

 CC-BY 4.0
CC-BY 4.0
1. Introduction
Recently, the depletion of non-renewable fossil fuels is an important factor that has focused attention on renewable energy resources [1]. Biodiesel is believed to be an attractive alternative to diesel fuels; however, dangerous gases emitted by combustion diesel engines are primarily responsible for environmental problems and diseases [2]. Biodiesel is obtained by transesterification reaction of triglycerides with methanol (low molecular weight alcohols) in presence of catalyst (base). In such a catalytic process, excess amount of glycerol (GL) is produced as main by-product (equivalent to 10 weight % of the total product) of biodiesel and it has similar properties as diesel fuel [3, 4, 5]. It is necessary to enhance the scope of the conversion of GL into value-added products to increase the economy of biodiesel manufacture. The GL is utilized as raw material for the production of value-added compounds namely pharmaceuticals, bio-plastic, surfactants, cosmetics, lubricants, food additive and also in fuel industries [6, 7]. Selective oxidation of glycerol to glyceric acid [8], dehydration to acrolien [9], synthesis of hydroxyquinoline by Skraup method [10] and hydrolysis to propylene glycol [11] are some of the possible alternative platforms.
The mixture of glycerol ethers, namely mono-tertiary butyl ether glycerols (2-usiners) (m-TBGe), higher glycerol ethers i.e. di-tertiary butyl ether glycerols (2 usiners) (d-TBGe) and tri-tertiary butyl ether glycerol (t-TBGe) were obtained by catalytic etherification of GL with t-butyl alcohol (TBuA). The m-TBGe has many applications, namely as reactive intermediates in the pharmaceutical industry, agrochemicals and surfactants and cannot be used as components of engine fuels [12]. Higher glycerol ethers (d-TBGe and t-TBGe) can be used as oxygenated fuel additive and also directly blended into fuel due to high oxygen content of d-TBGe (23.5%) and t-TBGe (18.5%), together with high octane numbers (but m-TBGe are much less miscible in diesel fuel) [13]. The particles of organic matter from diesel fuel, namely compounds of hydrogen and carbon, carbon oxide (CO) and unregulated emissions including aldehydes in the exhaust gases are drastically decreased by adding higher ethers [14]. Hence, over the last few years, the synthesis of higher ethers from etherification of GL with TBuA has gained a lot of interest. Etherification of GL with TBuA or isobutylene has been reported under the action of acid catalysts [13, 15, 16]. The use of TBuA in substitution of isobutylene keeps away the need of solvents (such as dioxane or dimethyl sulfoxide) to dissolve GL and the mass transfer limitation occurrence associated with the complex three-phase system [14].
Various catalysts have been studied to synthesize higher ethers from etherification of GL with TBuA under heterogeneous and homogeneous catalytic conditions. Major drawbacks of homogeneous catalysts are toxicity, irrecoverable nature, low yields, harsh reaction conditions, corrosions, prolonged reaction times, less selectivity, occurrence of side reactions and difficult separation of catalyst. In addition, from the economic and environmental point of view, a catalyst (heterogeneous solid acid) has a crucial role in numerous industrial applications as compared to homogeneous acids. The synthesis of higher ethers has been studied with heterogeneous acid catalysts, namely sulfonated hybrid silicas, H-Beta zeolites, sulfonated carbon black acid and fluorinated 𝛽-zeolites [16, 17, 18, 19, 20]. Those catalysts have shown higher percentage of GL conversion but less selectivity towards higher ethers at high temperature. A better catalytic activity is therefore required for that type of reaction. In the present work, an amorphous mesoporous aluminophosphate (AlPO) catalyst has demonstrated excellent catalytic activity for the synthesis of higher ethers with high selectivity.
It should be noted that successful replacement of eco-unfriendly acid catalyst (homogeneous) by the synthesis of amorphous mesoporous aluminophosphate (AlPO) as well as metal-aluminophosphate (m-AlPO) solid acid, is a promising approach in the next decades for several acid-catalysed reactions such as acetylation, esterification, hydrogenation, alkylation, hydroxylation, transesterification, rearrangement oxidation, hydrogenolysis etc. Those many organic applications were performed effectively to synthesize organic fine chemicals with m-AlPO catalyst due to its mesoporous nature such as vast pore size and extreme surface area [21, 22, 23, 24, 25, 26, 27, 28, 29, 30].
AlPO catalysts are a type of inorganic solid material, formed from alternating tedrahedral units (AlO and PO) connected by a shared oxygen atom and exhibit electronically neutral framework. The first AlPO molecular sieve was synthesized by Wilson and co-workers [31]. The AlPO sample with aluminium to phosphorous ratio equal to 1:1 is neutral in nature. The incorporation of transition metals (V, Fe, Co, Ni, Mo, W and Cu) into the framework of AlPO molecular sieves can generate catalytic basic or redox or acidic active sites on account of the replacement of both P and Al or only P/Al by the transition metal. Thus, many of the organic reactions carried out over modified forms of AlPO catalytic material provoked great interest as solid acid catalyst in recent years [24, 32, 33].
The previously reported experimental methods to synthesize mesoporous aluminophosphate are complex in nature, time consuming and demand multiple steps by the addition of template [34] or surfactants [35, 36, 37]. In the present work, we report the synthesis of a thermally stable mesoporous aluminophosphate solid acid catalyst by simple physical mixing through one-step method without the use of templates. Our recent work on amorphous AlPO is employed as an effective heterogeneous solid acid catalyst in acetylation reaction [32].
In the current work, we focused on the selective synthesis of higher ethers from etherification reaction between GL and TBuA over solid acid catalysts namely AlPO, FeAlPO, ZrAlPO, CuAlPO and ZnAlPO. Those catalytic materials were primed by specific fashion (co-precipitation approach) and characterized through a variety of techniques like Braunauer–Emmett–Teller (BET) for surface area, Ammonia-Temperature Programmed Desorption (NH3-TPD) for strength of surface acid sites, Fourier Transform Infrared Spectroscopy (FT-IR) for functionality, Powder X-ray Diffraction (PXRD) for crystallinity, N2 adsorption–desorption for pore volume/pore diameter, Scanning Electron Microscope (SEM)/Transmission Electron Microscope (TEM) for surface morphology, Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-OES) for elemental analysis, 27Al and 31P NMR for atomic nuclei environment and Energy Dispersive X-ray Analysis (EDAX) for elemental composition. Optimization study is performed for attaining highest GL conversion and higher ether selectivity. In addition, a comparative study of microwave-facilitated synthesis with ultrasonic and conventional heating method is also proposed. A kinetic measurement was conducted to evaluate the Ea (energy of activation) and Tc (temperature coefficient) of FeAlPO, ZrAlPO, CuAlPO and ZnAlPO catalysts. Reaction mechanism is reported by reactant pre-adsorption method and reusability test of solid acid catalyst is also shown.
2. Experimental
2.1. Chemicals
Commercial t-butyl alcohol (CH3)3COH and glycerol (C3H8O3) were purchased from Sigma Aldrich India Limited. Some important inorganic chemicals were endowed by M/S LOBA Chemie India Ltd, namely aluminium nitrate nanohydrate (Al(NO3)3⋅9H2O), zirconium nitrate (Zr(NO3)4), copper nitrate (Cu(NO3)2), iron nitrate (Fe(NO3)3), zinc nitrate (Zn(NO3)2) and ortho-phosphoric acid (H3PO4).
