


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Synthesis of the acridinium benzaldehyde 3⋅PF6 and the bisacridinium–porphyrin conjugate 1⋅2PF6.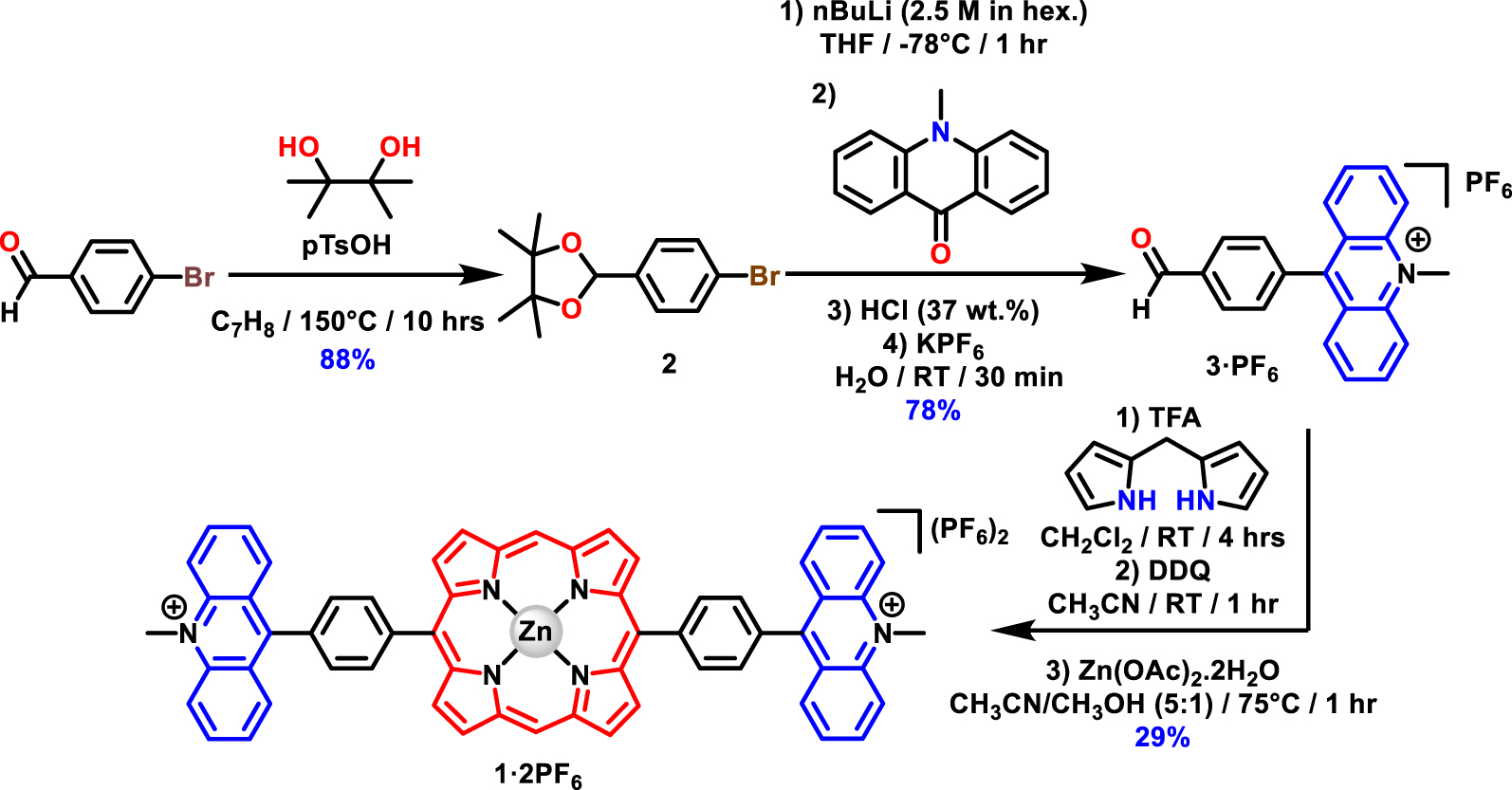
Porphyrins are versatile compounds that have found applications in various domains such as catalysis, materials science and medicine [1,2,3,4,5,6,7,8]. Their properties which stand from their extended π-conjugated system and metal chelating capacity are easily tunable by functionalization of their meso or β-pyrrolic positions. Porphyrins associated to acridinium units have not received much attention yet despite the perspectives opened by such association. Indeed, N-substituted acridiniums are multi-responsive units which modify their chemical and electronic features following chemical or redox stimuli [9]. N-substituted acridiniums and protonated acridines are also π-acceptors and as such, were incorporated in the host structure of various systems or used as a guest molecule for molecular recognition. As guest molecules, acridiniums were stabilized through charge transfer interactions in the cavities of bis-porphyrin systems based on clips or macrocyclic structures [10,11,12]. In some host–guest complexes, fast photoinduced electron transfer between the non-covalently linked porphyrin donor and acridinium acceptor units was demonstrated [13,14,15]. Regarding covalent porphyrin–acridinium associations, a free-base porphyrin connected to two acridiniums was reported as a fluorescent detector of superoxide anion. In this system, a fluorescence enhancement of the triad was detected following a two-electron reduction of the acridinium units by the superoxide anion [16].
