

1 Introduction
Since the 1960s, lanthanides have gained a growing importance because of their broad variety of technological applications and use in the industry. These elements are incorporated in materials for catalysis, optics, magnets and electronics [1], in metallurgy and in medicine, where they are used as contrast agents for magnetic resonance imaging, as luminescent probes for diagnostics and as optical imaging of cells [2,3]. They are also connected to radioactive wastes because they are present, together with actinides, in acidic effluents from where they need to be extracted.
Macrocyclic ligands have been extensively developed concerning lanthanide coordination chemistry, mainly those possessing a high number of donor atoms, as lanthanide ions require a coordination number of eight or nine. Among them, calixarenes and their derivatives have been largely used in metal cation binding in the last three decades. In particular, the capacity of carbonyl-containing substituents on the lower rim of calixarenes to bind monovalent, divalent and trivalent cations has been widely reported [4,5]. The increasing interest in the interaction between lanthanides and calixarenes is due to their ability to efficiently and selectively complex and extract those elements. To bind hard lanthanide cations, residues with hard oxygen donor atoms, such as phosphine oxides [6,7], carboxylic acids [8–10] or amides [8,11,12], have been introduced in the calixarene frameworks. Ketone moieties have also been incorporated in either the upper or the lower rims of these macrocycles [13,14].
We have been conducting a systematic study on the binding properties of dihomooxacalix[4]arenes (calix[4]arene analogues in which one CH2 bridge is replaced by one CH2OCH2 group) [15] bearing carbonyl groups (ester [16,17], ketone [18,19] and amide [20,21]) at the lower rim towards several cations with different valences. Herein, we complete this study, presenting our results concerning the lanthanide ion binding by dihomooxa ketone ligands.
This article presents the binding properties of four tetraketone (methyl 1a [22], tert-butyl 1b [22], adamantyl 1c [22,23] and phenyl 1d [24]) derivatives of p-tert-butyldihomooxacalix[4]arene (Fig. 1) towards several lanthanide cations. These properties have been assessed by extraction studies of metal picrates from an aqueous solution into dichloromethane and stability constant measurements in acetonitrile based on UV absorption spectrophotometry. The affinity of the ligands for selected cations (La3+, Eu3+ and Yb3+) has also been investigated by proton NMR experiments. Molecular dynamics (MD) and potential of mean force (PMF) simulations were performed in pure acetonitrile and dichloromethane solutions and at a dichloromethane/water interface to bring further structural and energetic insights into the binding and extraction processes. Tetraketone (methyl 2a, adamantyl 2c and phenyl 2d) derivatives of p-tert-butylcalix[4]arene (Fig. 1) were also studied in the present work, and the results of all ligands were compared and discussed in terms of the substituent groups attached to the ketone function, as well as the size and conformational effects of the macrocycles.
Structural formulae of calixarenes.
2 Results and discussion
2.1 Extraction studies
The ionophoric properties of tetraketones 1a-d and 2a, 2c and 2d towards lanthanide metal cations were evaluated by the standard picrate extraction method [25]. The results, expressed as a percentage of cation extracted (% E), are reported in Table 1. All the ketones had already been synthesised and obtained in the cone conformation, except methyl ketone 1a that was obtained in a partial cone conformation.
Extraction percentage of lanthanide picrates into CH2Cl2 at 25 °Ca.
| La3+ | Ce3+ | Pr3+ | Nd3+ | Sm3+ | Eu3+ | Gd3+ | Dy3+ | Er3+ | Yb3+ | |
| Ionic radiusb/Å | 1.03 | 1.01 | 0.99 | 0.98 | 0.96 | 0.95 | 0.94 | 0.91 | 0.89 | 0.87 |
| 1a | 4.9 | 4.5 | 3.3 | 4.6 | 4.5 | 5.0 | 4.4 | 5.8 | 5.6 | 5.1 |
| 1b | 6.8 | 6.4 | 7.3 | 8.4 | 12.3 | 8.6 | 19.1 | 7.0 | 11.6 | 18.0 |
| 1cc | 9.3 | 7.7 | 6.0 | 8.9 | 14 | 7.4 | 23 | 7.9 | 12 | 21 |
| 1d | 8.9 | 6.0 | 6.7 | 7.3 | 10.5 | 5.7 | 13.4 | 7.5 | 7.8 | 16.0 |
| 2a | 5.3 | 5.2 | 5.2 | 6.2 | 5.6 | 4.9 | 4.5 | 5.1 | 5.9 | 5.3 |
| 2c | 6.8 | 6.3 | 4.5 | 5.5 | 6.4 | 5.0 | 5.1 | 4.8 | 4.9 | 4.6 |
| 2d | 5.3 | 5.6 | 4.1 | 6.6 | 8.2 | 8.5 | 5.2 | 5.6 | 7.1 | 8.1 |
a Values with uncertainties less than 5%.