2.2. AlPO and m-AlPO catalysts preparation
The classic AlPO and m-AlPO (m: Fe, Zr, Cu and Zn) catalysts were prepared as earlier reported single step co-precipitation method. The m-AlPO catalysts go on to be prepared by mixing aluminium nitrate (aluminium origin) and corresponding metal nitrate salts (metal origin) in 500 ml de-ionized water followed by 85% O-phosphoric acid (phosphorous origin) in the desired proportion 0.95:0.025:1 (Al:m:P) and stirring mechanically for 1 h at 60 °C. Precipitating agent (28% aqueous ammonia solution) is added drop-by-drop in the resulting homogeneous clear solution from micro-burette until potential of hydrogen (pH) reaches 9. The resultant product was filtered, diluted with de-ionized H2O and evaporated at 120 °C (overnight). It should be noted that the samples (dried) were powdered, lastly further calcined for 5 h at 550 °C. The samples were abbreviated as FeAlPO, ZrAlPO, CuAlPO and ZnAlPO. Similar steps were followed for the preparation of clear aluminophosphate sample without adding metal nitrate salt in a desired molar ratio 0.95(Al):1(P). The obtained sample is abbreviated as AlPO.
2.3. Characterization
The solid samples were characterized by their powder X-ray diffraction (PXRD) pattern, using a X-ray diffractometer (Philips X’pert Pro) in 2𝜃 range 10° to 80° (graphite crystal monochromator) assembled with a Ni-filtered Cu K𝛼 source of wavelength (𝜆) at 1.5418 Å, functioning at 50 kV and 40 mA. The particle size of all samples which were decided by the equation of Debye–Scherrer makes use of full-width at half-maximum strategy (FWHM). The FT-IR spectra were recorded by a Nicolet IR 200 spectrophotometer. The analyses were executed by dried KBr pellet technique scan at 4 cm−1 spectral determination ranging from 400–4000 cm−1.
SEM studies of all catalytic materials were recorded by Carl Zeiss (Ultra 55-Oxford) operated at 30 kV accelerating voltage and EDAX spectra of samples obtained by Inca (Oxford instrument). TEM pictures of all effective samples were obtained from electron microscope (PHILIPS CM-200) under accelerating voltage of 20–200 kV. The 27Al NMR and 31P NMR spectra of CuAlPO sample were recorded on a Bruker DSX-300 instrument with spinning angle from 0° to 90° in the temperature range of 100 K–372 K and spinning speeds in the order of 10 kHz–12 kHz.
The metal content of m-AlPO samples were determined by the action of ICP-Optical Emission Spectrometer (ICP-OES) operating procedure (Thermo iCP-6000 apparatus). The pore volume, surface area and pore diameter of entire materials continued to be calculated by using high speed gas sorption analyser instrument (NOVA-1000 Quanta chrome). Previously, the solid acid catalysts were degassed for 5 h at 250 °C. The pore size distributions and pore volume were determined by N2 adsorption–desorption isotherms and P∕Po (relative pressure) using BJH model respectively. NH3-TPD (Pulse Chemisorb 2705) technique and simple n-BA back titration approach were used to calculate acidity of the samples.
2.4. General reaction procedure
Below we describe three different heating systems for the etherification of glycerol (GL) with t-butyl alcohol (TBuA).
2.4.1. Conventional heating approach (CVN)
In a typical reaction, about 80 mg of catalyst is added to a mixture of GL (1 mmol) and TBuA (4 mmol) in round bottom glass vessel (25 ml) adapted with a magnetic stirrer (hot plate). Once the reaction is completed, the reaction vessel is cooled, the reaction mixture is centrifuged then filtrated and the pellet is washed with solvent (ethyl acetate) to recover the catalyst. The conversion (%) and selectivity (%) were determined with a Mayura gas chromatograph adapted with a flame ionization detector (FID) using capillary Inno wax (16, 19, 20) and also by GC–MS (Varian) equipped with a 100% ethylene glycol capillary column Supelcowax 10, according to the characterization products reported by Jamróz et al. [17].
2.4.2. Microwave heating approach (MWV)
The reaction was carried out by taking reaction mixture i.e. 80 mg of catalyst, GL (1 mmol) and TBuA (4 mmol) in 10 ml reaction vessel tube equipped with magnetic stirrer (REMI), which was then irradiated under Labmate microwave reactor (450 W) set up with temperature cooling system at a certain temperature (40 °C to 60 °C) for a specific period of time (5 min to 20 min). After completion of the reaction, reactor vessel was allowed to cool rapidly by flowing compressed N2 gas to microwave cavity for five min.
2.4.3. Ultrasonic heating approach (ULS)
The etherification reaction was performed under ultrasonic horns operating at 24 kHz frequency and 400 W power output (Labmatrix manufacturing LLP, Digital Sonifier). The power amplitude was balanced in the range 40–200 W. The reaction mixture of GL (1 mmol), TBuA (4 mmol) and 80 mg of solid acid catalyst was poured into a round-bottomed vessel (25 ml). This mixture was kept in an ultrasonic bath to begin the reaction.
The conversion (%) and selectivity (%) were calculated by the following equations:
3. Experimental discussion
3.1. Samples characterization
Characterization of samples for their entire (physico-chemical) properties was made using PXRD, BET, NH3-TPD, N2 adsorption–desorption isotherms, ICP-OES, FT-IR, SEM/TEM, NMR and EDAX.
3.1.1. BET
The values of BET surface area (SA), pore volume (PV), pore diameter (PD), total surface acidity (TSA) and acid site distributions of AlPO, FeAlPO, ZrAlPO, CuAlPO and ZnAlPO are displayed in Table 1.
Physico-chemical properties of solid acids
| Catalyst | BET surface area (m2/g) | Pore volume (cc/g) | Pore diameter (nm) | Metal content (%)∗ | Acidity (mmol/g) | |||
|---|---|---|---|---|---|---|---|---|
| Weak (100–200 °C) | Medium (200–300 °C) | Strong (300–450 °C) | TSA | |||||
| AlPO | 89 | 0.08 | 5.04 | — | 0.0712 | 0.0280 | — | 0.096 (0.093) |
| FeAlPO | 189 | 0.18 | 11.48 | 0.042 | 0.0215 | 0.1035 | 0.0645 | 0.190 (0.188) |
| ZrAlPO | 168 | 0.12 | 7.50 | 0.035 | 0.0828 | 0.0127 | 0.0436 | 0.140 (0.142) |
| CuAlPO | 181 | 0.28 | 16.27 | 0.044 | 0.0213 | 0.0473 | 0.1621 | 0.243 (0.245) |
| ZnAlPO | 171 | 0.20 | 13.58 | 0.031 | 0.0116 | 0.0609 | 0.1442 | 0.216 (0.228) |
Note: Numbers in the parenthesis correspond to the total surface acidity (TSA) values obtained by n-butyl amine back titration method, ∗ analysed by ICP-OES technique.
All the solid samples were found to have specific surface area in the range 88–188 m2/g. The surface areas of catalytic materials are in the order of AlPO < ZrAlPO < ZnAlPO < CuAlPO < FeAlPO. This order underscores that m-AlPO has higher surface area than pure AlPO. Among these, the maximum surface area is attained by FeAlPO catalyst. Generally, heterogeneous catalytic reactions occur on the surface of the catalysts and the reaction rate is mostly in proportion to catalyst surface area. A small changes in the values of SA, PV and average PD was observed in m-AlPO because of the strong metal interaction in the form of oxide or phosphate with AlPO material. One of the most noticeable effects of metal incorporation into AlPO sample is an enormous increase in PV and PD values from 0.08 to 0.28 cc/g and from 5.04 to 16.27 nm respectively. Those increases in PV, PD, SA clearly indicated a change in the type of pore size from microporous to mesoporous. The CuAlPO attained highest surface acidity due to highest PV and PD as well as acid sites distribution with dissimilar acid strength (strong, weak and moderate acidic sites). The pore size distribution of all the catalytic materials were approximately arranged with a mesoporous nature. The PV vs PD values were plotted for AlPO and m-AlPO solid samples (Figure 1).