Herein, the synthesis of a Zn(II) porphyrin trans-substituted with 9-phenyl-N-methyl-acridinium units is reported. The electrochemistry, absorption and emission properties of the conjugate are investigated as well as its photophysics, by means of ultrafast transient absorption spectroscopy. Computational studies have been performed to provide insights into the frontier molecular orbitals involved in the electronic transitions of the conjugate.
2. Results and discussion
The synthesis of the bisacridinium-Zn(II) porphyrin 1⋅2PF6 was inspired from Fukuzumi and coworkers [16]. It is based on a 2 + 2 condensation methodology developed by the Lindsey group to access trans-A2B2-type porphyrins [17]. It started from commercially available 4-bromobenzaldehyde and was performed in four synthetic steps (Scheme 1). The aldehyde functional group was first protected in the presence of pinacol (1.2 equiv.) in toluene [18]. The obtained 2-(4-bromophenyl)-4,4,5,5-tetramethyl-1,3-dioxolane (2) was reacted with nBuLi (0.8 equiv.) in THF at −78 °C followed by addition of N-methyl-acridin-9(10H)-one (1 equiv.), obtained in one step from commercially available compounds [19]. After acidification of the reaction mixture using HCl (37 wt%), the freshly formed 9-(4-formylphenyl)-10-methylacridin-10-ium chloride (3⋅Cl) was converted to the corresponding hexafluorophosphate salt (3⋅PF6) by anion metathesis and isolated in 78% yield. The key intermediate 3⋅PF6 was then reacted under Lindsey conditions in the presence of di(1H-pyrrol-2-yl)methane (1 equiv.) [20], and trifluoroacetic acid (TFA, 0.6 equiv.) in CH2Cl2. After aromatization of the porphyrinoid using DDQ (3 equiv.), metalation of the free base porphyrin was undertaken using Zn(OAc)2⋅2H2O (0.5 equiv.). After purification, the porphyrin–acridinium conjugate 1⋅2PF6 was isolated in 29% yield.
3. Electrochemistry
Cyclic voltammograms (CV) were measured at 298 K under argon atmosphere, using tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte. It is worthwhile to note that due to the limited solubility of 1⋅2PF6 in CH3CN in the presence of supporting electrolyte, DMF was used to perform the experiments (Figure 1). In the cathodic regime, a reversible wave (Ered = −0.475 V versus SCE) corresponding to the reduction of the acridinium units followed by two reduction waves of half intensity (Ered = −1.34 V versus SCE and Ered = −1.43 V versus SCE) were recorded [21]. The observed ratio in intensity between the reduction processes suggests that the first reduction involves the reduction of both acridinium moieties at the same potential since no electronic coupling between these fragments is expected. The first reversible reduction process of the acridinium units allowed for the calculation of the half-wave reduction potential E1∕2 = −0.442 V versus SCE. One of the two last reduction processes is related to the second electron injection to one of the acridinium fragments leading to a carbanionic species. Formation of this species is supported by the reduced intensity of the re-oxidation peak of the first reduction wave (−0.409 V versus SCE) leading to an EC mechanism (electron transfer-chemical). This carbanion is known to be highly reactive and capable of reacting with solvent molecules. The obtained CV in the anodic regime (Figure 1) consists of an irreversible anodic wave (Eox = +0.949 V versus SCE) attributed to the oxidation of the porphyrin moiety. Interestingly, this wave has the same intensity (I = 7.3 μA) as the first cathodic process thus indicating that an equal number of electrons is exchanged in both processes. This surprising bi-electronic oxidation of the porphyrin core was further supported by rotating disk electrode (RDE) experiments recorded in benzonitrile (see ESI, Figure S23). These data allow the estimation of the HOMO–LUMO gap to be 1.4 eV.