b R.D. Shannon, C.T. Prewitt, Acta Cryst. B25 (1969) 925; B26 (1970) 1046; data quoted in I. Marcus, Ion Properties, Marcel Dekker, New York, 1997, pp. 46–47.
c Data obtained from the study by Marcos et al. [23].
The results obtained show that ketones 1b-1d are modest extractants, showing preference for the heavy lanthanides Gd3+ and Yb3+. The three ketones present similar extraction trends, with the two former displaying relatively higher percentages than the latter. The presence of the tert-butyl and adamantyl groups in ligands 1b and 1c, respectively, more donating than the phenyl group (1d), leads to a higher basicity of the carbonyl oxygen in the former derivatives. Adamantyl ketone 1c is a slightly better phase-transfer agent than t-butyl ketone 1b (Gd3+ = 23 and 19.1% E; Yb3+ = 21 and 18% E, respectively). A similar behaviour had already been observed previously by us towards monovalent and divalent cations [18,19]. Methyl ketone 1a, in a partial cone conformation, is a very weak extractant (% E ranges from 3.3 to 5.8), being unable to discriminate among the lanthanide ions studied.
Concerning tetraketones 2a, 2c and 2d, they exhibit very low extractability too (% E ranges from 4.1 to 8.5), showing almost no discrimination for the ten lanthanide ions. The higher conformational flexibility of the dihomooxacalix[4]arene macrocycle compared to that of the calix[4]arene may allow for a more suitable arrangement of the ketone pendant arms around the lanthanide cations, which require high coordination numbers, and thus favours their extraction. With regards to other dihomooxa ligands, such as the analogue ethylamide derivative [24], these dihomooxa ketones show an opposite extraction profile.
2.2 UV-vis complexation studies
Complexation of five representative lanthanide ions (La3+, Pr3+, Eu3+, Gd3+ and Yb3+) by tetraketone ligands 1b, 1d and 2d was studied in acetonitrile by UV-vis absorption spectrophotometry. The spectral changes of the ligands upon stepwise addition of the metal triflates in solution allow the determination of the nature and the stability of the complexes formed. The stability constants (as log β) of the 1:1 complexes are presented in Table 2.
Stability constants (log β)a for 1:1 lanthanide complexes in acetonitrile (T = 25 °C; I = 10−2 M, Et4NClO4).
| Ligands | La3+ | Pr3+ | Eu3+ | Gd3+ | Yb3+ |
| 1b | 6.1 ± 0.6 | 5.9 ± 0.6 | 5.7 ± 0.3 | 6.0 ± 0.5 | 5.5 ± 0.5 |
| 1d | 6.0 ± 0.1 | 5.6 ± 0.7 | 4.9 ± 0.4 | 4.6 ± 0.9 | 4.8 ± 0.3 |
| 2d | 5.1 ± 0.2 | n.d. | n.d. | 5.0 ± 0.2 | 6.2 ± 0.2 |
a Confidence interval corresponding to ±σn−1, σn−1 being the standard deviation on the mean value of at least two experiments.
Some kinetic effects were observed, i.e. the time needed to reach the equilibrium was important with ligands 1b and 1d, in particular for Pr3+ (20 min) and to a lesser extent for the other lanthanide cations (between 5 and 10 min). In these cases, the experimental protocol had to be adapted either by waiting the time needed to reach the equilibrium between each addition or by preparing the mixtures in separated vials.