N2 adsorption–desorption isotherms of (a) AlPO, (b) FeAlPO, (c) ZrAlPO, (d) CuAlPO and (e) ZnAlPO.
N2 adsorption–desorption isotherms study has been used to characterize the type of porous nature (pore structure and pore size) of the samples. Table 1 represents the PD, PV and SA values of the samples. This type of experimental measurement of the solid samples belonging to type IV isotherms, in addition to well-defined H2 and H3 hysteresis loops with P∕Po (relative pressure) varying from 0.4–1 is shown in Figure 1. These results indicated the characteristics of mesoporous type of the catalytic materials [25]. All the samples namely ZrAlPO, FeAlPO, CuAlPO and ZnAlPO presented H2 hysteresis loop whereas AlPO presented H3 hysteresis loop. This obviously indicated that the categories of pore size are different. The H2 hysteresis loop between the relative pressures 0.4 to 1 indicated the capillary condensation taking place in narrow slit-like pores and confirms the large number of pores [38]. The CuAlPO showed highest PV and PD with the H2 hysteresis loop showing sharp rise in absorbed volume of nitrogen at P∕Po > 0.4 expressed by narrow pore size distribution. The relative pressure (P∕Po) at which hysteresis loop was shifted to higher value for the metal loaded AlPO sample compared to pure AlPO sample.
3.1.2. TSA
TSA values of all the samples were determined by ammonia-TPD analysis (Table 1). The ammonia utilized as probe molecule to study samples is of acidic character on account of its more basic strength, smaller molecular size and high stability. Figure 2 shows the NH3-TPD profiles of all samples. TSA values follow the order: AlPO < ZrAlPO < FeAlPO < ZnAlPO < CuAlPO. Surface acidity of m-AlPO was higher than AlPO. Different values of surface acidity and surface area were not correlated, but depends on the type of metal present in the AlPO. The surface acidity of CuAlPO was higher compared to others. Due to availability of P–OH and Al–OH bonds in AlPO molecular sieve, surface acidity is created [39]. The m-AlPO catalysts exhibited three desorption peaks at approximately 100–200 °C (weak acidic sites), 200–300 °C (moderate acidic sites) and 300–450 °C (strong acidic sites). The acidic sites produced by metal ion in AlPO are in the following order: divalent (m2+) > trivalent (m3+) > tetravalent (m4+). Anionic vacancy is formed by the substitution of pentavalent (P5+) or trivalent (Al3+) by divalent metals (m2+). These anionic vacancies accept the proton (H+), which may result in the formation of Brønsted acidic sites. Figure 2 shows broad desorption peak for CuAlPO at approximately 300–450 °C due to availability of more number of strong acidic sites compared to ZnAlPO in this work. CuAlPO catalyst shows more surface acidity compared to ZnAlPO (m2+) because of the formation of more number of Brønsted acidic sites. The acidic strength of FeAlPO (m3+) is lower for the sake of maintaining electro-neutrality by the substitution of trivalent (Al3+) in AlPO or it may also be due to the existence of less acidic sites on account of substitution of the pentavalent (P5+) in AlPO. In addition, the tetravalent metal acidic strength of ZrAlPO (m4+) is lower due to reduction in acidic sites due to decreased formation of anionic vacancies. The substitution of Al3+ by the tetravalent metal Zr (Zr4+), which in turn leads to the formation of excess positive charge, causes the formation of weak acid sites (Lewis) rather than strong acid sites (Brønsted) [40].
NH3-TPD profiles of (a) AlPO, (b) FeAlPO, (c) ZrAlPO, (d) CuAlPO and (e) ZnAlPO.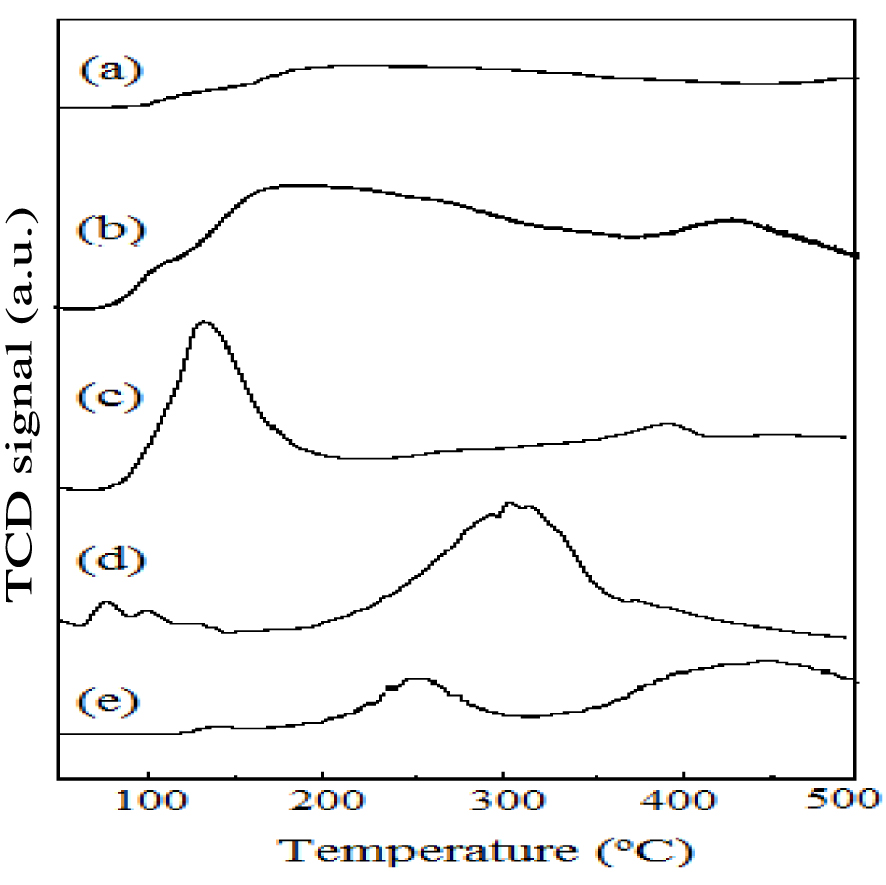
3.1.3. Powder-XRD
Figure 3 shows the PXRD patterns of all the catalytic materials in the range 2𝜃 = 10°–70°. One broad band at 2𝜃 = 15°–30° corresponds to the amorphous AlPO catalyst calcined at 823 K. A few weak angle diffraction bands at 2𝜃 = 20.4, 21.6, 23.2, 35.8 correspond to the occupancy of two varieties of forms like tridymite (20.4, 21.6, 23.2) and 𝛼-cristobalite (35.8) in m-AlPO solid samples [41, 42]. These weak angle diffraction bands sustain the mesoporous nature of the samples [43]. The m-AlPO catalytic materials which are prepared in hot condition exhibits weak angle diffraction bands whereas in cold condition broad bands are shown. In addition, clear cut small particle size and high surface area samples are attained in hot condition compared to the samples prepared in cold [40, 27]. Thus, amorphous and mesoporous nature of the catalytic materials have been clearly shown by weak angle diffraction peaks of PXRD patterns.
