

1 Introduction
Polyfluorinated benzylamines are widely used as pharmaceuticals, agrochemicals, and imaging materials (Fig. 1).[1] As an example, Taradenacin, which is an antimuscarinic agent, contains a 3,4,5-trifluorobenzylamine group. The difluorinated benzylamine larotrectinib is a drug for the treatment of cancer, and the baloxavir marboxil is an antiviral medication for treatment of influenza A and influenza B. Pexmetinib, which includes a 3-fluorobenzylamine group, is an antiinflammatory drug, and trelagliptin is a drug used for the treatment of type 2 diabetes. Previous methods for the synthesis of fluorinated aromatics, e.g., Balz−Schiemann reaction, often required harsh reaction conditions,[2] which limit the scope of this transformation.[3] More recently, fluoroaromatics were prepared from aryl (pseudo)halides or aryl metallic derivatives via transition metal–catalyzed fluorinations.[4] However, this strategy required to use prefunctionalized starting materials. The C–H bond fluorination has also been reported but required the preinstallation of directing groups and/or the use of expensive fluorine sources.[5] The discovery of simple and efficient methods to access to fluorinated molecules with broad molecular diversity remains an important challenge for both academic research groups and chemical companies.
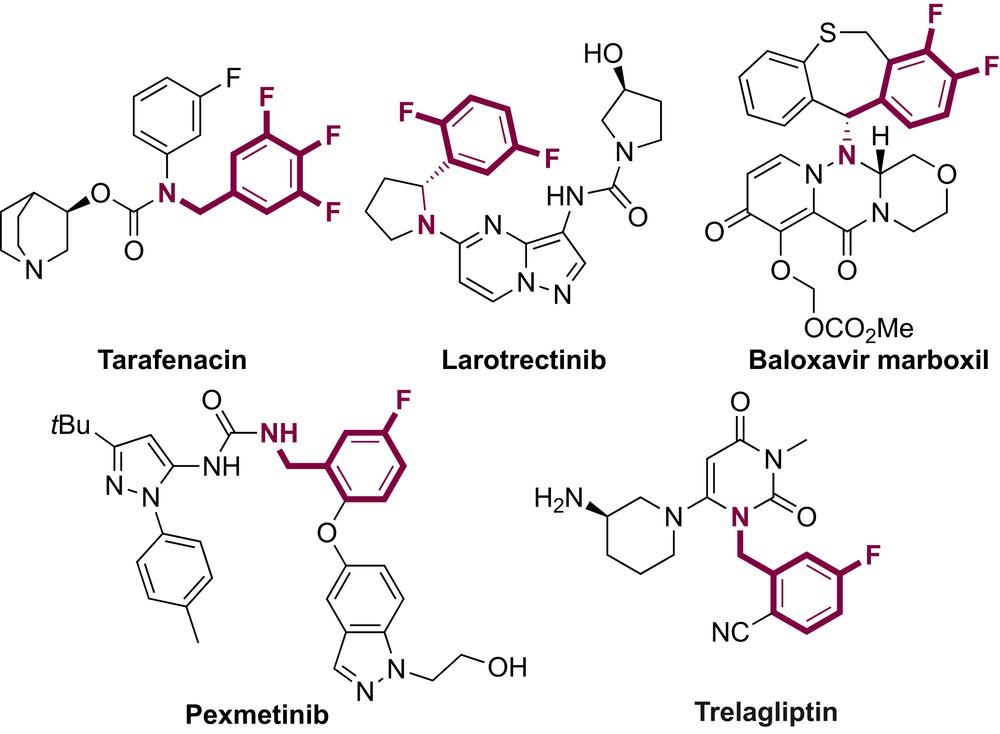
Relevant pharmaceuticals containing a (poly)fluorinated benzylamine scaffold.
Recently, transition metal–catalyzed regioselective C–H bond functionalization has emerged as a suitable tool to access molecular diversity with a minimum number of manipulations.[6] In 2006, Fagnou et al.[7] reported the first example of palladium-catalyzed C–H bond arylation of (poly)fluorobenzene derivatives using aryl halides as coupling partners (Fig. 2a). The reaction generally took place at ortho-position of fluorine atoms allowing the preparation of fluorinated biphenyls in good yields from commercially available fluorinated raw materials. However, the regioselectivity can be affected by the presence of an additional substituent leading to mixtures of para- or meta-fluorobiphenyl derivatives.[8] We have shown that using Pd/KOPiv/DMA as a catalytic system, (poly)fluorobenzene bearing a benzoxazole,[9] a pyridine,[10] or an amide[11] group reacted preferentially at the C–H bond flanked by two fluorine atoms (Fig. 2b). To the best of our knowledge, no example of Pd-catalyzed C–H bond arylation of polyfluorinated benzylamines was reported so far. Such substrates are challenging because previous reports have shown that benzylamine derivatives such as sulfinyl isobutyramide,[12] picolinamide,[13] and pyrazine-2-carboxamide preferentially react at the ortho-position of the methylamine group (Fig. 2c).

Pd-catalyzed C–H bond arylation of polyfluorinated benzylamines.
Herein, starting from a set of N-methyl-N-(polyfluorobenzyl)acetamides and an N-methyl-N-(polyfluorobenzyl)benzamide as reactants, we report (i) on the reactivity and regioselectivity for the direct arylation of benzene units containing both fluoro and N-methyl tertiary amide substituents and (ii) on the access to a variety of N-protected-methylamine–substituted (poly)fluorobiphenyls using a variety of aryl bromides as coupling partners (Fig. 2d).
2 Result and discussion
We first investigated the reactivity of nonprotected (2,3,5,6-tetrafluorophenyl)methanamine (a) in Pd-catalyzed C–H bond arylation with 4-bromobenzonitrile (Table 1, entry 1). Using our previous reaction conditions for C–H bond arylation of polyfluorobenzene derivatives [namely, 2.5 mol% of Pd(OAc)2 associated with 2 equivalents of KOAc in DMA at 150 °C], the desired biphenyl product 1a was not obtained. A similar reactivity trend was observed with N-(2,3,5,6-tetrafluorobenzyl)acetamide (b) (Table 1, entry 2). The lack of reactivity of these two substrates might be explained by the presence of NH function, which could inhibit the palladium activity. Therefore, we decided to investigate the reactivity of N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c). We were pleased to find that under the same reaction conditions, the desired biphenyl product 1c was obtained in 45% yield (Table 1, entry 3). Then, we explored the effect of several reaction parameters (i.e., base, palladium sources and solvent). The use of K2CO3 as base inhibited the reaction, while the use of PivOK as base slightly improved the yield of 1c to 58% (Table 1, entries 4 and 5). In both cases, we observed partial conversions of 4-bromobenzonitrile; therefore, other sources of palladium were tested. No improvement was observed using 2.5 mol% PdCl2(CH3CN)2, whereas the use of a palladium diphosphine catalyst [PdCl(C3H5)(dppb)] gives 1c in 89% yield with a full conversion of the aryl bromide (Table 1, entries 6 and 7). A partial conversation was obtained using only 1 mol% of this Pd catalyst (Table 1, entry 8). No improvement was observed by changing DMA to DMF or xylene (Table 1, entries 9 and 10).