Important spectral variations were obtained with all ligands, as shown, for example, in Fig. 2 for Pr3+ with 1b. These ligands present high affinities for all the lanthanides studied, displaying stability constants for the ML (metal:ligand) complexes that range from 4.6 to 6.2 log units. t-Bu ketone 1b forms more stable species than Ph ketone 1d, which is consistent with the donor property of the tert-butyl group. Ph ketone 1d is, however, a more selective ligand, showing a variation of almost 1.4 log units between the highest and the lowest log β values, in contrast to only 0.6 log units for ketone 1b. Both dihomooxa ketones exhibit a similar behaviour, with preference for the light lanthanides (La3+ and Pr3+). The studies with Ph ketone 2d allowed us to determine the influence of the oxygen bridge and consequently of the size and the flexibility of the macrocycles on the cation complexation. The results with ligand 2d showed a reverse behaviour in respect to its dihomooxa homologous, as 2d displayed a preference for the heavy and smaller lanthanide Yb3+.
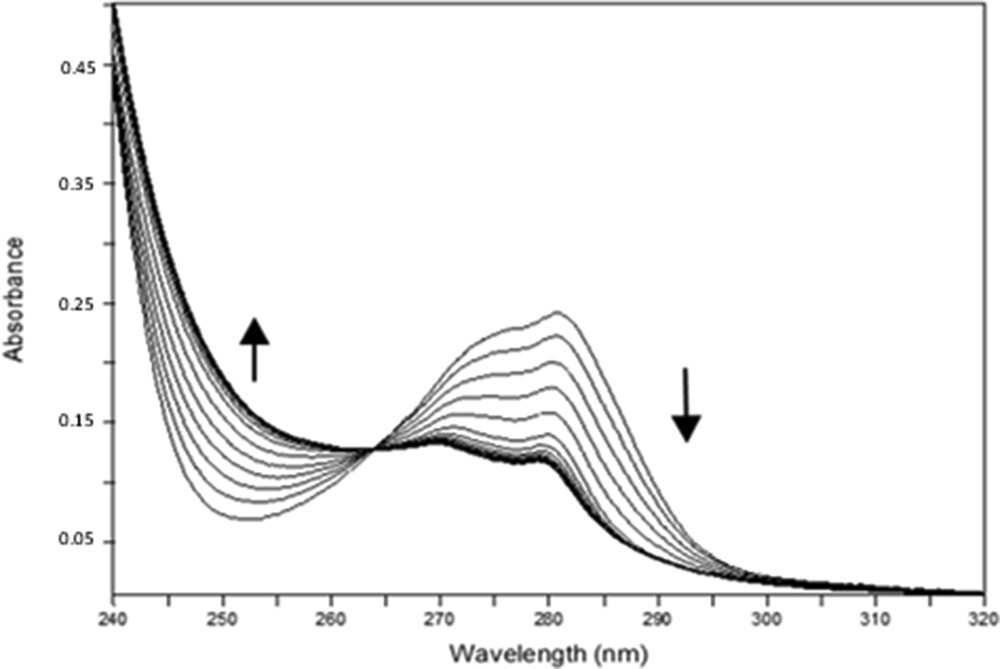
Spectral variations corresponding to the complexation of Pr3+ with 1b in acetonitrile at 25 °C (CL = 6.63×10−5 M; 0 ≤ R = CM/CL ≤ 1.97); I = 10−2 M Et4NClO4.
The comparison with the analogue ethylamide [24] brings out that these dihomooxa ketones, although weaker binders, are more selective ligands, mainly Ph ketone 1d.
2.3 Proton NMR binding studies
To obtain further information on the binding behaviour of ketones 1a-d and 2a, 2c and 2d, specifically concerning the binding sites, proton NMR titrations were performed in CDCl3/CD3OD with these ligands and La3+, Eu3+ and Yb3+ cations, representatives of the light, middle and heavy categories of the lanthanide series, respectively. Variable amounts of the salts were added to the ligands, and the proton spectra were recorded after each addition.