PXRD patters of (a) AlPO, (b) FeAlPO, (c) ZrAlPO, (d) CuAlPO and (e) ZnAlPO.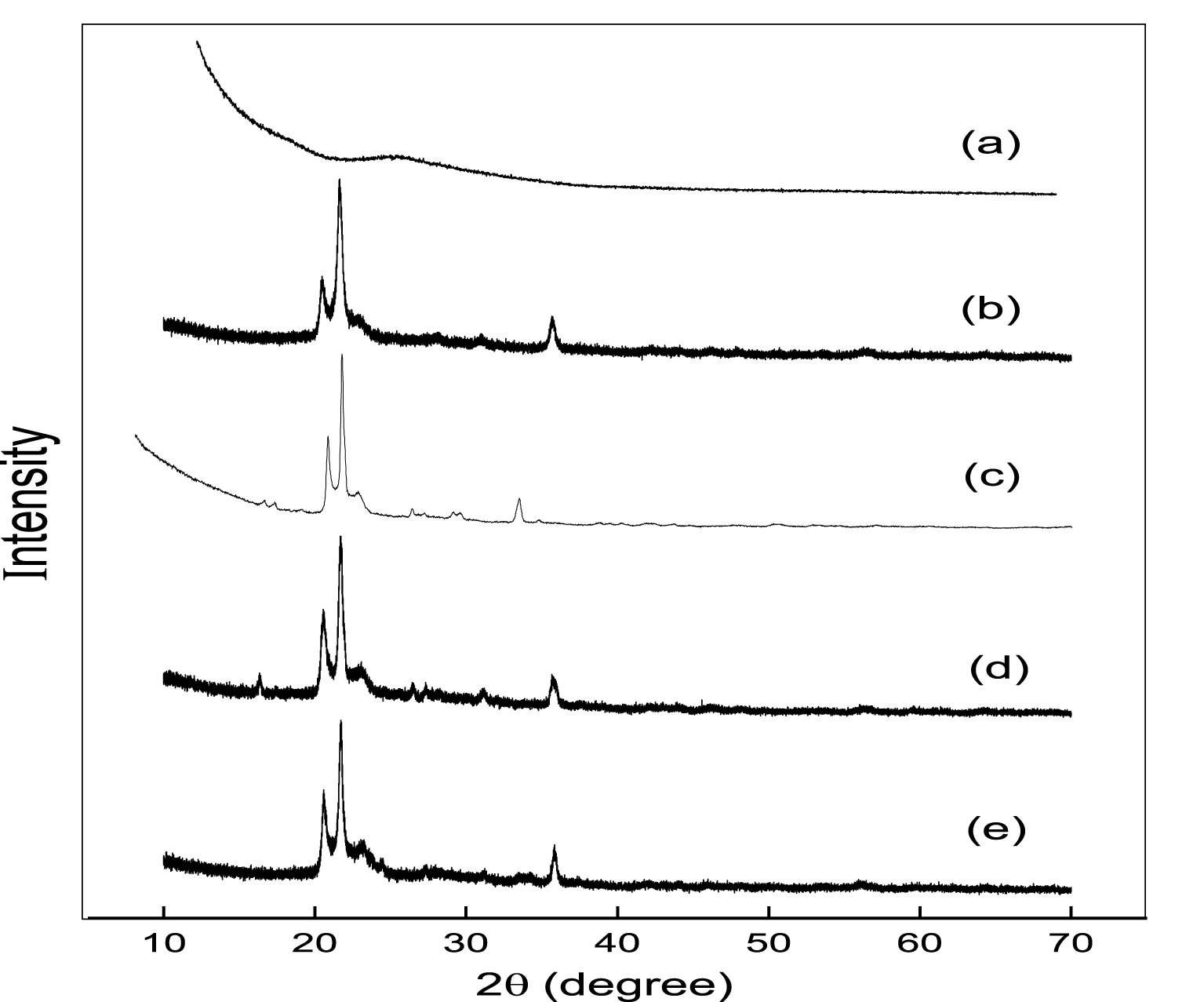
The particle sizes (D) of all the samples were calculated from full-width at half-maximum intensity (FWHM) according to the Debye–Scherrer equation (from PXRD) [44].
i.e., particle size (D in nm), full-width at half-maximum (FWHM = 𝛽), wavelength of X-ray (𝜆= 0.154 nm), diffraction angle (𝜃) and dimensionless factor (k = 0.94) which is nearly equal to unity. As observed in Table 2, average particle sizes of m-AlPO solid samples were found to be in the range of 10–21 nm, it is clear that the samples are of mesoporous nature.
Average particle size (Da) of m-AlPOs by Debye–Scherrer method from XRD-analysis
| Peak position of catalyst (2𝜃) | FWHM (𝛽) | Particle size D (nm) | Average particle size Da (nm) | |
|---|---|---|---|---|
| FeAlPO | 20.44684 | 0.52139 | 16.18 | 16.38 |
| 21.77819 | 0.52142 | 16.21 | ||
| 35.66911 | 0.52079 | 16.74 | ||
| ZrAlPO | 20.51329 | 0.83539 | 10.1 | 10.23 |
| 21.57997 | 0.83532 | 10.12 | ||
| 35.86045 | 0.83461 | 10.45 | ||
| CuAlPO | 20.55912 | 0.41714 | 20.22 | 20.49 |
| 21.89047 | 0.41683 | 20.28 | ||
| 35.78139 | 0.41593 | 20.97 | ||
| ZnAlPO | 20.6714 | 0.50192 | 16.81 | 17.014 |
| 21.68194 | 0.50231 | 16.83 | ||
| 35.78139 | 0.50131 | 17.4 | ||
3.1.4. Fourier transform-IR
Figure 4 shows the FT-IR peaks of calcined AlPO and m-AlPO solid samples, documented in the specified scale (400–4000 cm−1). Except AlPO, there was appearance of similar broad absorption bands fluctuating between 3150–3500 cm−1 and 1630–1640 cm−1 which were characteristic of the –OH stretching as well as bending mode vibrations of hydroxyl group. The hydroxyl group (–OH) is coordinated with aluminium atom, loaded metal atom (Zr/Fe/Cu/Zn) and phosphorous atom [45]. It is to be noted that the broad band in the range of 1110–1130 cm−1 present for all the catalysts is on account of triply degenerate phosphate P–O–P asymmetric stretching mode of vibration. The peaks were observed at 730 cm−1 for triply degenerate P–O–P symmetric stretching vibration and another at 490 cm−1 for tetrahedral (PO) unit of O–P–O bending vibration [41].