Reactivity of (2,3,5,6-tetrafluorophenyl)methanamine (a), N-(2,3,5,6-tetrafluorobenzyl)acetamide (b), and N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c) in Pd-Catalyzed C–H bond arylation with 4-bromobenzonitrile.
| Entry | Reactant a, b, or c | [Pd] catalyst | Base | Yield (%) |
| 1 | a | Pd(OAc)2 | KOAc | 0 |
| 2 | b | Pd(OAc)2 | KOAc | 0 |
| 3 | c | Pd(OAc)2 | KOAc | 45 |
| 4 | c | Pd(OAc)2 | K2CO3 | 0 |
| 5 | c | Pd(OAc)2 | PivOK | 58 |
| 6 | c | PdCl2(CH3CN)2 | PivOK | 41 |
| 7 | c | PdCl(C3H5)(dppb) | PivOK | 89 |
| 8a | c | PdCl(C3H5)(dppb) | PivOK | 76 |
| 9b | c | PdCl(C3H5)(dppb) | PivOK | 21 |
| 10c | c | PdCl(C3H5)(dppb) | PivOK | 0 |
a Using 1 mol% of Pd catalyst.
b In dimethylformamide (DMF).
c In xylene.
Having determined the best conditions to arylate the C–H bond of N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c), we turned our attention to the scope of aryl bromides (Scheme 1). Other aryl bromides bearing an electron-withdrawing substituent at the para-positions – such as ethyl ester and formyl groups – well reacted to afford the biphenyl products 2 and 3 in 87% and 84% yield, respectively. The reaction was more sluggish when 4-bromoanisole was used, as the desired acetamide 4 was isolated in only 51% yield because of a partial conversion of this electron-rich aryl bromide. Similarly, the coupling between c and 3-bromotoluene led to the formation of 5 in 53% yield. The reaction was not very sensitive to steric factors as 2-bromobenzaldehyde and 2-bromobenzonitrile reacted well with c to give the biphenyl products 6 and 7 in 87% and 78% yield, respectively. The reaction also tolerated the use of nitrogen-containing heteroaryl bromides such as 3-bromoquinoline and 3-bromopyridine, as the products 8 and 9 were isolated in good to excellent yields.
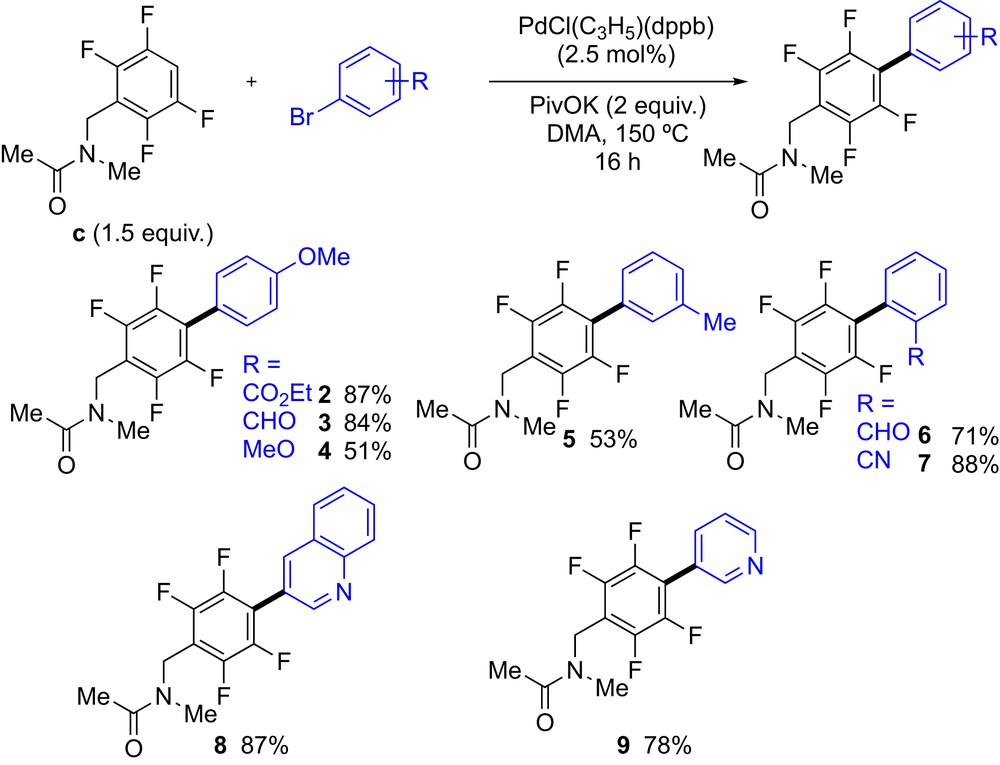
Scope of (hetero)aryl bromides in Pd-catalyzed C–H bond arylation of N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c).
Then, we investigated the reactivity of N-methyl-N-(2,3,5-trifluorobenzyl)acetamide (d) in Pd-catalyzed C–H bond arylation with aryl bromides (Scheme 2). Although this substrate bears two C–H bonds, the reaction regioselectively occurred only at the C–H bond flanked by the two-fluorine atoms, affording the biphenyls 10 and 11 in good yields from 4-bromobenzonitrile and ethyl 4-bromobenzoate, respectively. A similar regioselectivity has been previously observed with 6-substituted 1,2,4-trifluorobenzene derivatives.[7a, 10] This regioselectivity might be assigned to a lower Gibbs free energies of activation (ΔG≠) of the cleavage of C–H bond adjacent to two fluorine atoms in concerted metalation deprotonation (CMD) process.[14]

Scope of aryl bromides in Pd-catalyzed C–H bond arylation of N-methyl-N-(2,3,5-trifluorobenzyl)acetamide (d).