Two different situations were found after the addition of La triflate to the ligands. No complexation was observed in the cases of dihomooxa Me ketone 1a and the three ketones 2a, 2c and 2d, while with the other dihomooxa ketones 1b–d titrations induced broadening and chemical shift variation in the peaks, as shown in Fig. 3 for the titration of Ad ketone 1c. Although the broad proton signals prevent a quantitative analysis of the data, it was possible to record downfield shift variations between 0.13 and 0.28 ppm for the aromatic protons of 1c after the addition of 1 equiv of La triflate. These results indicate a slow exchange rate between the free and the complexed species on the NMR time scale at room temperature. After subsequent addition of the salt (up to 2–3 equiv), the peaks remain broad but unchanged. This behaviour seems to indicate a strong affinity of the ligands for La3+.

500 MHz 1H NMR spectra of Ad ketone 1c in CDCl3 at 25 °C: (a) free ligand, (b) upon addition of 0.5 and (c) 1 equiv of La triflate.
All the tetraketones exhibited similar behaviour towards Eu3+. Small chemical shift variations were observed upon the addition of an excess of salt (3 equiv), indicating weak complexation with this cation.
Concerning Yb3+ complexation, the titrations with all the ketones produced broad peaks that become sharp after the addition of an excess of the salt, as shown in Fig. 4 for t-Bu ketone 1b. These results should indicate interaction with the cation, although no meaningful upfield or downfield shift variations of the peaks were observed, as expected from the binding of a paramagnetic metal ion, such as Yb3+ [26].
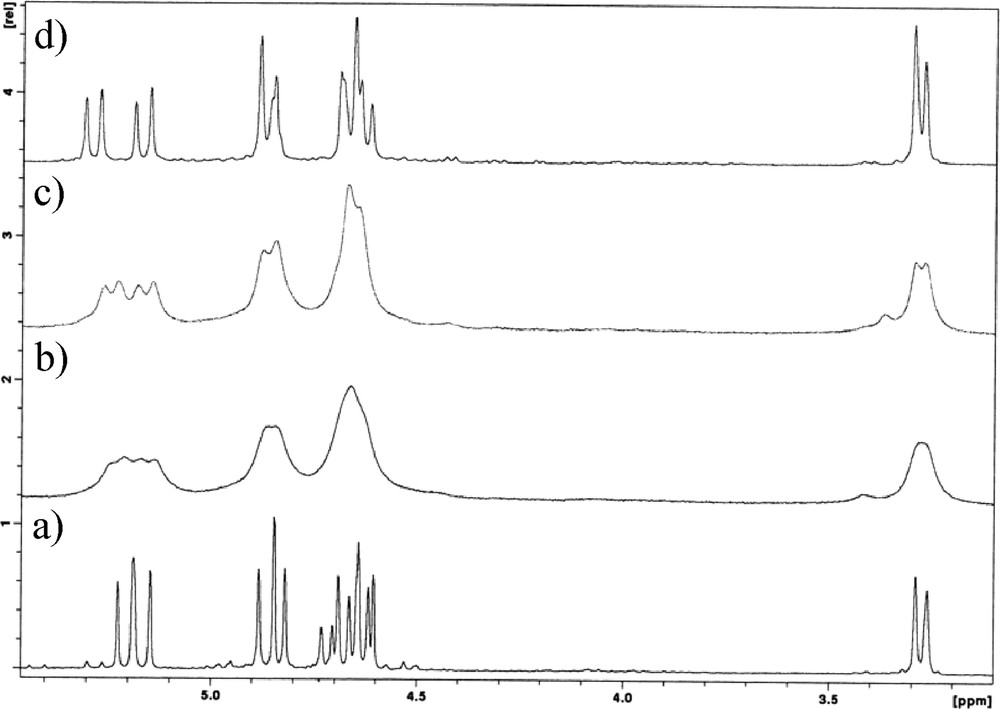
Methylene region of the 1H NMR spectra of t-Bu ketone 1b in CDCl3, 25 °C and 500 MHz: (a) free ligand, upon addition of (b) 0.5, (c) 1 equiv and (d) 3 equiv of Yb triflate.
In general, the NMR results follow the same trend of those obtained before by UV spectrophotometric studies.
