytic reduction of CO2 at the Cu/CuxO/Ni electrode (Cat). The reduction took place according to Eqs. 1 and 2:
| Cat + e− → Cat− E | (1) |
| Cat− + CO2 → Cat + CO2•− C′ | (2) |
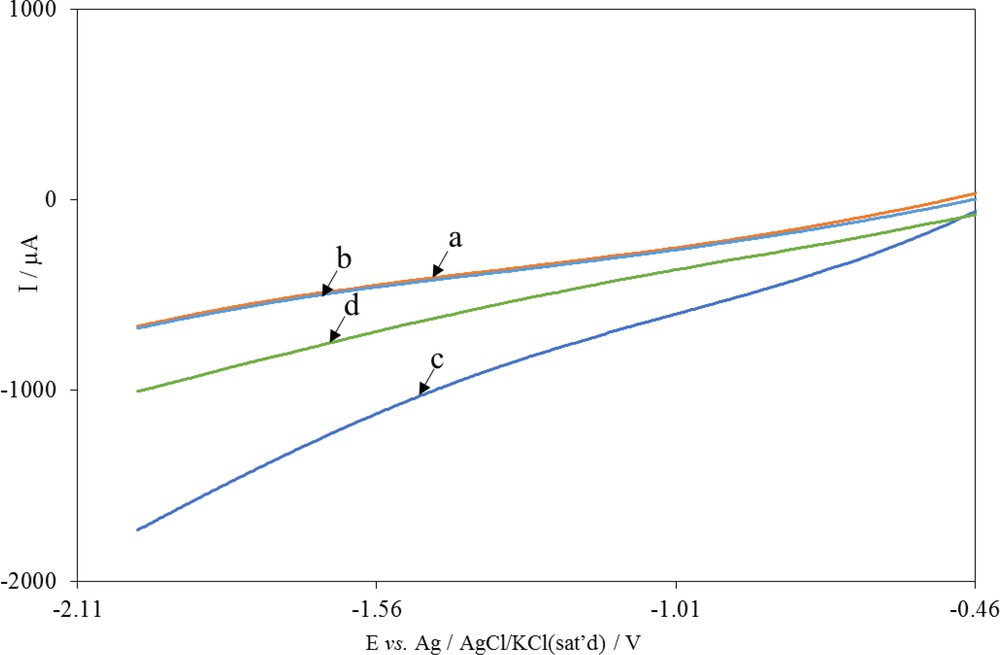
Linear-sweep voltammograms of Cu/CuxO/Ni electrode in ACN (0.1 M TBAP) solution after (a) removing O2, (b) addition of 1.0 mM pyridine, (c) addition of CO2 and (d) as (b) after addition of CO2. Scan rate: 0.1 V s−1. ACN, acetonitrile; TBAP, tetrabutylammonium perchlorate.
Voltammogram (d) belongs to the Cu/CuxO/Ni in a solution containing 1.0 mM of pyridine in the presence of CO2. As reported in the literature [33], electrocatalytic activated CO2 (CO2•−) transfers an electron to the aromatic ring of pyridine, leading to a radical anion of pyridine (Eq. 3) and the produced CO2 returns to the electrocatalytic cycle.
| Py + CO2•− → Py•− + CO2 (C′) | (3) |
A comparison of voltammograms (c) and (d) shows the electrocatalytic current decreases in the presence of pyridine. This suggests that some CO2•− do not participate in the electrocatalytic reduction of pyridine (Eq. 3) but react with Py•− (Eq. 4).
The produced pyridine radical anions can participate in two reactions as follows. Pyridine radical anions react with the radical anions of CO2•− to form dianions of isonicotinic acid, as an intermediate (Eq. 4). Then, isonicotinic acid is obtained through the oxidation of the intermediate (Eq. 5) [33].
| Py•− +CO2•− → PyCOO2− (C) | (4) |
| (5) |
In summary, the proposed EC'C′CC mechanism involves three steps including one-electron electrocatalytic reduction of CO2 (EC′) followed by reaction (3) to form Py•− (C′), chemical reaction of Py•− and CO2•− (C) as shown in reaction (4) and, finally, oxidation of the produced intermediate (PyCOO2−) to produce isonicotinic acid (C) in accordance with reaction (5).