We also explored the reactivity of N-(3,5-difluorobenzyl)-N-methylbenzamide (e) in Pd-catalyzed C–H bond arylation with aryl bromides (Scheme 3). In line with the previous reports on arylation of 1,3-difluorobenzene derivatives,[7a, 9−10] reaction between the difluorinated benzyl-N-methylbenzamide e and 4-bromobenzonitrile led to the single regioisomer 12 in 67% yield. The arylation again took place at the C–H bond flanked by the two fluorine atoms. The coupling reactions with other aryl bromides para-substituted by an electron withdrawing group (e.g., nitro, formyl, ethyl ester or benzoyl) afforded the 2,6-difluoro-[1,1′-biphenyl] derivatives 13–16 in 54–66% yields. From N-(3,5-difluorobenzyl)-N-methylbenzamide (e) and 2-bromobenzonitrile, the desired coupling product 17 was isolated in only 42% yield. Moreover, 3-bromoquinoline was successfully used as aryl source to provide an efficient access to the 2-(2,6-difluorophenyl)quinoline derivative 18 in 58% yield.
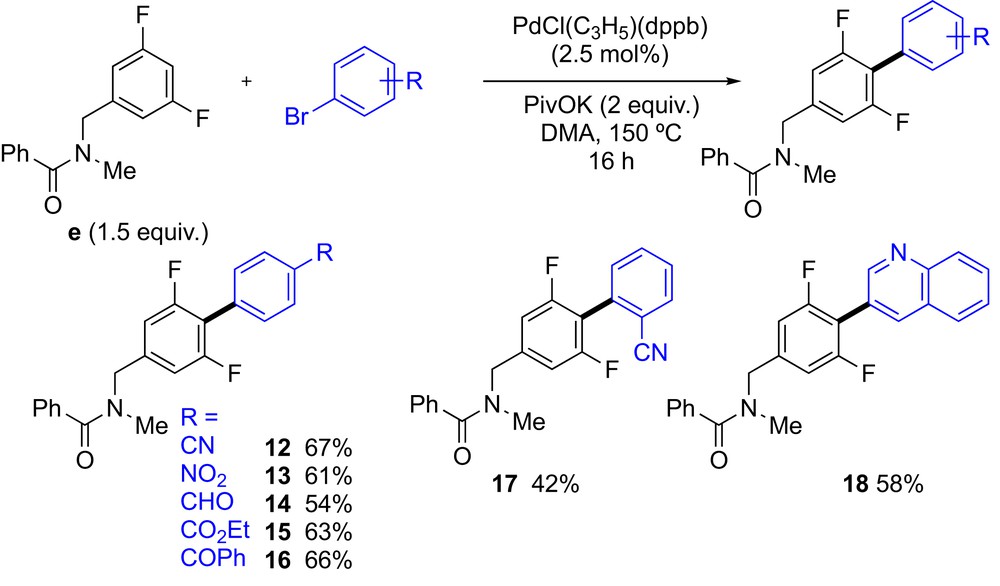
Scope of (hetero)aryl bromides in Pd-catalyzed C–H bond arylation of N-(3,5-difluorobenzyl)-N-methylbenzamide (e).
The reactivity and regioselectivity was not sensitive to the substitution pattern of the fluorine atoms, provided that the C–H bond remained flanked by two fluorine atoms (Scheme 4). As an example, N-(2,4-difluorobenzyl)-N-methylacetamide (f) underwent C–H bond arylation in the presence of 2.5 mol% of Pd(OAc)2 associated with 2 equivalents of KOAc in DMA at 150 °C. The biphenyl products 19–21 were regioselectively obtained in 55–76% yields from 4-bromobenzonitrile, 1-bromo-4-nitrobenzene, and 3-bromobenzonitrile. Again, 3-bromoquinoline displayed good reactivity, affording the 3-arylquinoline 22 in 58% yield.

Scope of (hetero)aryl bromides in Pd-catalyzed C–H bond arylation of N-(2,4-difluorobenzyl)-N-methylacetamide (f).
3 Conclusion
In summary, we have demonstrated that Pd-catalyzed direct arylation of electron-deficient arenes such as 1,2,4,5-tetrafluorobenzyl, 1,2,4-trifluorobenzyl, or 1,3-difluorobenzyl, bearing N-methylacetamide or N-methylbenzamide groups, always reacted via the cleavage of the C–H bond flanked by two fluorine atoms, whereas the C–H bonds at ortho-position of N-methylacetamide or N-methylbenzamide remained untouched. These fluorine-directed C–H bond functionalizations proceed with an easy-to-handle diphosphine Pd catalyst and PivOK as base in DMA. This procedure tolerates a wide variety of substituents on the aryl bromides such as nitro, cyano, ester, ketone, formyl, and heteroaryl bromides with pyridinyl or quinolyl units. The reaction is not limited to activated aryl bromides, as electron-donating substituents such as methoxy and methyl are also tolerated. Owing to the importance of polyfluorinated benzylamines and polyfluorinated biphenyls in pharmaceutical and material sciences, this methodology could find applications in late-stage modifications via regioselective C–H bond arylation to discover new active fluorinated molecules.
4 Experimental section
4.1 General
All reactions were carried out under argon atmosphere with standard Schlenk-tube techniques. High-performance liquid chromatography (HPLC)–grade DMA was stored under argon and used without further purification. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AV III 400 MHz NMR spectrometer equipped with BBFO probe head. Chemical shifts (d) were reported in parts per million relative to residual chloroform (7.26 ppm for 1H; 77.0 ppm for 13C), and constants were reported in Hertz. 1H NMR assignment abbreviations were the following: singlet (s), doublet (d), triplet (t), quartet (q), doublet of doublets (dd), doublet of triplets (dt), and multiplet (m). All reagents were weighed and handled in air.
4.2 Preparation of the PdCl(C3H5)(dppb) catalyst
An oven-dried 40-mL Schlenk tube equipped with a magnetic stirring bar under argon atmosphere was charged with [Pd(C3H5)Cl]2 (182 mg, 0.5 mmol) and dppb (426 mg, 1 mmol). Ten milliliters of anhydrous dichloromethane were added; then, the solution was stirred at room temperature for 20 minutes. The solvent was removed in vacuum. The powder was used without purification. (31P NMR 381 MHz, CDCl3) δ = 19.3 (s).[15]
4.3 General procedure A for synthesis of N-methyl-N-(polyfluorobenzyl)acetamides (c-d and f) and N-(3,5-difluorobenzyl)-N-methylbenzamide (e)
To a DMF (10 mL) solution of sodium hydride (60% dispersion in mineral oil) (0.719 g, 0.431 g corrected for mineral oil, 18 mmol, 1.2 equiv) a solution of N-methylacetamide (1.22 g, 16.8 mmol, 1.12 equiv) or N-methylbenzamide (2.27 g, 16.8 mmol, 1.12 equiv) in DMF (8 mL) was slowly added. After 30 min at room temperature, polyfluorobenzylbromide (15 mmol, 1 equiv) was added concurrently over 1 h. A water bath was used to maintain the temperature below 40 °C. The resulting mixture was stirred overnight at room temperature and then poured into a mixture of 20% NH4Cl (50 mL) and Et2O (50 mL). The layers were separated, and the aqueous layer was extracted with Et2O (2 x 50 mL). The organic layers were combined and washed with H2O (3 x 50 mL) and then brine (50 mL). The organic layer was dried (Na2SO4), and the solvent was removed to afford a residue. The crude mixture was purified by silica column chromatography to afford the desired arylated product.