2.4 MD simulations
2.4.1 Solvation in pure acetonitrile and dichloromethane
Simulation studies were mainly focused on the comparison between dihomooxacalix[4]arene and calix[4]arene derivatives. Thus, the affinity of Ph ketones 1d and 2d for La3+ and Yb3+ cations was obtained through MD simulations. These cations, representative of the light and heavy lanthanides, were chosen because of the highest difference on their E % and log β values. Picrate and triflate counterions were added to neutralise the chemical systems and to reproduce the extraction and complexation experiments in dichloromethane and acetonitrile, respectively. Dichloromethane solutions mimic the final organic phase once the extraction has taken place. Final structures obtained are given in Fig. 5, and interaction energies and radial distribution functions performed during the simulations are presented in Table 3 and in Fig. S1, respectively.

Final snapshots of M3+⊂1d and M3+⊂2d complexes in acetonitrile (left) and dichloromethane (right). Final views of the full simulation boxes and snapshots of the structures and solvation of the calixarenes.
Interaction energies (kcal mol−1) in acetonitrile solution and gas phase.
| MDa acetonitrile solution | MDa vacuum | QMb vacuum | |||
| M3+ | ligand | M3+⊂ligand | 3X− | solute | solventc |
| La3+⊂1d | −498 | −544 | −311 | −534 | −589.9 |
| La3+⊂2d | −508 | −576 | −240 | −521 | −581.9 |
| Yb3+⊂1d | −467 | −813 | −219 | −649 | −669.2 |
| Yb3+⊂2d | −577 | −617 | −229 | −656 | −672.4 |
a Calculated during the last 5 ns of dynamics; fluctuations are about 8 kcal mol−1.
b QM B3LYP/6-31G(d,p) optimisations.
c Solute = [M3+⊂ligand⋯3X−]; X− = triflate anion.
Simulations show some clear differences in the structures and solvation of the M3+⊂1d and M3+⊂2d complexes and significant interactions between the M3+ cations and the counterions.
The M3+⊂1d complexes are asymmetrical owing to the presence of the supplementary oxygen atom, while 2d leads to more symmetrical complexes with an equivalent participation of the oxygen atoms. The position of the La3+ cation within the cavity formed by the four phenoxy and the four carbonyl oxygen atoms of both ligands in acetonitrile and dichloromethane solvents is more centred when compared to that of Yb3+. As clearly illustrated by the snapshots of Fig. 5, this smaller cation interacts with the ligands preferentially via the carbonyl oxygen atoms, which leads to a lower position in the cavity. However, in the case of Yb3+⊂2d in dichloromethane, the cation is perfectly centred in the cavity.
For both calixarenes, the coordination spheres of the lanthanide cations are completed by triflate anions and solvent molecules in acetonitrile (via the nitrogen atom of the cyano groups) and picrate anions in dichloromethane. It is important to note that, on average, there are only two triflates around the complexes, which results in the formation of a +1 charged association. In dichloromethane solutions, three picrate anions sit into the first sphere of coordination of the lanthanide ions and interact with the M3+⊂calixarene complexes, thus forming a neutral species. M3+⋯calixarene and M3+⋯anions radial distribution functions (RDFs; Fig. S1) calculated during the last 5 ns of dynamics clearly show the differences of interaction between the ligands, the cations and the anions. The number of oxygen atoms around La3+ varies from 9.0 to 11.0 and around Yb3+ from 7.6 to 10.0. The first peak of the RDFs clearly show that the OC=O⋯Yb3+ distances are always shorter than OC=O···La3+ distances (2.4 vs 2.9), being consistent with the fact that La3+ has a higher radius than Yb3+. This tendency is the same for the counterions. Moreover, for both cations, the picrate anions are always further away than the triflate anions. In all but one case, the upper rim of the calixarenes is filled with a solvent molecule (Fig. 5). This trapped molecule interacts with the complexed cation via an Ncyano⋯M3+ or Cl⋯M3+ interaction.