Cyclic voltammograms (DMF, WE: Pt, CE: Pt, RE: Hg/Hg2Cl2/KCl sat., 100 mV⋅s−1) of a solution of 1⋅2PF6 (c = 1 × 10−3 mol⋅L−1) in the presence of TBAPF6 as supporting electrolyte (c = 0.1 mol⋅L−1).
Emission data for models and conjugate in CH3CN
| RT | 77 K | ||||||
|---|---|---|---|---|---|---|---|
| 𝜆max (nm)a | 𝜙emb | 𝜏 (ns)c | 𝜆max (nm)a | 𝜏 (ns)c | E (eV)d | ||
| 3⋅PF6 | 3⋅PF | 515 | 0.045 | 1.73 | 471, 500 | 2.70 (20%); 16.6 (80%) | 2.63 |
| Zn-bP | Zn-bP1 | 588, 638 | 0.035 | 2.30 | 605, 648 | 2.31 | 2.05 |
| Zn-bP3 | 794 | — | 1.56 | ||||
| 1⋅2PF6 | 590, 642e | <1.0 × 10−4e | — | — | — | ||
aEmission maxima from corrected spectra. bFluorescence quantum yields, measured with reference to TPP (meso-tetraphenylporphyrin) in aerated toluene as a standard for the porphyrin units and with reference to Coumarin 153 in ethanol for the acridinium units. cFluorescence lifetimes in the nanosecond range, excitation at 465 nm for Zn-bP and at 368 nm for 3⋅PF6. dEnergy of the excited state determined as the energy of the 0–0 emission band collected at 77 K. eUpon selective excitation of the Zn-porphyrin at 543 nm, the yield is below the minimum value measurable with steady-state experiments, i.e. 1.0 × 10−4.
4. Photophysical properties
The photophysical properties of the conjugate 1⋅2PF6 were characterized in CH3CN. Model compounds Zn-bisphenyl porphyrin (Zn-bP) and acridinium benzaldehyde (3⋅PF6) (see ESI, Scheme S1) were also studied in the same solvent for comparison purposes.
The absorption spectrum of the conjugate is presented in Figure 2, compared with the spectra of its models Zn-bP and 3⋅PF6 and their weighted sum. It can be observed that the spectrum of 1⋅2PF6 correlates reasonably well with the sum in the 280–400 nm region, while the Q-bands of the porphyrin (500–600 nm) are slightly red-shifted with respect to the model porphyrin, as well as the Soret band (400–450 nm) which also shows a decrease in intensity and a broadening. Theoretical analysis demonstrates that the latter absorption features are given by the contribution of three electronic π–π∗ transitions either centered on the porphyrin or on the acridinium cores (see below).
Absorption spectra of bisacridinium–porphyrin conjugate 1⋅2PF6 (blue), model compounds Zn-bP (red) and 3⋅PF6 (black) and the sum of the spectrum of Zn-bP with twice the spectrum of 3⋅PF6 (green dotted) in CH3CN.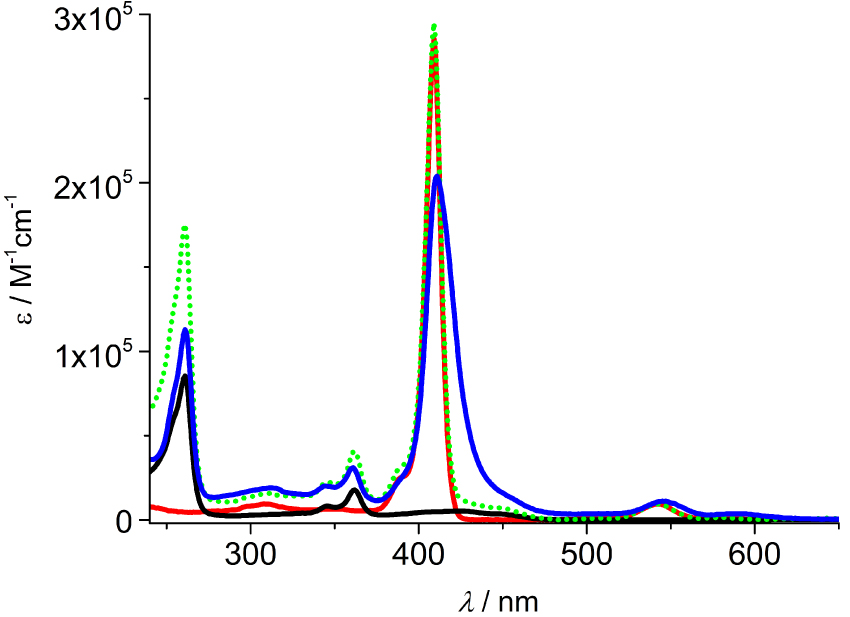
Uncorrected emission spectra of CH3CN solutions of 1⋅2PF6 (blue) and models 3⋅PF6 (black) and Zn-bP (red), isoabsorbing at 262 nm (A262 = 0.1). 𝜆exc = 262 nm. The blue spectrum has been multiplied by a factor of 100.