4.4 N-Methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c)
Following the general procedure A using 3-(bromomethyl)-1,2,4,5-tetrafluorobenzene (3.64 g, 15 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 70-30) to afford the desired compound c (3.25 g, 92%) as a yellow solid (Mp = 85–97 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 7.08–6.86 (m, 1H), 4.80 & 3.55 (s, 2H), 2.91 & 2.73 (s, 3H), 2.15 & 1.98 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 170.4 & 170.2, 145.7 (dm, J = 250.4 Hz), 145.1 & 145.7 (dm, J = 250.4 Hz), 116.5 & 115.6 (t, J = 17.0 Hz), 106.2 & 105.2 (t, J = 22.7 Hz), 42.3 & 39.2, 35.8 & 32.0, 21.4 & 20.9. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C10H9F4NO (235.18): C 51.07, H 3.86; found: C 51.21, H 4.03.
4.5 N-Methyl-N-(2,3,5-trifluorobenzyl)acetamide (d)
Following the general procedure A using 3-(bromomethyl)-2,4,5-trifluorobenzene (3.38 g, 15 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 70-30) to afford the desired compound d (3.06 g, 94%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 6.88–6.56 (m, 2H), 4.55 & 4.52 (s, 2H), 2.95 & 2.87 (s, 3H), 2.08 & 2.07 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 171.0 & 170.8, 157.8 (dm, J = 250.1 Hz), 150.1 (dm, J = 250.1 Hz), 145.5 (dm, J = 245.8 Hz), 127.8 (dd, J = 8.6, 13.8 Hz), 111.1 & 109.2 (td, J = 3.2, 24.1 Hz), 104.4 (dd, J = 20.9, 27.6 Hz), 48.0 & 44.0, 36.1 & 33.6, 21.6 & 21.1. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C10H10F3NO (217.19): C 55.30, H 4.64; found: C 55.48, H 4.87.
4.6 N-(3,5-Difluorobenzyl)-N-methylbenzamide (e)
Following the general procedure A using 1-(bromomethyl)-3,5-difluorobenzene (3.11 g, 15 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 70-30) to afford the desired compound e (3.60 g, 92%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 7.44–7.28 (m, 6H), 6.87–6.80 (m, 1H), 6.78–6.69 (m, 1H), 4.71 & 4.47 (s, 2H), 2.94 & 2.86 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 172.5 & 172.0, 163.5 (dm, J = 249.1 Hz), 141.2 (m), 135.8, 130.0, 128.7, 127.2, 110.7(m), 103.2 (m), 50.5, 37.4. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C15H13F2NO (261.27): C 68.96, H 5.02; found: C 69.19, H 6.15.
4.7 N-(2,4-Difluorobenzyl)-N-methylacetamide (f)
Following the general procedure A using 1-(bromomethyl)-2,4-difluorobenzene (3.11 g, 15 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 70-30) to afford the desired compound f (2.69 g, 90%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 7.31 & 7.11 (q, J = 6.9, 7.6 Hz, 1H), 6.92–6.72 (m, 2H), 4.57 & 4.51 (s, 2H), 2.96 & 2.90 (s, 3H), 2.15 & 2.11 9s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 170.6 (2), 162.1 &161.8 (dd, J = 14.2, 248.1 Hz), 160.6 & 161.0 (dd, J = 11.8, 248.2 Hz), 131.2 (m), 129.0 (dd, J = 5.8, 9.7 Hz), 128.0 & 126.6, 120.1 & 119.3 (d, J = 3.7 Hz), 111.2 & 111.1 (d, J = 21.1 Hz), 103.9 & 103.2 (t, J = 25.7 Hz), 47.5 & 43.3, 35.5 & 32.9, 21.3 & 20.8. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C10H11F2NO (199.20): C 60.30, H 5.57; found: C 60.45, H 5.29.
4.8 General procedure B for synthesis of fluorinated biphenyls
To a 25 mL oven dried Schlenk tube, (polyfluorobenzyl)acetamide (0.75 mmol), (hetero)arylbromide (0.5 mmol), PivOK (140 mg, 1 mmol), DMA (2 mL) and PdCl(C3H5)(dppb) (7.7 mg, 0.012 mmol) were successively added. The reaction mixture was evacuated by vacuum-argon cycles (5 times) and stirred at 150 °C (oil bath temperature) for 16 h (see tables and schemes). After cooling the reaction at room temperature and concentration, the crude mixture was purified by silica column chromatography to afford the desired arylated product.
4.9 N-((4′-Cyano-2,3,5,6-tetrafluoro-[1,1′-biphenyl]-4-yl)methyl)-N-methylacetamide (1c)
Following the general procedure B using N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c) (176 mg, 0.75 mmol) and 4-bromobenzonitrile (91 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 70-30) to afford the desired compound 1c (150 mg, 89%) as a white solid (Mp = 130–132 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 7.81–7.74 (m, 2H), 7.61–7.54 (m, 2H), 4.79 & 4.69 (s, 2H), 3.01 & 2.90 (s, 3H), 2.29 & 2.11 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 170.8 & 170.5, 147.7 (dm, J = 252.2 Hz), 143.5 (dm, J = 246.2 Hz), 132.5 & 132.4, 132.1 & 131.7 (brs), 131.0, 118.3, 118.2 & 118.0, 116.5 & 115.3 (t, J = 17.2 Hz), 113.5 & 113.2, 42.7 & 40.1, 36.6 & 32.6, 21.8 & 21.3. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C17H12F4N2O (336.29): C 60.72, H 3.60; found: C 60.89, H 3.56.
4.10 Ethyl 2′,3′,5′,6′-tetrafluoro-4'-((N-methylacetamido)methyl)-[1,1′-biphenyl]-4-carboxylate (2)
Following the general procedure B using N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c) (176 mg, 0.75 mmol) and ethyl 4-bromobenzoate (115 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 60-40) to afford the desired compound 2 (167 mg, 87%) as a yellow solid (Mp = 102–105 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 8.17–8.08 (m, 2H), 7.55–7.44 (m, 2H), 4.74 & 4.66 (s, 2H), 4.39 (q, J = 7.1 Hz, 2H), 3.05 & 2.99 (s, 3H), 2.11 & 2.06 (s, 3H), 1.38 (t, J = 6.7 Hz, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 170.7 & 170.5, 166.1 & 166.0, 145.8 (dm, J = 252.2 Hz), 143.7 (dm, J = 247.8 Hz), 131.8 & 131.4 (brs), 131.5 & 131.2, 130.2, 129.9, 129.8, 120.3 & 119.3 (t, J = 16.4 Hz), 115.6 & 114.6 (t, J = 17.2 Hz), 61.4 & 61.3, 42.6 & 39.8, 36.3 & 32.6, 21.8 & 21.3, 14.4. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C19H17F4NO3 (383.35): C 59.53, H 4.47; found: C 59.62, H 4.71.