The interaction energies between the lanthanide cations and the two ligands in gas phase and in solution were calculated, and the results are reported in Table 3. Cation…ligand interaction energies calculated in vacuum by quantum mechanics (QM) and MD simulations are consistent and follow the same order. These energies are higher for the Yb3+ cation than for La3+ in agreement with the shorter OC=O⋯M3+ distances observed before. For the majority of the cases studied, these values follow the same trend of the stability constants. In solution, the cation⋯ligand energies are lower; the variations are quite low for the La3+ cation (from 13 to 36 kcal mol−1) and much higher for Yb3+ (from 79 to 182 kcal mol−1). These differences are compensated by a better interaction between the Yb3+⊂ligand and the triflate anions.
2.4.2 Free energy calculations at the dichloromethane/water interface
To get further insights of the extraction process, PMF calculations were performed at the dichloromethane/water interface. The free energy profiles were calculated for M3+⊂calixarene interface crossing for both 1d and 2d with picrate as a counter ion. Free energies were calculated as a function of the z-position of the complex (Fig. 6). The initial positions of the calixarenes were obtained after 10 ns of MD runs at the dichloromethane/water interface. The complexed calixarenes sit into the organic phase or at the interface between the two liquids (Fig. 6).
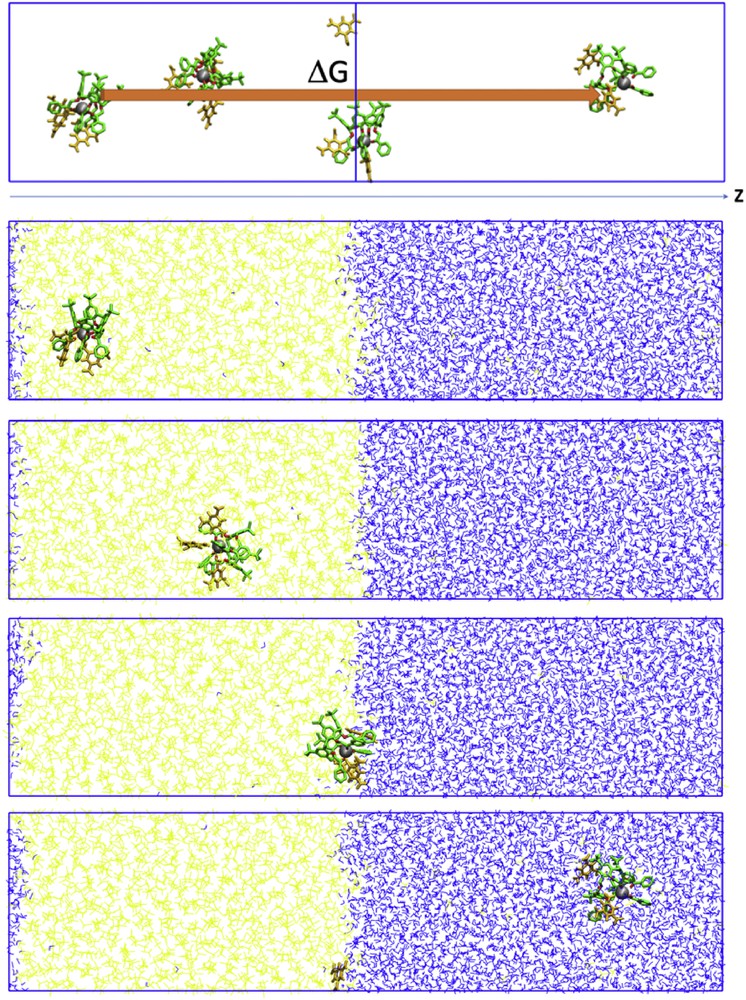
Yb3+⊂2d···3Pic− system at a CH2Cl2/water interface (CH2Cl2 represented in yellow and water in blue). Scheme of the procedure of the PMF calculations (top) and different snapshots during the dynamics of interface crossing (bottom). A precise description of the PMF calculation can be found in the Supporting Information.