On the second track, two pyridine radical anions react together and produce a 4,4′-bipyridine dimer, as shown in Eqs. 6 and 7 [33].
| 2Py•− → Py–Py2- (C) | (6) |
| (7) |
Based on this explanation, the electrochemical mechanism of 4,4′-bipyridine is the same as that of isonicotinic acid.
To determine the main product of the reaction and to understand the mechanism of the pyridine radical anion reaction, controlled potential coulometry was performed at −1.50 V in a solution containing CO2 and pyridine at the Cu/CuxO/Ni electrode, as described in section 3. Then, the coulometric products were separated and characterized by FTIR, 1H, and 13C NMR spectroscopy. The spectral characteristics (see Figs. S1-S4 and Supporting Information) confirmed isonicotinic acid and 4,4′-bipyridine as the final products with yields of 80% and 20%, respectively. Therefore, it is logical to conclude that the radical anion of pyridine reacts with CO2•− and leads to the production of isonicotinic acid as the main product. The coulometry current of pyridine in the presence of CO2 was also recorded for a long time (10 h) as in Fig. 7. The steady current density (average ≈1.32 mA cm−2) implies that no obvious deactivation of Cu/CuxO/Ni occurred during the coulometry.
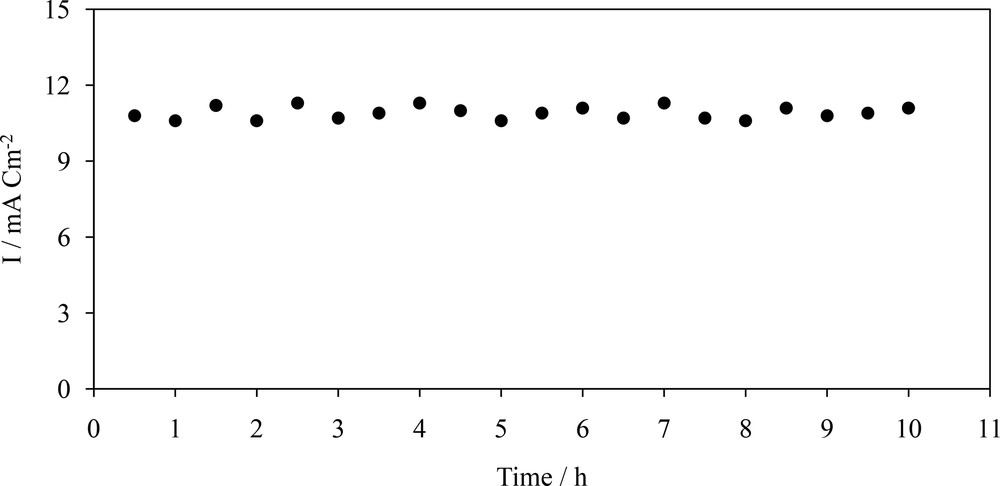
Controlled potential coulometry of pyridine in the presence of CO2 at the Cu/CuxO/Ni electrode surface over a period of 10 h. Potential step: −1.5 V.
4 Conclusion
A Cu/CuxO/Ni electrode was fabricated as a new electrocatalyst, and the electrochemical reduction of CO2 was carried out by means of that electrode at a low overpotential in an acetonitrile solvent. The Cu/CuxO/Ni electrode was prepared by decorating the surface of a copper foam with CuxO nanosheets and Ni nanorods (Cu/CuxO/Ni). As the results showed, the simultaneous use of Ni and CuO–Cu2O (CuxO), rather than each one alone, in an electrode increases its electrocatalytic activity for the reduction of CO2. CO2•−, as the product of the electrocatalytic reduction of CO2, was used for the electrosynthesis of isonicotinic acid in mild conditions. The spectral characteristics of the products indicated that, beside isonicotinic acid as the main product with the yield of 80%, 4, 4′-bipyridine was formed with the yield of 20%. CO2•− had a dual role in the electrosynthesis of isonicotinic acid. First, it induced the indirect electrocatalytic reduction of pyridine to pyridine radical anions. Second, it reacted with pyridine radical anions to form isonicotinic acid based on an EC'C′CC mechanism. The pyridine radical anions reacted with themselves to form 4, 4′-bipyridine based on the same mechanism. Furthermore, as the data obtained through coulometry showed, long-term use of the proposed electrode does not lead to any noticeable loss of activity for CO2 electroreduction.
Acknowledgements
The authors are grateful for the support of the Iran National Science Foundation (INSF), Iran, under Grant No. 96004700.