4.11 N-Methyl-N-((2,3,5,6-tetrafluoro-4′-formyl-[1,1′-biphenyl]-4-yl)methyl)acetamide (3)
Following the general procedure B using N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c) (176 mg, 0.75 mmol) and 4-bromobenzaldehyde (93 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 50-50) to afford the desired compound 3 (143 mg, 84%) as a yellow solid (Mp = 93–95 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 10.10 & 10.08 (s, 1H), 8.05–7.96 (m, 2H), 7.70–7.60 (m, 2H), 4.76 & 4.69 (s, 2H), 3.09 & 2.92 (s, 3H), 2.31 & 2.13 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 191.7 & 191.6, 170.8 & 170.6, 145.8 (dm, J = 252.2 Hz), 143.9 (dm, J = 249.1 Hz), 136.8 & 136.6, 133.5 & 133.0, 131.0 (m), 129.9 & 129.9, 118.9 & 116.1 (t, J = 16.3 Hz), 42.7 & 40.0, 36.5 & 32.6, 21.8 & 21.3. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C17H13F4NO2 (339.29): C 60.18, H 3.86; found: C 60.02, H 4.06.
4.12 N-Methyl-N-((2,3,5,6-tetrafluoro-4′-methoxy-[1,1′-biphenyl]-4-yl)methyl)acetamide (4)
Following the general procedure B using N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c) (176 mg, 0.75 mmol) and 4-bromoanisole (94 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 70-30) to afford the desired compound 4 (87 mg, 51%) as a white solid (Mp = 108–110 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 7.44–7.37 (m, 2H), 7.06–6.99 (m, 2H), 4.76 & 4.66 (s, 2H), 3.86 & 3.86 (s, 3H), 3.04 & 2.90 (s, 3H), 2.30 & 2.12 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 170.6 & 170.5, 145.7 (dm, J = 252.2 Hz), 143.8 (dm, J = 251.1 Hz), 131.4 (m), 120.0 (t, J = 16.5 Hz), 119.4, 119.0, 114.2, 114.1, 114.0 & 113.8, 55.4 & 44.3, 42.5 & 39.3, 36.0 & 32.4, 21.8 & 21.2. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C17H15F4NO2 (341.31): C 59.83, H 4.43; found: C 59.98, H 4.21.
4.13 N-Methyl-N-((2,3,5,6-tetrafluoro-3′-methyl-[1,1′-biphenyl]-4-yl)methyl)acetamide (5)
Following the general procedure B using N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c) (176 mg, 0.75 mmol) and 3-bromotoluene (85 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 60-40) to afford the desired compound 5 (86 mg, 53%) as a yellow solid (Mp = 48–50 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 7.49–7.33 (m, 1H), 7.29–7.21 (m, 3H), 4.76 & 4.66 (s, 2H), 3.04 & 2.91 (s, 3H), 2.40 (s, 3H), 2.30 & 2.12 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 170.6 & 170.4, 145.7 (dm, J = 246.9 Hz), 143.8 (dm, J = 245.6 Hz), 138.5 & 128.3, 130.6 (m), 130.2 (m), 128.7 & 128.6, 128.6 & 128.5, 127.2 & 126.8, 121.1 & 120.4 (t, J = 16.9 Hz), 114.5 & 113.5 (t, J = 17.3 Hz), 42.5 & 39.5, 36.0 & 32.4, 21.7 & 21.4, 21.6 & 21.2. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C17H15F4NO (325.31): C 62.77, H 4.65; found: C 62.81, H 4.86.
4.14 N-Methyl-N-((2,3,5,6-tetrafluoro-2′-formyl-[1,1′-biphenyl]-4-yl)methyl)acetamide (6)
Following the general procedure B using N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c) (176 mg, 0.75 mmol) and 2-bromobenzaldehyde (93 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 40–60) to afford the desired compound 6 (120 mg, 71%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 9.93 & 9.92 (s, 1H), 8.03 (d, J = 7.6 Hz, 1H), 7.78–7.59 (m, 2H), 7.44–7.35 (m, 1H), 4.78 & 4.70- (s, 2H), 3.08 &2.93 (s, 3H), 2.30 & 2.13 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 190.5, 170.8 & 170.6, 145.5 (dm, J = 249.3 Hz), 143.8 (dm, J = 247.8 Hz), 134.3, 134.1 (m), 132.1 & 131.8, 130.9, 130.4 & 130.2, 127.9 & 127.9 (t, J = 2.9 Hz), 118.7 & 117.2 (d, J = 17.8 Hz), 116.1 & 114.7 (d, J = 18.3 Hz), 42.7 & 39.8, 36.4 & 32.6, 21.8 & 21.1. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C17H13F4NO2 (339.29): C 60.18, H 3.86; found: C 60.22, H 4.01.
4.15 N-((2′-Cyano-2,3,5,6-tetrafluoro-[1,1′-biphenyl]-4-yl)methyl)-N-methylacetamide (7)
Following the general procedure B using N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c) (176 mg, 0.75 mmol) and 2-bromobenzonitrile (91 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 70-30) to afford the desired compound 7 (148 mg, 88%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 7.89–7.81 (m, 1H), 7.79–7.70 (m, 1H), 7.66–7.58 (m, 1H), 7.53–7.44 (m, 1H), 4.79 & 4.71 (s, 2H), 3.08 & 2.93 (s, 3H), 2.17 & 2.14 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 170.8 & 170.6, 145.7 (dm, J = 249.3 Hz), 143.5 (dm, J = 247.8 Hz), 133.5 (m), 133.1 (m), 131.7 & 131.6, 131.2 (t, J = 3.3 Hz), 130.2, 130.0, 117.5, 117.0 (m), 114.0, 42.8 & 29.9, 36.5 & 32.7, 21.9 & 21.4. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C17H12F4N2O (336.29): C 60.72, H 3.60; found: C 60.91, H 3.82.