FT-IR spectra of (a) AlPO, (b) FeAlPO, (c) ZrAlPO, (d) CuAlPO and (e) ZnAlPO.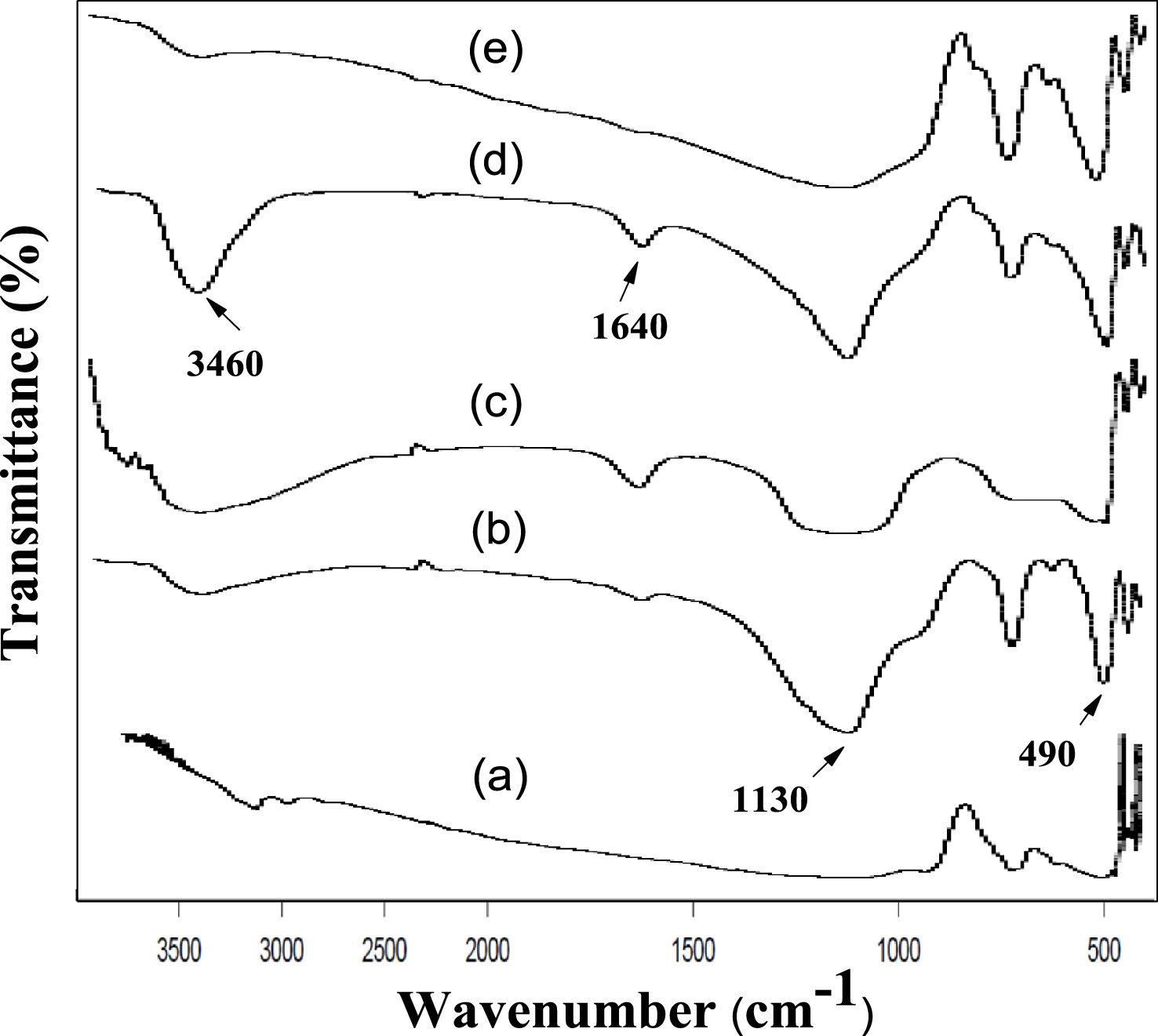
3.1.5. NMR studies
The 31P and 27Al NMR techniques were used only for CuAlPO (Figure 5) due to its superior catalytic activity.
31P and 27Al NMR spectra of CuAlPO.
The 31P NMR spectrum exhibits signal at chemical shift −26.6 ppm which is due to the existence of P–O–Al bonds from phosphorous atom in P(–OAl)4 tetrahedral unit. The peaks in the region of −18 to −13 ppm are described by the condensation of phosphorous atom integrated in a tetrahedral environment with water or hydroxyl groups [46, 47].
The 27Al NMR spectrum shows two peaks by aluminium species i.e., sharp peak at 38.1 ppm and broad peak at −13.5 ppm due to existence of tetrahedral (Al(OP)4) environment and octahedral (Al(OP)x(OH)6−x) environment respectively. It is recommended that the tetrahedral Al species coordinates with four OP molecules and octahedral Al species bonding with OP and OH molecules [46, 47].
3.1.6. SEM, TEM and EDAX studies
As seen from Figure 6, the SEM analysis of AlPO and m-AlPO samples describes the non-uniform size of particles with spherical morphology, with its irregular porosity nature. The agglomerated particles were observed in all the samples.
SEM images of (a) AlPO, (b) FeAlPO, (c) ZrAlPO, (d) CuAlPO and (e) ZnAlPO.
The morphology and size of AlPO and m-AlPO catalysts were characterized by TEM. The particles have spherical geometry with non-uniform nature. The average particle size is around 16–32 nm as shown in Figure 7. The particle boundary was not determined accurately because of the strong co-ordination among the neighbouring molecules as well as due to the amorphous type of the particles. Thus, the particle size and amorphous nature of the samples are in agreement with XRD results.
TEM images of (a) AlPO, (b) FeAlPO, (c) ZrAlPO, (d) CuAlPO and (e) ZnAlPO.
The elemental composition of AlPO and m-AlPO catalysts was determined by EDAX technique. As seen from Figure 8, EDAX spectra and images of every catalyst showed the presence of major elements namely Al, P and O. It is clearly indicated that metal compositions (i.e., Fe, Cu, Zn and Zr) are not seen in m-AlPO catalysts due to the low metal content (less than 1%). Metal content less than 1% is not detected by EDAX (Inca) technique.
EDAX spectra of (a) AlPO, (b) FeAlPO, (c) ZrAlPO, (d) CuAlPO and (e) ZnAlPO.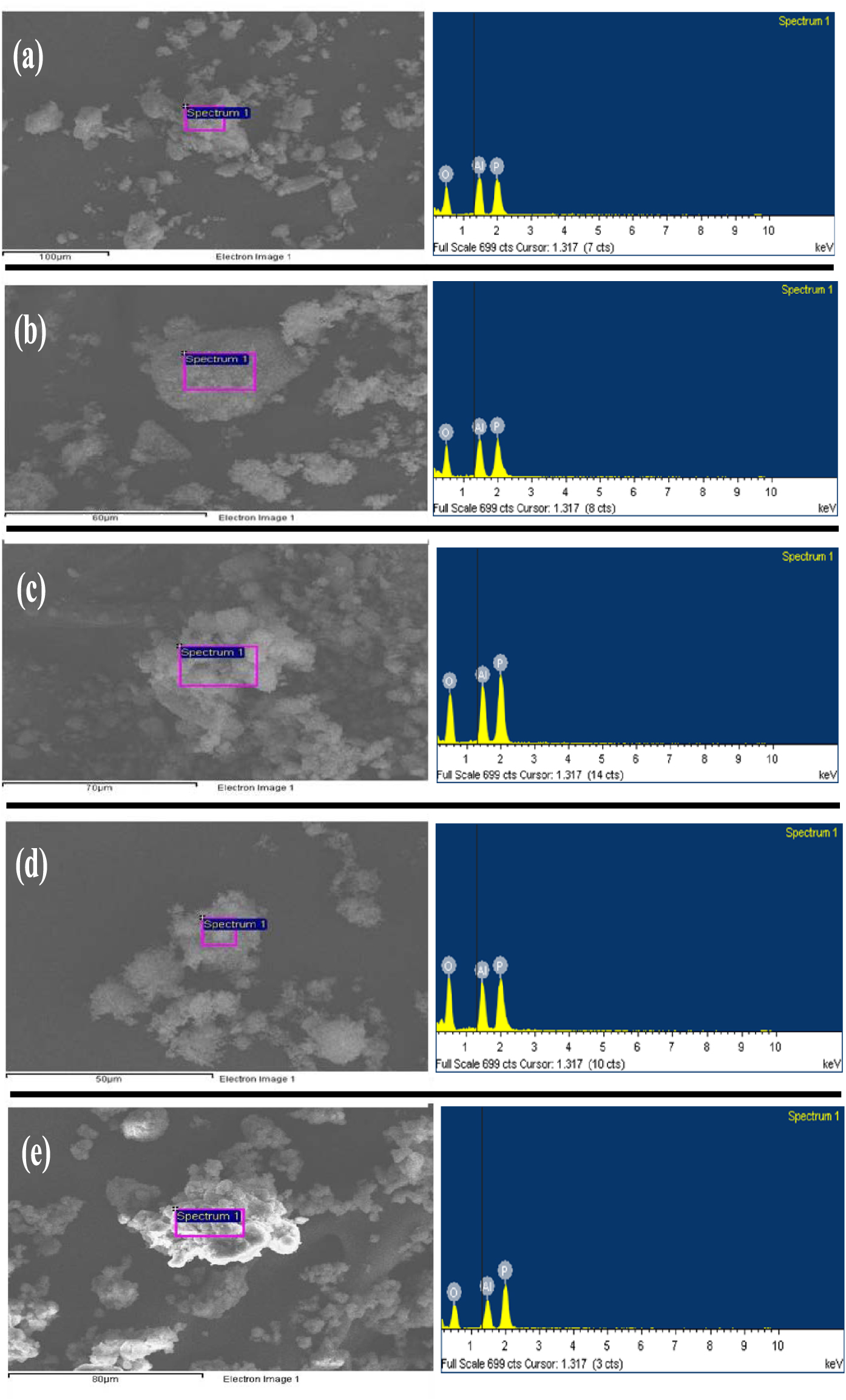
3.1.7. ICP-OES system
Atomic (transition metal) mole percentage values for Fe (0.042%), Zr (0.035%), Cu (0.044%) and Zn (0.031%) in m-AlPOs were found in good agreement with theoretical values.