Calculated free energy profiles are reported in Fig. 7; although these values are unknown, it is clear from extraction experiments that the lanthanide cations are extracted and thus prefer the organic phase. The transfer to the organic phase is found to be very similar for La3+ and Yb3+ cations and is almost independent of the nature of the calixarene used which is consistent with the moderate extraction rate of ligands 1d and 2d. To retain the neutrality of the source and receiving phases, the cation's charge is compensated by counterions to be extracted. Free energy of transfer shows a clear energetic preference of both M3+⊂calixarene for dichloromethane, as well as small differences in the extraction of La3+ and Yb3+ by 1d and 2d. PMF curves nicely follow the extraction percentages measured experimentally: The free energy calculated for Yb3+⊂1d (full blue curve, Fig. 7) shows no minima at the interface and a large energy gap between water and the organic phase, which indicates that the extraction process is spontaneous. The same conclusion applies to La3+⊂1d, with an energy gap a little bit smaller (full red curve, Fig. 7). The two curves for ligand 2d show a small energy minimum of about 3–6 kcal mol−1 at the interface. This indicates that the M3+⊂2d species should be trapped and accumulates at the interface, being the diffusion to dichloromethane harder. This observation can also explain the smaller extraction percentages obtained for this ligand compared to those of 1d.
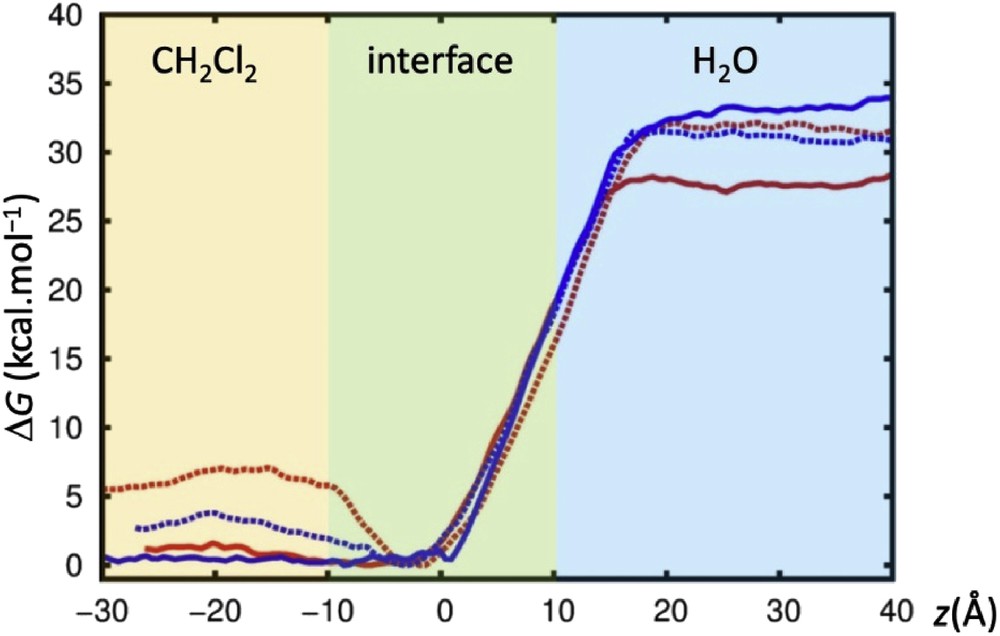
PMF calculations crossing the CH2Cl2/H2O interface with picrate anions. Free energies ΔG (kcal mol−1) as a function of the distance Z (Å). In red: La3+⊂1d (full), La3+⊂2d (dotted); in blue: Yb3+⊂1d (full), Yb3+⊂2d (dotted).
3 Conclusions
Extraction studies from an aqueous solution into CH2Cl2 have indicated that dihomooxa ketones 1b–1d, in the cone conformation, are modest extractants for the lanthanides, displaying some preference for Gd3+ and Yb3+ cations (% E = 23 and 21, respectively, for 1c). In the case of classical calix[4]arenes, ketones 2a, 2c, and 2d presented low percentages of extraction (% E ≤ 8.5), with almost no discrimination among the ten lanthanides studied. The stability constants obtained in acetonitrile have shown however that both types of ligands are good binders, with some selectivity for La3+ in the case of ketones 1b and 1d (log β = 6.1 and 6.0, respectively) and for Yb3+ in the case of 2d (log β = 6.2). NMR titration results indicated a similar binding trend as that observed by UV studies. Phenyl ketone 1d showed to be a slightly weaker ligand than tert-butyl 1b and adamantyl 1c, being in agreement with the basicity of their carbonyl oxygen atoms.