4.16 N-Methyl-N-(2,3,5,6-tetrafluoro-4-(quinolin-3-yl)benzyl)acetamide (8)
Following the general procedure B using N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c) (176 mg, 0.75 mmol) and 3-bromoquinoline (104 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 60-40) to afford the desired compound 8 (158 mg, 87%) as yellow solid (Mp = 104–106 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 9.01–8.93 (m, 1H), 8.34–8.26 (m, 1H), 8.19–8.10 (m, 1H), 7.93–7.86 (m, 1H), 7.83–7.75 (m, 1H), 7.66–7.56 (m, 1H), 4.78 & 4.70 (s, 2H), 3.09 & 2.93 (s, 3H), 2.31 & 2.13 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 170.7 & 170.4, 150.4 & 150.2 (t, J = 2.7 Hz), 147.9 & 147.8, 145.8 (dm, J = 250.6 Hz), 144.0 (dm, J = 247.8 Hz), 137.7 & 137.7, 130.9 & 130.7, 129.4 & 129.4, 128.2, 127.5, 127.4 (m), 120.9 & 120.5, 117.9 & 116.9 (t, J = 16.6 Hz), 116.0 & 114.8 (t, J = 17.2 Hz), 42.6 & 39.8, 36.3 & 32.5, 21.7 & 21.2. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C19H14F4N2O (362.33): C 62.98, H 3.89; found: C 63.12, H 4.09.
4.17 N-Methyl-N-(2,3,5,6-tetrafluoro-4-(pyridin-3-yl)benzyl)acetamide (9)
Following the general procedure B using N-methyl-N-(2,3,5,6-tetrafluorobenzyl)acetamide (c) (176 mg, 0.75 mmol) and 3-bromopyridine (79 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 70-30) to afford the desired compound 9 (122 mg, 78%) as a yellow solid (Mp = 84–86 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 1H NMR (400 MHz, CDCl3) δ 8.73–8.68 (m, 1H), 8.68–8.65 (m, 1H), 7.83–7.74 (m, 1H), 7.47–7.36 (m, 1H), 4.78 & 4.68 (s, 2H), 3.07 & 2.90 (s, 3H), 2.29 & 2.11 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 170.8 & 170.5, 150.5 (m), 150.3, 145.8 (dm, J = 250.6 Hz), 143.8 (dm, J = 247.8 Hz), 137.5, 124.0, 123.7 & 123.6, 117.8 & 116.8 (t, J = 16.6 Hz), 116.1 & 114.9 (t, J = 17.2 Hz), 42.7 & 39.9, 26.4 & 32.6, 21.8 & 21.1. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C15H12F4N2O (312.27): C 57.70, H 3.87; found: C 57.89, H 3.62.
4.18 N-((4′-Cyano-2,3,6-trifluoro-[1,1′-biphenyl]-4-yl)methyl)-N-methylacetamide (10)
Following the general procedure B using N-methyl-N-(2,3,5-trifluorobenzyl)acetamide (d) (163 mg, 0.75 mmol) and 4-bromobenzonitrile (91 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 70-30) to afford the desired compound 10 (126 mg, 79%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 7.80–7.70 (m, 2H), 7.63–7.43 (m, 2H), 7.05–6.76 (m, 1H), 4.66 & 4.63 (s, 2H), 3.08 & 2.00 (s, 3H), 2.17 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 171.3 & 171.1, 154.8 (dm, J = 247.4 Hz), 147.6 (ddd, J = 7.1, 15.2, 252.2 Hz), 146.0 (ddd, J = 3.7, 13.9, 245.0 Hz), 133.2 & 132.8, 132.3 & 132.2, 131.0 (m), 127.7 (dd, J = 8.8, 14.2 Hz), 118.5 & 118.4, 117.2 (dd, J = 14.7, 20.2 Hz), 112.9 & 112.6, 111.6 & 109.5 (td, J = 3.4, 25.5 Hz), 48.2 & 44.5, 36.6 & 34.0, 21.8 & 21.4. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C17H13F3N2O (318.29): C 64.15, H 4.12; found: C 64.21, H 4.01.
4.19 Ethyl 2′,3′,6′-trifluoro-4'-((N-methylacetamido)methyl)-[1,1′-biphenyl]-4-carboxylate (11)
Following the general procedure B using N-methyl-N-(2,3,5-trifluorobenzyl)acetamide (d) (163 mg, 0.75 mmol) and ethyl 4-bromobenzoate (115 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 70-30) to afford the desired compound 11 (135 mg, 75%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 8.16–8.07 (m, 2H), 7.57–7.46 (m, 2H), 6.94 & 6.78 (ddd, J = 2.1, 5.4, 9.7 Hz, 1H), 4.66 & 4.62 (s, 2H), 4.40 (q, J = 5.8, 6.6 Hz, 2H), 3.06 & 2.99 (s, 3H), 2.17 (s, 3H), 1.40 (t, J = 7.1 Hz, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 171.3 & 171.0, 166.2, & 166.1, 155.0 (dm, J = 249.4 Hz), 147.7 (ddd, J = 7.5, 14.9, 251.4 Hz), 146.0 (ddd, J = 3.7, 13.9, 244.3 Hz), 132.9 & 132.5, 130.9 & 130.7, 130.2 (m), 129.7 & 129.6, 126.8 & 126.2 (dd, J = 8.8, 14.1 Hz), 118.2 (dd, J = 15.0, 20.5 Hz), 111.4 & 109.4 (td, J = 3.4, 25.6 Hz), 61.3, 48.2 & 44.3, 36.5 & 34.0, 21.8 & 21.4, 14.3. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C19H18F3NO3 (365.35): C 62.46, H 4.97; found: C 62.58, H 5.10.
4.20 N-((4′-Cyano-2,6-difluoro-[1,1′-biphenyl]-4-yl)methyl)-N-methylbenzamide (12)
Following the general procedure B using N-(3,5-difluorobenzyl)-N-methylbenzamide (e) (196 mg, 0.75 mmol) and 4-bromobenzonitrile (91 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 60-30) to afford the desired compound 12 (121 mg, 67%) as a white solid (Mp = 123–125 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 7.75 (d, J = 8.5 Hz, 2H), 7.59 (d, J = 8.2 Hz, 2H), 7.51–7.42 (m, 5H), 7.08–6.79 (m, 2H), 4.77 & 4.55 (s, 2H), 3.09 & 2.99 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 171.7, 159.8 (d, J = 253.4 Hz), 140.9, 135.4, 131.9, 131.0, 129.9, 128.5, 127.0, 118.5, 111.9, 111.4, 50.3, 37.5. Elemental analysis: calcd (%) for C22H16F2N2O (362.38): C 72.92, H 4.45; found: C 73.08, H 4.21.