3.2. Catalytic activity studies (in etherification of GL)
The catalytic activity of AlPO, FeAlPO, ZrAlPO, CuAlPO and ZnAlPO were evaluated by the liquid phase etherification (Scheme 1) of GL with TBuA under three heating methods. There are five isomer units obtained in the reaction mixture depending on the degree of etherification i.e., two isomer units of m-TBGe (product A), two isomer units of d-TBGe (product B) and one isomer unit of t-TBGe (product C). In the present work, the performance of etherification reaction was carried out through three heating methods to explore the chance of enhancing the desirable GL conversion with excellent product selectivity towards diesel-miscible oxygenates such as product B and C (higher ethers), which is attractive for fuel applications. Many reaction parameters, viz. nature of various samples, sample weight, molar ratio, heating methods, temperature and time were determined to optimize the reaction conditions and the results obtained are described below.
Etherification of GL with TBuA to synthesize biofuel additives (higher ethers).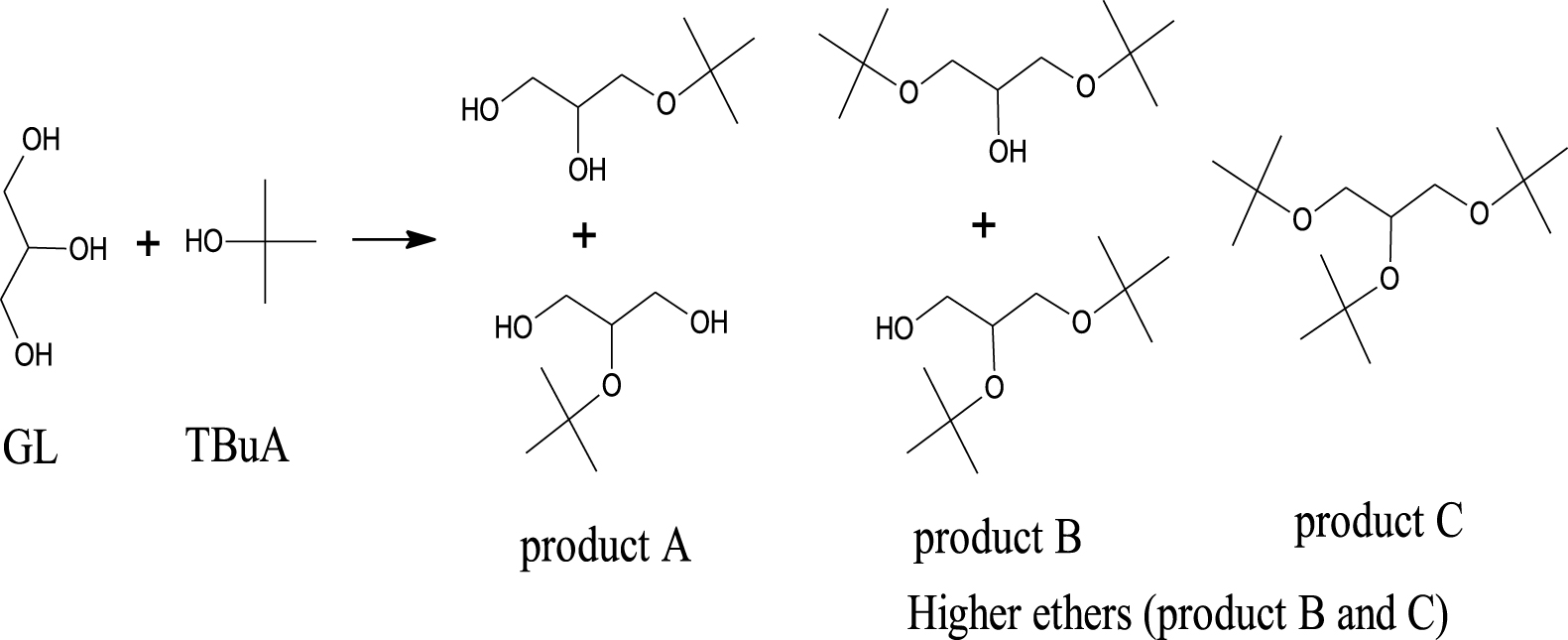
3.2.1. Effect of various samples on GL conversion and higher ethers selectivity by three heating methods (MWV, ULS and CVN)
The results of GL conversion and higher ethers selectivity were obtained with different set of reaction conditions by various solid acid catalysts namely AlPO, FeAlPO, ZrAlPO, CuAlPO and ZnAlPO in accordance with three heating methods as illustrated in Figures S1, S2, S3 and Table 3. Figures S1, S2 and S3 represent the reaction performed using three different heating methods MWV, ULS and CVN respectively. The data clearly show that MWV is excellent and effective compared to ULS and CVN with respect to percentage of GL conversion and selectivity towards higher ethers.
Comparison of optimized reaction parameters between three methods over CuAlPO solid acid with respect to the results of etherification of GL with TBuA
| Heating method | 1 (min) | 2 (°C) | 3 (g) | 4 (x:y) | 5 (%) | Ethers selectivity (%) | 10 (%) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 7 | 8 | 9 (7 + 8) | |||||||
| MWV | 15 | 50 | 0.06 | 1:4 | 98 | 31.4 | 18.7 | 49.6 | 68.3 | 66.93 |
| ULS | 60 | 80 | 0.10 | 1:6 | 90 | 56.1 | 10.5 | 33.2 | 43.7 | 39.33 |
| CVN | 180 | 100 | 0.10 | 1:12 | 75 | 83.1 | 12.6 | 4.3 | 16.9 | 12.68 |
Note: 1 is reaction time, 2 is reaction temperature, 3 is catalyst weight, 4 is molar ratio of GL(x):TBuA(y), 5 is GL conversion, 6 is product A, 7 is product B, 8 is product C, 9 is higher ethers, 10 is yield of higher ethers.
In these experiments, all the used catalysts showed good catalytic activity for the percentage of GL conversion but among them CuAlPO is the best catalyst as related to selectivity percentage of higher ethers due to its stronger acid sites. Overall the percentage of selectivity towards higher ethers (products B and C) over various solid acid catalysts under MWV heating method was found to increase in the following order: AlPO (only 18.3% of product B) < ZrAlPO (53.3%) < FeAlPO (54.3%) < ZnAlPO (56%) < CuAlPO (68.3%). This order clearly suggests that the sample’s catalytic activity mainly depends on the properties, namely acidic nature as well as porosity of the samples. The selectivity percentage towards higher ethers (bulky ethers) is favoured by strength of an acid as well as porous nature of the catalyst [48]. There was no appreciable selectivity to higher ether i.e., product C in the case of AlPO catalyst due to absence of strong acid sites.