Luminescence measurements were carried out both at room temperature and at 77 K in CH3CN. At room temperature, model 3⋅PF6 shows a broad emission spectrum peaking at 515 nm, with a quantum yield of 0.045 and an excited state lifetime of 1.73 ns, while Zn-bP shows features typical of Zn-porphyrins, with maxima at 588 and 638 nm, 𝜙fl = 0.035 and 𝜏 = 2.30 ns (Figure S24 and Table 1).
Conversely, the conjugate 1⋅2PF6 is weakly emissive, displaying a strong quenching of both the acridinium and the porphyrin units. Selective excitation of the porphyrin component in 1⋅2PF6 at 543 nm, led to evaluate an emission quantum yield below 10−4, i.e. reduced to less than 0.3% the yield of model Zn-bP (Table 1). In order to estimate the quenching of the acridinium unit in 1⋅2PF6, excitation of isoabsorbing solutions of the conjugate, the porphyrin model and acridinium benzaldehyde model at 262 nm, wavelength at which prevalent excitation of the acridinium units (ca. 96%) is achieved, was carried out and the results are shown in Figure 3. The residual porphyrin fluorescence in 1⋅2PF6 is far below the 4% of the model emission, percentage which would correspond to the direct excitation of the porphyrin unit in the array, confirming the quenching already observed. Moreover, the weak residual acridinium emission points to a quenching higher than 99% of these units in the array. The observed features indicate that a very fast and efficient photoinduced process depopulates the lowest singlet excited states of both components in the array, likely an electron transfer reaction. This hypothesis is confirmed by transient absorption analysis, as discussed in detail below.
Energy level diagram and kinetics of the photoinduced processes occurring in the conjugate 1⋅2PF6 in CH3CN. The singlet and triplet energy levels are taken from data of the present paper and from literature [22]. The energy of the charge separated state (1.39 eV) has been approximated as Eox–Ered (with Eox and Ered as “redox energy”, expressed in eV), by considering the oxidation potential of the porphyrin unit (Eox = +0.949 V versus SCE) and the reduction potential of the acridinium unit (E1∕2 = −0.442 V versus SCE) measured in DMF, whose polarity is similar to that of CH3CN.
The electron transfer reaction does not exclude a possible energy transfer from the acridinium to the porphyrin singlet excited states, a process thermodynamically allowed (Scheme 2) and supported by a non-zero overlap between the emission spectrum of the acridinium unit and the absorption spectrum of the porphyrin component (Figures 2 and S24). In order to investigate this event, the excitation spectrum of 1⋅2PF6 was collected at 660 nm, wavelength at which only the emission of the porphyrin component is present and compared to the absorption spectrum (Figure S25). Even if noisy, due to the very weak emission, the excitation spectrum clearly contains the absorption bands of the porphyrin and lacks the characteristic absorption peaks of the acridinium unit at 262 and 362 nm. This outcome leads to conclude that an energy transfer process from the acridinium units to the central porphyrin is not occurring in the conjugate, probably due to the competition with the ultrafast electron transfer process.
Emission measurements conducted at 77 K showed fluorescence spectra with higher vibrational resolution for the models 3⋅PF6 and Zn-bP (Figure S26). In Zn-bP, moreover, phosphorescence emission at 794 nm is clearly observed. The conjugate 1⋅2PF6 appears to be weakly emissive also at 77 K and its spectrum is hardly detectable due to scattering issues that affect these measurements. This outcome leads to deduce that the decrease in temperature is not affecting the efficiency of the quenching process occurring within the components of the array.
In order to get insights into the fast photoinduced processes occurring within the conjugate, pump-probe transient absorption measurements with femtosecond resolution were performed on 1⋅2PF6 and its relevant models.
Two excitation wavelengths were chosen: 560 nm, where a selective population of the porphyrin singlet is achieved and 360 nm, where the acridinium units are prevalently excited (the peak at 262 nm of the acridinium is not experimentally accessible).