4.21 N-((2,6-Difluoro-4′-nitro-[1,1′-biphenyl]-4-yl)methyl)-N-methylbenzamide (13)
Following the general procedure B using N-(3,5-difluorobenzyl)-N-methylbenzamide (e) (196 mg, 0.75 mmol) and 1-bromo-4-nitrobenzene (101 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 60-40) to afford the desired compound 13 (117 mg, 61%) as yellow solid (Mp = 150–152 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 8.31 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 8.7 Hz, 2H), 7.52–7.40 (m, 5H), 7.09–6.83 (m, 2H), 4.77 & 4.57 (brs, 2H), 3.08 & 3.00 (brs, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 172.0, 160.1 (d, J = 249.2 Hz), 147.7, 141.2, 135.6, 131.4, 130.2, 128.7, 127.2 (m), 124.1, 123.7, 123.6, 111.4 (m), 50.6, 37.3. Elemental analysis: calcd (%) for C21H16F2N2O3 (362.38): C 65.97, H 4.22; found: C 66.12, H 4.45.
4.22 N-((2,6-Difluoro-4′-formyl-[1,1′-biphenyl]-4-yl)methyl)-N-methylbenzamide (14)
Following the general procedure B using N-(3,5-difluorobenzyl)-N-methylbenzamide (e) (196 mg, 0.75 mmol) and 4-bromobenzaldehyde (93 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 70-30) to afford the desired compound 14 (98 mg, 54%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 10.07 (s, 1H), 7.97 (d, J = 8.3 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 7.56–7.37 (m, 5H), 7.12–6.76 (m, 2H), 4.77 & 4.55 (s, 2H), 3.08 & 2.99 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 191.9, 172.9, 160.2 (d, J = 252.3 Hz), 140.6, 138.3, 136.0, 135.8 (d, J = 11.3 Hz), 131.1, 130.2, 130.2 (d, J = 7.9 Hz), 129.7, 128.7, 127.1, 126.8, 111.4, 103.0, 52.9, 37.6. Elemental analysis: calcd (%) for C22H17F2NO2 (365.38): C 72.32, H 4.69; found: C 72.21, H 4.56.
4.23 Ethyl 2′,6′-difluoro-4'-((N-methylbenzamido)methyl)-[1,1′-biphenyl]-4-carboxylate (15)
Following the general procedure B using N-(3,5-difluorobenzyl)-N-methylbenzamide (e) (196 mg, 0.75 mmol) and ethyl 4-bromobenzoate (115 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 60-40) to afford the desired compound 15 (129 mg, 63%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 8.13 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.2 Hz, 2H), 7.48–7.41 (m, 5H), 7.06–6.81 (m, 2H), 4.76 & 4.60 (s, 2H), 4.41 (q, J = 7.1 Hz, 2H), 3.09 & 2.98 (s, 3H), 1.41 (t, J = 7.1 Hz, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 171.9, 166.4, 160.3 (d, J = 251.8 Hz), 140.2, 135.8, 133.6, 130.4, 130.1, 128.6, 128.7, 127.2 (brm), 111.2 (m), 61.2, 50.5, 37.6, 14.5. Elemental analysis: calcd (%) for C24H21F2NO3 (409.43): C 70.41, H 5.17; found: C 70.58, H 5.29.
4.24 N-((4′-Benzoyl-2,6-difluoro-[1,1′-biphenyl]-4-yl)methyl)-N-methylbenzamide (16)
Following the general procedure B using N-(3,5-difluorobenzyl)-N-methylbenzamide (e) (196 mg, 0.75 mmol) and 4-bromobenzophenone (131 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 60-40) to afford the desired compound 16 (145 mg, 66%) as a brown oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 7.96–7.80 (m, 4H), 7.68–7.56 (m, 4H), 7.56–7.39 (m, 6H), 6.95 (d, J = 6.4 Hz, 2H), 4.78 & 4.68 (s, 2H), 3.09 & 2.99 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 196.2, 171.9, 160.2 (d, J = 250.2 Hz), 140.3, 137.5, 137.3, 135.7, 135.5, 133.3, 132.6, 130.3, 130.1, 130.1, 130.0, 128.6, 128.4, 127.2, 111.2 (m), 54.7 & 50.5, 37.6 & 33.5. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C28H21F2NO2 (441.48): C 76.18, H 4.79; found: C 75.98, H 4.51.
4.25 N-((2′-Cyano-2,6-difluoro-[1,1′-biphenyl]-4-yl)methyl)-N-methylbenzamide (17)
Following the general procedure B using N-(3,5-difluorobenzyl)-N-methylbenzamide (e) (196 mg, 0.75 mmol) and 2-bromobenzonitrile (91 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 60-40) to afford the desired compound 17 (76 mg, 42%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 7.80 (d, J = 7.6 Hz, 1H), 7.69 (td, J = 1.3, 7.7 Hz, 1H), 7.54 (dt, J = 1.0, 7.6 Hz, 1H), 7.49 (d, J = 7.7 Hz, 2H), 7.46–7.41 (m, 4H), 7.09–6.86 (m, 2H), 4.79 & 4.56 (s, 2H), 3.09 & 2.00 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 172.5 & 172.1, 160.2 (d, J = 254.7 Hz), 141.7, 135.6, 133.2, 132.7, 131.8, 130.1, 129.1, 128.7, 127.2, 117.7, 114.1, 111.8 (m), 110.2 (m), 54.7 & 50.5, 37.7 & 31.1. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C22H16F2N2O (362.38): C 72.92, H 4.45; found: C 73.13, H 4.58.
4.26 N-(3,5-Difluoro-4-(quinolin-3-yl)benzyl)-N-methylbenzamide (18)
Following the general procedure B using N-(3,5-difluorobenzyl)-N-methylbenzamide (e) (196 mg, 0.75 mmol) and 3-bromoquinoline (104 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 60-40) to afford the desired compound 18 (113 mg, 58%) as a yellow solid (Mp = 154–156 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 9.00 (s, 1H), 8.30 (s, 1H), 8.16 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 7.9 Hz, 1H), 7.81–7.73 (m, 1H), 7.64–7.57 (m, 1H), 7.54–7.39 (m, 5H), 7.11–6.84 (m, 2H), 4.80 & 4.58 (s, 2H), 3.11 & 3.01 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 171.8 (brs), 160.4 (d, J = 252.0 Hz), 151.1, 147.5 & 147.4, 140.5 (brs), 137.4, 135.6 & 135.3, 130.1, 130.0, 129.3 & 129.2, 128.5, 128.1 & 127.9, 127.6, 127.3 & 127.0, 122.4, 114.1 (t, J = 21.2 Hz), 111.4 (d, J = 24.7 Hz), 110.3 (m), 54.4 & 50.4, 37.4 & 33.4. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C24H18F2N2O (388.42): C 74.21, H 4.67; found: C 74.28, H 4.51.