MD and PMF simulations performed for ligands 1d and 2d with La3+ and Yb3+ cations in acetonitrile and dichloromethane, and at a CH2Cl2/water interface, were consistent with the experimental results. The calculations showed features of each ligand (1d vs 2d) and the role of counterions in the complexation. Calculated free energy profiles for M3+⊂calixarene interface crossing demonstrated that its transfer to the organic phase is energetically favourable. Overall, simulations provided explanations, at the microscopic level, that contribute to a better understanding of the differences between ligands 1d and 2d observed experimentally.
4 Experimental
4.1 Extraction studies
After mutual saturation of the solvents, equal volumes (5 mL) of aqueous solutions of metal picrates (2.5 × 10−4 M) and solutions of the calixarenes (2.5 × 10−4 M) in CH2Cl2 were vigorously shaken for 2 min and then thermostated in a water bath with mechanical stirring at 25 °C overnight. After complete phase separation, the absorbance A of picrate ions in the aqueous phase was determined spectrophotometrically (λmax = 354 nm). For each cation-calixarene system the absorbance measurements were repeated at least four times. Blank experiments without calixarene were run under the same conditions, yielding an absorbance A0. The percentage of cation extracted was calculated as the ratio 100 × (A0 – A)/A0. The details of the metal picrate preparation have already been described [23].
4.2 Determination of stability constants
The apparent overall stability constants β, defined as the concentration ratio [MLn+]/([Mn+][L]) (where Mn+ = cation and L = ligand), were determined in acetonitrile (Chromasolv@gradient grade for HPLC ≥99.9%, used without further purification; Sigma-Aldrich) by absorption spectrophotometry. The spectra were recorded between 240 and 350 nm using a Shimadzu UV-2401 PC spectrophotometer using quartz cells (Hellma) with an optical path length of 1 cm. Successive additions of the metal salt solution were made to 2.5 mL of a ligand solution (CL = 1.8×10−5 – 8.5×10−5 M) directly in the spectrophotometric cell. When kinetic effects were observed, solutions were prepared in separated vials and measured after the time needed to reach the equilibrium. The experiments were performed at 25 °C and constant ionic strength provided by 10−2 M Et4NClO4 (98%; Acros Organics). The spectral changes were interpreted using the numerical program SpecFit [27]. The metal salts used were triflates: La(CF3SO3)3,2.7H2O (99%; Alfa Aesar); Pr(CF3SO3)3,3.6H2O(98%; Alfa Aesar); Eu(CF3SO3)3,3.2H2O (98%; Alfa Aesar); Gd(CF3SO3)3,13H2O (98%; Alfa Aesar); Yb(CF3SO3)3,1.4H2O (99.99%; Sigma-Aldrich). All these salts were dried under vacuum for at least 24 h before use. The concentrations of their stock solutions were standardised by complexometry titrations with ethylenediaminetetraacetic acid using the xylenol orange as a coloured indicator [28].
4.3 Proton NMR titrations
Several aliquots (up to 3 equiv) of the salt solutions in CD3OD were added to CDCl3 solutions (5 × 10−3 M) of the ligands directly in the NMR tube. The salts used were La, Eu and Yb triflates. Owing to the low solubility of Eu triflate in MeOH, it was necessary to decrease the concentration of the ligands (2.5 × 10−4 M) and of the salt accordingly. The spectra were recorded on a Bruker Avance III 500 Spectrometer after each addition of the salts. The temperature of the NMR probe was kept constant at 25 °C.
4.4 MD simulations
The solutions have been simulated by classical MD simulations using the Amber18 software [29], in which the potential energy U is empirically represented by a sum of bond and angle harmonic deformations, of dihedral torsions, and by pairwise additive 1-6-12 (electrostatic + van der Waals) interactions between nonbonded atoms (Eq. 1):
| (1) |
A full description of the simulation parameters, PMF procedure and analysis is given in the Supporting Information.
Acknowledgements
The authors thank Fundação para a ciência e a Tecnologia, Project ref. UID/QUI/00100/2013.