Transient absorbance of (a) Zn-bP and (b) 1⋅2PF6 in CH3CN at different delays. Excitation at 560 nm (A560 = 0.1, 0.2 cm optical path, 2 μJ/pulse).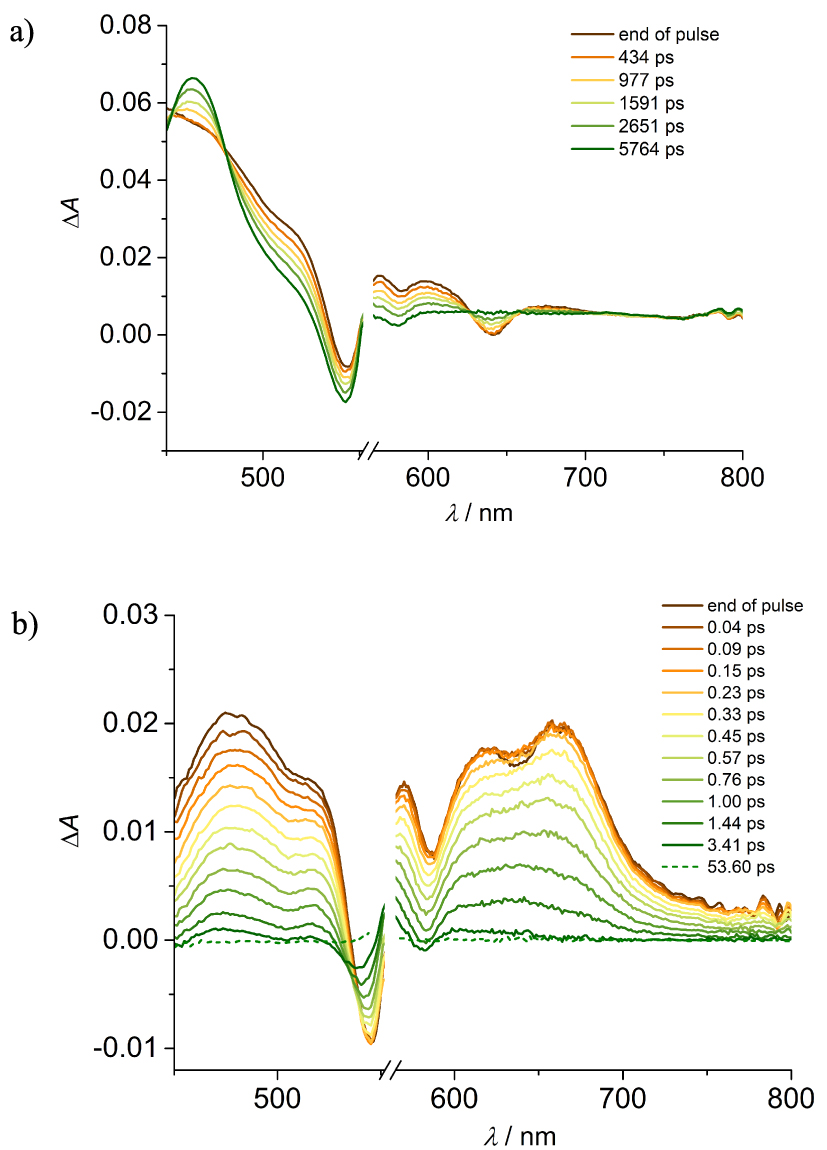
Transient absorbance of (a) 3⋅PF6 and (b) 1⋅2PF6 in CH3CN at different delays. Excitation at 360 nm (A360 = 0.2, 0.2 cm optical path, 2 μJ/pulse).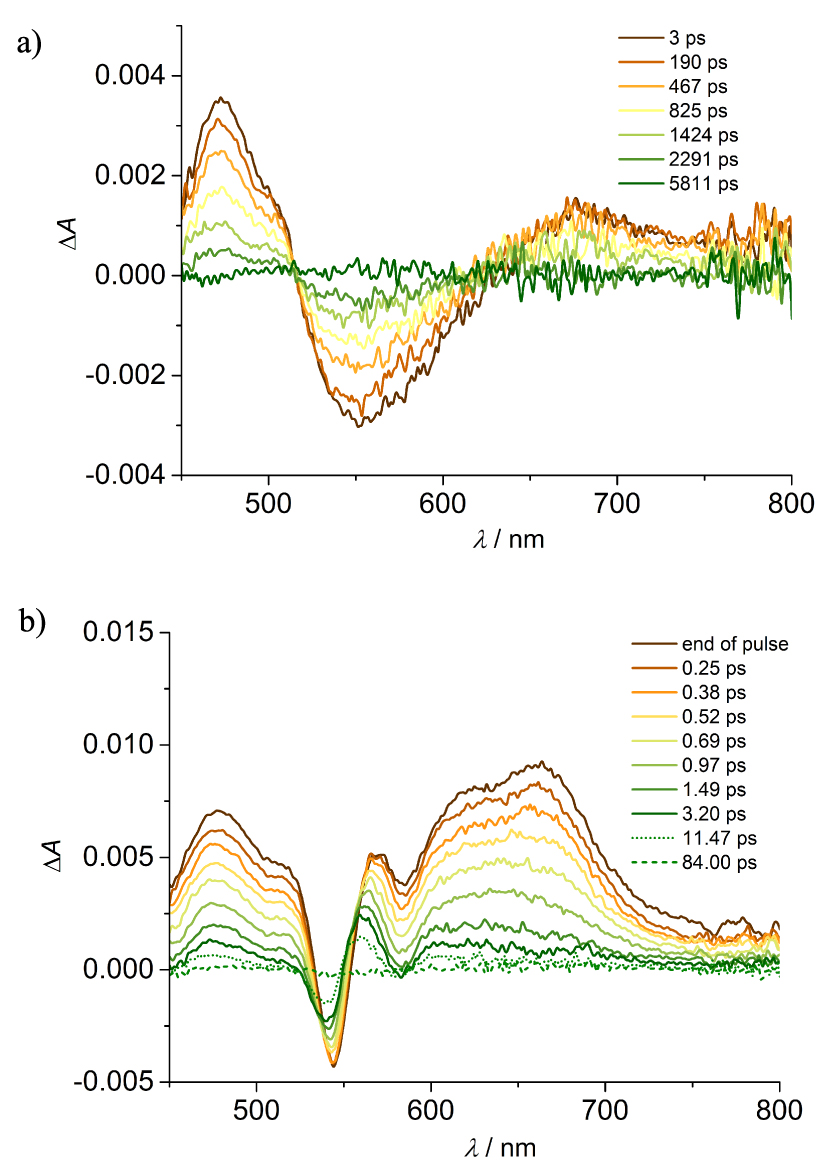
The time evolution of the transient spectrum of model Zn-bP upon excitation at 560 nm is reported in Figure 4(a). The initial spectrum with a positive band below 530 nm, ground state bleaching at 542 and 580 nm and stimulated emission at 640 nm evolves into a new spectrum with a positive maximum at 463 nm, with clear isosbestic points. The kinetics reflects the fluorescence lifetime of the molecule (the time profiles are reported in Figure S27), i.e. 2.3 ns; the final spectrum is thus attributed to the triplet state and the process is assigned to S1→T1 intersystem crossing.
The scenario is completely different for the conjugate 1⋅2PF6 (Figure 4(b)). The end of pulse spectrum shows maxima at 480, 520, 620 and 662 nm, as well as bleaching bands at 545 and 586 nm. In the region 620–660 nm an ultrafast signal rise, of the order of the time resolution of the system, is observed. This spectrum quickly decays, with a lifetime of 0.65 ps (Figure S28a). The observed transient spectrum can be safely ascribed to the charge separated state P+–Ac⋅ (P: porphyrin, Ac: acridinium), since the bands between 600 and 700 nm well resembles those reported for a Zn-porphyrin cation, [23 ] and bands at ca. 480 nm and 520 nm have been reported for the reduced species of N-alkyl substituted acridinium compounds [14 , 15 , 24 , 25 ]. The singlet excited state of the porphyrin is thus depopulated in ca. 0.3 ps to yield the charge separated state, which in turn lives less than 1 ps. It can be noticed that the decay of the spectrum shows a second component, accounting for a very small fraction of the signal (ca. 2%), with a lifetime of ca. 32 ps (Figure S28b). This second component can be tentatively ascribed to a slower charge recombination occurring in a different conformation of the array, or deriving from another minimum of the first excited singlet potential energy surface. This is consistent with the molecular flexibility in the excited state highlighted by the theoretical results (see below).