Many authors have reported that the etherification reaction was performed only on the nature of the acidic sites. Gonzalez et al. [19] showed that modified fluorinated 𝛽 zeolites have the highest yield towards higher ethers (37%) after 24 h at 348 K due to strong Brønsted acid sites. Saxena et al. [48] reported that on account of strong acid sites the BEA zeolites showed the highest (99.1%) selectivity towards higher ethers at high temperature (110 °C) after 4 h by conducting both batch reactor and fixed bed reactor experiments. Celdeira et al. [49] showed that the sulfonated nobia AS100 catalyst showed maximum selectivity to higher ethers (38.6%) at 120 °C after 5 h by etherification of glycerol. Srinivas et al. [50] showed that caesium-exchanged tungstophosphoric acid 20C1TS had the best catalytic activity for highest selectivity to higher ethers (44%) at 100 °C after 60 min. Similarly various other acid catalysts used for etherification of GL with TBuA are listed in Table 4. In the present work, 68.3% selectivity towards higher ethers was achieved by CuAlPO catalyst in a short period of time at 50 °C. Therefore, CuAlPO catalyst was preferred for further optimization of reaction conditions under MWV heating method.
Comparison of catalytic results for the etherification of GL with TBuA over various acid catalysts
| Sl. no. | Catalyst [Ref.] | Selectivity | 1 (%) | 2 | 3 (°C) | 4 (min) | 5 (wt%) | 6 (cycles) | |
|---|---|---|---|---|---|---|---|---|---|
| X (%) | Y (%) | ||||||||
| 1 | 20TZ [56] | 84 | 12.4 | 84 | 1:12 | 120 | 60 | 0.5 g | 4 |
| 2 | BC 10:1-S2h [20] | 52 | 22 | 100 | 1:4 | 120 | 360 | 5 | 8 |
| 3 | AS100 [49] | 60.3 | 38.6 | 95 | 1:4 | 120 | 300 | 5 | 4 |
| 4 | 20C1TS [50] | 56 | 44 | 91 | 1:12 | 100 | 60 | 0.5 | 4 |
| 5 | BEA Zeolites [48] | 0.9 | 99.1 | 98 | 1:4 | 110 | 600 | 5 g | 3 |
| 6 | Amberlyst 15 [57] | 87 | — | 96 | 1:4 | 90 | 180 | 5 | — |
| 7 | Montmorillonite K10 [18] | 92 | 4 | — | 1:4 | 83 | 1440 | 6 | — |
| 8 | MFI Nano-Zeolites [14] | 76.1 | 23.9 | 83 | — | 120 | 720 | — | — |
| 9 | Fluorinated 𝛽-Zeolites [19] | 63 | 37 | 75 | 0.25 | 75 | 1440 | 5 | — |
| 10 | A-15 [15] | 68 | 28.2 | 93.6 | 1:5 | 70 | 360 | 7.5 | — |
| 11 | S50TS50O [16] | 71 | 28 | 98 | 1:4 | 75 | 1020 | 5 | — |
| 12 | CuAlPO (present work) | 31.8 | 68.3 | 98 | 1:4 | 50 | 15 | 0.08 g | 7 |
Note: X = product A, Y (Higher ethers) = product B and C, 1 = GL conversion, 2 = Molar ratio (GL:TBuA), 3 = Temperature, 4 = Time, 5 = Catalyst weight, 6 = Reusability.
3.2.2. Comparison between MWV, ULT and CVN heating method for etherification reaction over CuAlPO catalyst
The effect of variation of the reaction conditions under three different heating methods on the percentage of GL conversion and selectivity to higher ethers by etherification of GL is discussed here. Data in Table 3 clearly indicate that the effect of three different heating methods on the selectivity of higher ethers (%) are in the order: CVN < ULS < MWV.
The catalytic reaction is performed extremely slowly and much less (16.9%) selectivity to higher ether is obtained in CVN heating method due to the hotness being circulated first to the vessel wall, heating jackets and finally to used reactants. Hence, this method is considered as an unfit and slow method for the shipping of energy to the main reaction media.
ULS-facilitated heating technique showed higher values in relation to the selectivity towards higher ethers (43.7%) compared to CVN heating. As a result ULS heating process can increase the reaction speed by simply raising the interface mixing [51].
MWV heating method was found to be superior (68.3%) to ULS and CVN heating method with respect to the selectivity to higher ethers. The enrichment in the rate of formation of products by rapid internal heating of reactants was highly efficient to increase the active interactions between the active site of the catalyst surface and reactant molecule under microwave energy. Without catalyst, the power of microwave (energy) under MWV heating failed to start the etherification even though polar reactants were used. Further, MWV heating method was a highly efficient technique to accelerate the reaction rate by reducing the initiating time from hours to minutes. Moreover, the polar acid sites on the surface of the catalyst play a vital role in this reaction. Therefore, the etherification reaction by CuAlPO catalyst under MWV irradiation is the best technique to explore the product selectivity towards diesel miscible oxygenates i.e., fuel additives such as product B and C and also the application of microwave energy (faster rate) is a promising route compared to regular heat treatment (slower rate) [52, 53].
3.2.3. Effect of temperature
Figure S4 shows the results obtained for GL conversion (%) and selectivity (%) towards higher ethers from catalytic reaction between GL with TBuA at temperatures ranging from 40 °C to 60 °C (15 min) over CuAlPO using MWV heating method. It should be noted that the percentage of GL conversion as well as selectivity to higher ethers increase with temperature. At 40 °C, 92% of GL conversion and 58.9% of higher ethers (products B and C) selectivity were observed. As temperature is increased to 50 °C, GL conversion (%) and selectivity (%) were increased to 98% and 68% respectively. At 50 °C, selectivity of higher ethers increased at the expense of product A. Not much change in the % of GL conversion and selectivity towards higher ethers was observed by further increasing temperature up to 60 °C. Hence, the optimized temperature factor on etherification under MWV heating treatment was set to 50 °C. Previous studies reported that above 90 °C, GL was not totally converted because of de-etherification reaction [54]; polymeric products formed by dimerization of isobutene were also observed above 110 °C [50]. In the present work, the etherification reaction showed maximum selectivity towards higher ethers (68%) at 50 °C.
3.2.4. Effect of molar ratio
Impact of molar proportion (GL:TBuA) on GL conversion (%) and higher ethers selectivity (%) at molar ratios ranging from 1:3–1:5 was studied (Figure S5). Molar ratio is an important parameter to achieve highest GL conversion and higher selectivity. The results show that percentage of GL conversion as well as selectivity towards higher ethers was increased from 90% to 98% and from 64.7% to 68.3% respectively by varying the molar ratio from 1:3 to 1:4. It is clearly suggested that the results are varied on the molar ratio of the reactants. Beyond 1:4 molar ratios, the decrease in higher ethers selectivity (from 68.3% to 57.5%) and also slight decrease in GL conversion (95%) were observed. This decline in selectivity and conversion is due to blockage of active acidic sites on the surface of the catalyst and also due to the fact that the bulky nature of TBuA hinders the etherification reaction of GL [20]. Hence, 1:4 molar ratios were considered as optimal in the GL etherification reaction.