4.27 N-((4′-Cyano-2,6-difluoro-[1,1′-biphenyl]-3-yl)methyl)-N-methylacetamide (19)
Following the general procedure B using N-(2,4-difluorobenzyl)-N-methylacetamide (f) (149 mg, 0.75 mmol) and 4-bromobenzonitrile (91 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 50-50) to afford the desired compound 19 (114 mg, 76%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 7.80–7.70 (m, 2H), 7.64–7.54 (m, 2H), 7.37 & 7.17 (td, J = 6.4, 8.4 Hz, 1H), 7.05 & 6.98 (td, J = 1.3, 9.0 Hz, 1H), 4.62 & 4.57 (s, 2H), 3.05 & 2.95 (s, 3H), 2.17 & 2.14 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 171.1 & 171.0, 158.9 (dd, J = 6.4, 250.0 Hz), 157.7 (dd, J = 6.5, 249.6 Hz), 134.2 & 133.7, 132.5–132.1 (m), 131.4–130.9 (m), 128.6 (dd, J = 6.3, 10.1 Hz), 121.1 & 120.4 (dd, J = 3.8, 16.6 Hz), 118.7 & 118.6, 116.5 (t, J = 18.7 Hz), 112.3 & 112.0 (d, J = 3.9 Hz), 112.2 (m), 48.4 & 44.5 (d, J = 4.5 Hz), 36.4 & 33.6 (d, J = 1.3 Hz), 21.9 & 21.4. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C17H14F2N2O (300.31): C 67.99, H 4.70; found: C 68.23, H 4.67.
4.28 N-((2,6-Difluoro-4′-nitro-[1,1′-biphenyl]-3-yl)methyl)-N-methylacetamide (20)
Following the general procedure B using N-(2,4-difluorobenzyl)-N-methylacetamide (f) (149 mg, 0.75 mmol) and 1-bromo-4-nitrobenzene (101 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 40–60) to afford the desired compound 20 (88 mg, 55%) as a brown solid (Mp = 90–93 °C): 1H NMR (400 MHz, CDCl3) δ (ppm) 8.34–8.28 (m, 2H), 7.64 (d, J = 8.7 Hz, 2H), 7.39 &7.20 (td, J = 6.4, 8.4 Hz, 1H), 7.07 & 7.00 (t, J = 8.8 Hz, 1H), 4.63 & 4.59 (s, 2H), 3.04 & 2.96 (s, 3H), 2.17 & 2.15 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 171.2, 159.0 (dm, J = 250.4 Hz), 157.8 (dm, J = 251.6 Hz), 147.8 & 147.7, 136.2 & 135.6, 131.5, 128.7 (m), 123.6 (m), 121.3 (d, J = 16.7 Hz), 116.1 (t, J = 18.6 Hz), 112.2 (m), 48.4 & 44.5, 36.5 & 33.7, 21.9 & 21.5. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C16H14F2N2O3 (320.30): C 60.00, H 4.41; found: C 60.29, H 4.42.
4.29 N-((3′-Cyano-2,6-difluoro-[1,1′-biphenyl]-3-yl)methyl)-N-methylacetamide (21)
Following the general procedure B using N-(2,4-difluorobenzyl)-N-methylacetamide (f) (149 mg, 0.75 mmol) and 3-bromobenzonitrile (91 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 50-50) to afford the desired compound 21 (98 mg, 65%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 7.76 (s, 1H), 7.74–7.65 (m, 2H), 7.63–7.52 (m, 1H), 7.38 & 7.17 (td, J = 6.5, 8.4 Hz, 1H), 7.05 & 6.99 (td, J = 1.3, 9.0 Hz, 1H), 4.63 & 4.58 (s, 2H), 3.03 & 2.96 (s, 3H), 2.17 & 2.15 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 171.2 & 171.1, 159.0 (dd, J = 6.4, 249.7 Hz), 157.8 (dd, J = 6.5, 249.4 Hz), 134.9 & 134.8, 134.0 & 133.9, 132.1 & 131.9, 131.1 (dd, J = 6.3, 10.1 Hz), 133.7 & 130.3, 129.4 & 129.3, 128.4 (dd, J = 6.3, 10.1 Hz), 121.1 & 120.4 (dd, J = 3.8, 16.6 Hz), 118.6 & 118.4, 116.1 (t, J = 18.8 Hz), 113.0 & 112.8, 112.3 & 112.1 (d, J = 3.9 Hz), 112.2 & 112.0 (d, J = 3.9 Hz), 48.4 & 44.5 (d, J = 4.5 Hz), 36.4 & 33.7 (d, J = 1.4 Hz), 21.9 & 21.4. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C17H14F2N2O (300.31): C 67.99, H 4.70; found: C 67.91, H 4.65.
4.30 N-(2,4-Difluoro-3-(quinolin-3-yl)benzyl)-N-methylacetamide (22)
Following the general procedure B using N-(2,4-difluorobenzyl)-N-methylacetamide (f) (149 mg, 0.75 mmol) and 3-bromoquinoline (104 mg, 0.5 mmol), the residue was purified by flash chromatography on silica gel (heptane-EtOAc, 50-50) to afford the desired compound 22 (95 mg, 58%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ (ppm) 9.30 (d, J = 2.3 Hz, 1H), 8.47 (d, J = 2.1 Hz, 1H), 8.19 (d, J = 8.5 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.78 (ddd, J = 1.4, 6.9, 8.4 Hz, 1H), 7.71–7.58 (m, 1H), 7.39–7.28 & 7.18–7.08 (m, 1H), 6.95–6.73 (m, 1H), 4.58 & 4.51 (s, 2H), 2.97 & 2.91 (s, 3H), 2.16 & 2.13 (s, 3H). Observed complexity is due to the mixture of rotamers. 13C NMR (100 MHz, CDCl3) δ (ppm) 171.0, 162.4 (dd, J = 11.8, 248.5 Hz), 161.2 (dd, J = 11.9, 248.3 Hz), 149.7 & 147.8, 134.0, 131.8 (dd, J = 5.9, 9.6 Hz), 130.9, 130.1, 129.5, 129.1 (dd, J = 5.8, 9.7 Hz), 128.2, 128.0, 127.6, 120.4 (d, J = 11.5 Hz), 111.8 & 111.7 (dd, J = 3.8, 21.1 Hz), 104.5 & 103.5 (t, J = 25.4 Hz), 48.1 & 43.9 (d, J = 3.8 Hz), 36.1 & 22.6, 21.9 & 21.4. Observed complexity is due to the mixture of rotamers. Elemental analysis: calcd (%) for C19H16F2N2O (326.35): C 69.93, H 4.94; found: C 70.22, H 5.28.
Acknowledgements
The authors thank the Algerian “Ministry of Higher Education and Scientific Research” for a fellowship to M.E.B. and the CNRS and Université Rennes-1 for financial support. H.-Y. H. thanks the China Scholarship Council for fellowships.