Transient absorption analysis with excitation at 360 nm has been performed for 1⋅2PF6 and models 3⋅PF6 and Zn-bP. In model acridinium benzaldehyde, 3⋅PF6, the initial formation of a signal with a risetime of 1 ps, ascribable to vibrational relaxation, is observed (Figure S29). The formed spectrum displays positive bands in the 450–510 nm and 630–800 nm regions, with maxima at 472 nm and 680 nm, and stimulated emission at 550 nm, red-shifted with respect to that detected from luminescence measurements due to the sum with the positive absorption bands. The signal decays with a lifetime of 1.6 ns, in good agreement with the fluorescence lifetime (Figures 5(a) and S30), and with defined isosbestic points, allowing to ascribe the process to S1→T1 intersystem crossing, even if the spectral features of the triplet are hardly detectable. Zn-bP shows a behaviour similar to that observed upon excitation at 560 nm, with intersystem crossing occurring in 2.3 ns, but preceded by an initial fast evolution of 1 ps, ascribable to internal conversion (Figures S31 and S32) [26].
The end of pulse spectrum of 1⋅2PF6 does not present any of the spectral features of the singlet excited states absorption of the respective components, but recall those observed upon excitation at 560 nm (Figure 5(b)) with clear features of the charge separated state. There is no evidence of signal formation, implying that the process, upon prevalent excitation of the acridinium component, occurs on an ultrafast scale (⩽0.3 ps). The spectrum evolves quickly, almost disappearing with 𝜏 = 0.70 ps. A minor component, accounting for ca. 5% of the decay and with a lifetime of 33 ps (Figures 5(b) and S33) is detected, in agreement with that observed upon excitation of the porphyrin unit.
The observed photoinduced electron transfer process is, indeed, thermodynamically allowed upon excitation of both the porphyrin (𝛥G = −0.66 eV) and the acridinium units (𝛥G = −1.24 eV), as indicated in Scheme 2 and, in the latter case, a higher energy gap accounts for the increased reaction rate, placing the reaction in normal Marcus region [27].
5. Computational studies
The structure of 1⋅2PF6 was first optimized without any symmetry and revealed that the phenyl rings are orthogonal to the porphyrin rings (88°) and to the acridinium moieties (88°). Interestingly, the frontier orbitals are mainly composed of two sets of degenerated orbitals localized on the porphyrin fragment for the HOMOs and on the acridinium units for the LUMOs (Figure S34). This observation unambiguously confirms the interpretation of the electrochemical investigation for the anodic and cathodic process involving the oxidation of the porphyrin and the reduction of the acridinium moieties, respectively. It is worthwhile to note that no contribution from the phenyl rings linking the porphyrin and acridinium units was observed (see ESI). The theoretical absorption spectrum (Figure 6) is composed of two intense absorbing bands at 332 and 406 nm and two weak absorbing bands at 512 and 610 nm, matching the experimental optical transitions observed at 361, 411, 545 and 586 nm (Figure S17).
Theoretical absorption spectrum of 1⋅2PF6 in CH3CN.
The nature of the electronic transitions of 1⋅2PF6 is presented in Figure 7. The main band at 406 nm results from the contribution of three electronic transitions at 397, 406, and 408 nm corresponding to π–π∗ excitations either centered on the porphyrin or on the acridinium cores (Figure S35). The band at 331 nm is attributed to two π–π∗ transitions centered on the acridinium moieties exclusively. Finally, the low energy domain (Figure 6 inset) is governed by two π–π∗ transitions centered on the porphyrin core at 512 and 510 nm respectively and by a charge transfer transition from the porphyrin to the acridinium core at 610 nm where the electron is delocalized over the two acridinium moieties. For this lowest excited singlet, two calculations were performed imposing a C2 symmetry in order to preserve the delocalization and a broken symmetry. Already in the symmetric structure, the geometry underwent significant distortions upon relaxation. First, a flattening of the structures was observed resulting in the decrease of the dihedral angles between the fragments (porphyrin-phenyl angle = 59.9° and acridinium-phenyl angle = 77.2°) compared to ground state. In this C2 optimized structure, this singlet emits at 693 nm thus corroborating the experimental data. Finally, breaking the symmetry leads to additional distortions and to localization of the electron in the excited state on one of the two acridiniums (Figure S37). This generates two minima separated by a very low barrier (302 cm−1) between which the exciton freely oscillates, thus giving rise to an average symmetrical conformer.
Character of the singlet states generating the lowest energy absorption band in the absorption spectra of 1⋅2PF6.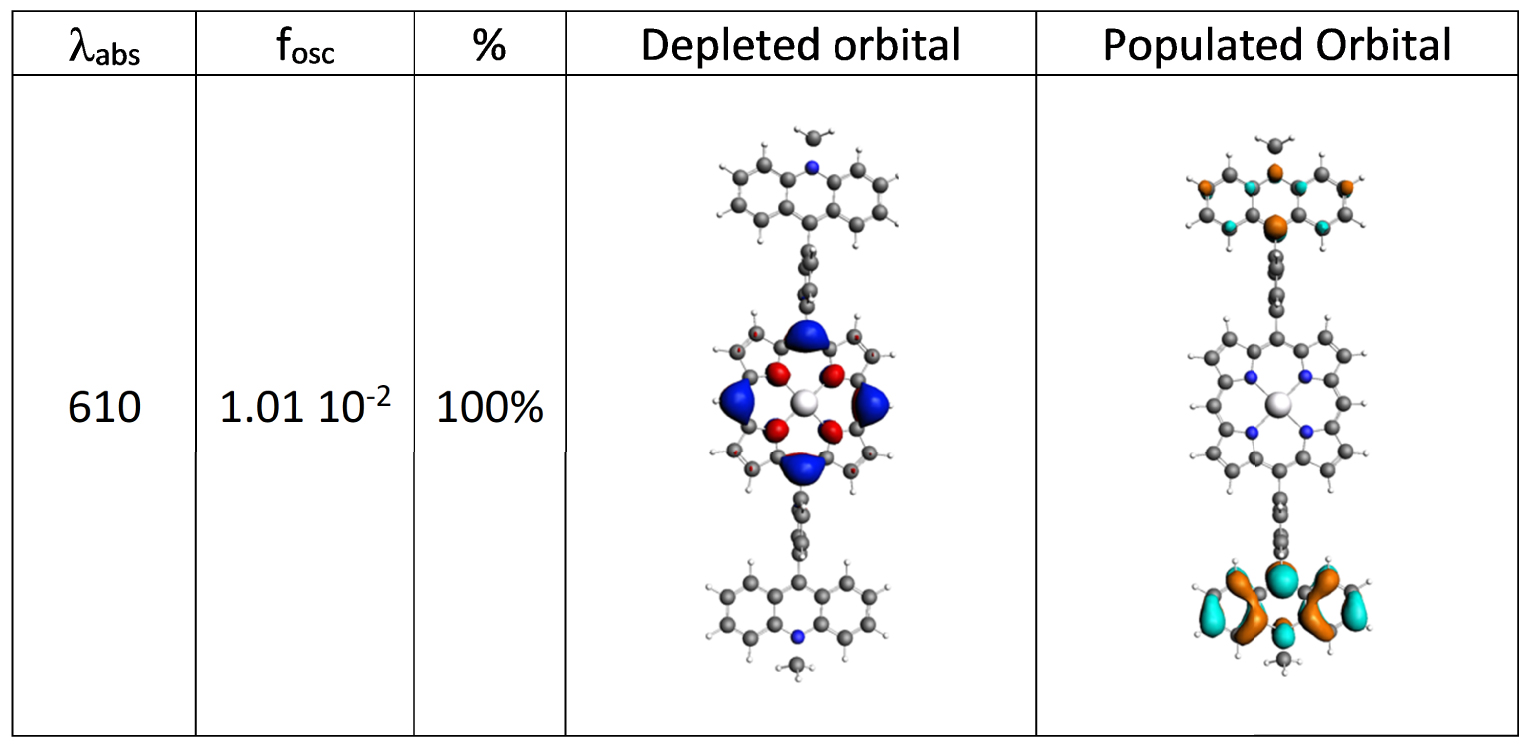
6. Conclusion
A bisacridinium-Zn(II) porphyrin conjugate has been successfully synthesized in four steps using a novel acridinium benzaldehyde intermediate. Electrochemical characterization via cyclic voltammetry has revealed that the one-electron reduction of the two acridiniums occurs simultaneously, indicating limited communication between the two acridiniums. Furthermore, the observed two-electron oxidation of the porphyrin is supported by computational analysis showing two near degenerate HOMOs for the porphyrin. Comparison of the absorption features of the conjugate with the acridinium benzaldehyde intermediate and the Zn-bisphenyl porphyrin model confirms that the acridiniums are non-innocent moieties, with a complex interplay of porphyrin and acridinium centered transitions accounting for the apparent red-shift of both the Soret and the Q-bands of the porphyrin core, as shown by theoretical calculations. The almost complete quenching of the emission of both components in the conjugate was ascribed to an ultrafast photoinduced electron transfer process. Transient absorption analysis revealed the formation of a charge separated state within 0.3 ps, and its fast decay with a lifetime below 1 ps. In addition, an energy transfer process from the acridinium units to the porphyrin core was excluded. The interesting ultrafast photophysics of the conjugate opens new perspectives for the development of acridinium–porphyrin architectures.