3.2.5. Effect of sample weight
Figure S6 shows the effect of samples’ weight on GL conversion (%) and selectivity (%) using CuAlPO catalyst under MWV heating. Without the catalyst, the etherification reaction does not take place, while significant rise in conversion and selectivity can be observed as the catalyst weight increases. GL conversion (%) and higher ethers selectivity (%) were increased from 89% to 98% and from 48.6% to 68% respectively while weight of sample is increased across 0.04–0.08 g. When the catalyst weight was further increased to 0.10 g there is slight decrease in GL conversion to 96% and also higher ethers selectivity to 65.9%. This is due to a certain amount of isobutene formation ( <1.5%) by dehydration of TBuA reactant [15, 50]. Therefore, 0.08 g was elected as optimized sample weight for the present work.
3.2.6. Effect of reaction time
Figure S7 shows the effect of reaction time on GL conversion (%) and higher ethers selectivity (%) using CuAPO catalyst under MWV heating technique. GL conversion (82%) and higher ether selectivity (54.2%) were found to be lowest at 5 min. Increase in GL conversion as well as selectivity towards higher ethers and decrease in product A selectivity as the time increases from 5 to 15 min was observed. The maximum selectivity towards higher ethers and GL conversion were observed at 15 min. Beyond 15 min, there is no appreciable change in the percentage of GL conversion and selectivity towards higher ethers. It clearly indicates that the etherification reaction reached equilibrium stage at 20 min. Hence, CuAlPO showed efficient catalytic activity for higher ethers selectivity at 15 min.
3.2.7. Effect of stirring speed
Figure S8 shows the higher ethers yield (%) obtained in the presence of CuAlPO catalyst under MWV heating method at three different speeds, i.e. 450, 650 and 850 rpm. This experiment was carried out in order to determine the possible external heat transfer in the following reaction conditions: 5, 10 and 15 min reaction time, 50 °C reaction temperature, 1:4 (GL:TBuA) molar ratio and 0.08 g catalyst weight. From the figure it is clearly observed that higher ethers yield (%) did not depend on the speed of agitation (in the range 450–850 rpm). Hence, it reflects the lack of heat moving via bulk liquid phase reaction mixture to the external surface of the catalyst [55].
3.3. Leaching test of catalyst
This test was carried out to check the catalyst heterogeneous form by conducting the etherification reaction between GL with TBuA under MWV heating method as shown in Figure 9. The reaction was carried out using 0.08 g CuAlPO or ZnAlPO (heterogeneous) catalysts for 20 min at 50 °C with 1:4 molar ratios of GL and TBuA. In order to indicate whether CuAlPO and ZnAlPO catalysts are precisely heterogeneous or not, the CuAlPO or ZnAlPO solid catalyst was separated from reaction media after 5 min by centrifugation followed by hot filtration. The experiment was further monitored up to 20 min under same reaction conditions. The GL conversion remains same after removal of catalyst, which reveals that the reaction was completely stopped. It clearly suggests that the etherification reaction over CuAlPO and ZnAlPO is truly heterogeneous.
Catalytic deactivation test for CuAlPO and ZnAlPO solid acid catalysts on GL etherification with TBuA under MWV heating method. (Reaction condition: molar ratio = 1:4 (GL:TBuA), catalyst weight = 0.08 g, reaction time = 15 min, reaction temperature = 50 °C.)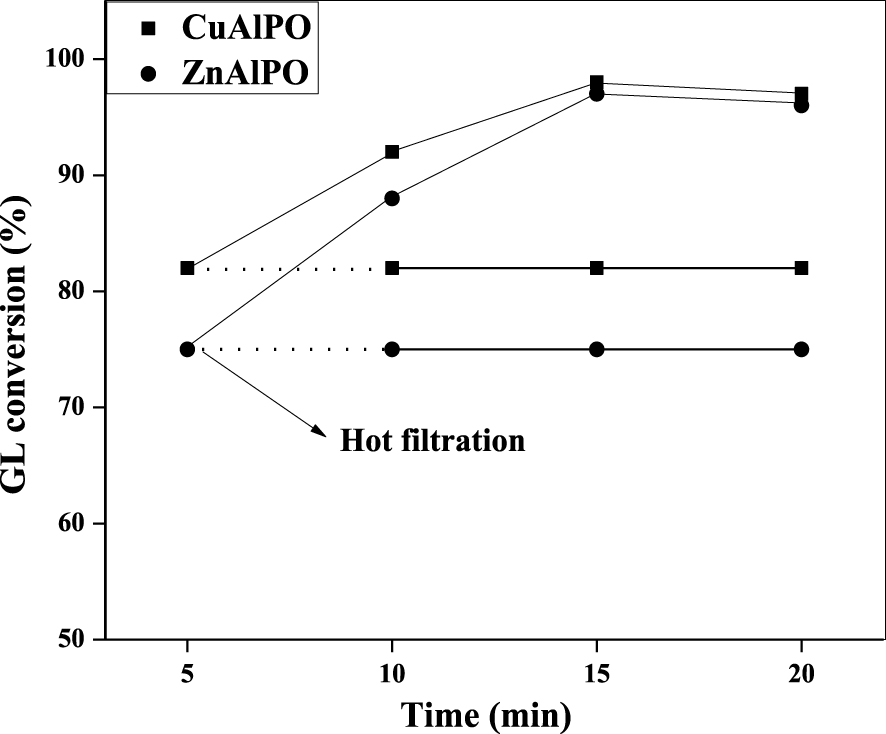
3.4. Comparison of catalytic activity with reports in previous literature
The comparison table (Table 4) suggests that the on-going work is highly useful in the case of some important parameters like time, temperature, reusability and weight of the sample. Very simple single step was followed in the preparation of the catalysts such as AlPO, FeAlPO, ZrAlPO, CuAlPO and ZnAlPO contrary to various catalysts reported in Table 4. Additionally, the important characteristic of catalyst (heterogeneous form) is the reusability action in which CuAlPO catalyst can be used up to seven consecutive cycles without loss of its catalytic activity.
3.5. Kinetic study
The establishment of the influence of heat and mass shifting limitations in the course of etherification between GL and TBuA by the kinetic examination was conducted. Kinetic studies were carried out under MWV heating method to evaluate the catalytic activity of FeAlPO, ZrAlPO, CuAlPO and ZnAlPO catalysts.
The values of reaction rate constant, activation energy (Ea) and temperature co-efficients (Tc) of FeAlPO, ZrAlPO, CuAlPO and ZnAlPO catalysts for etherification of GL with TBuA under MWV heating method
| Catalyst | Rate constant ( × 10−3 min−1) | Ea (kJ/mol) | Tc (k2∕k1) | |
|---|---|---|---|---|
| 313 K (k1) | 323 K (k2) | |||
| ZrAlPO | 34.3 | 43.3 | 19.58 | 1.26 |
| ZnAlPO | 42.4 | 51.8 | 18.84 | 1.22 |
| FeAlPO | 38.1 | 47.6 | 17.62 | 1.25 |
| CuAlPO | 54.0 | 66.0 | 16.87 | 1.22 |
The experiments were also conducted for the study of heat shifting limitations by varying the speed of agitation of the reaction mixture as shown in Figure S8. This clearly indicated that there was no influence of stirring rates on the higher ethers yield (%) and it reveals that the heat shifting limitation was completely absent. Hence, it may be recommended that heat shifting from bulk media (liquid reactant phase) to the external surface of the sample is absent [55].
A simple experiment was conducted to evaluate the mass shifting limitation by varying the catalyst weight. From Figure S6, the GL conversion (%) and higher ethers selectivity (%) increase linearly by increase in sample weight. Thus, it opposes the mass moving factor between the bulk phase of liquid reactant and outer surface of the sample, which was negligibly small. Madon et al. [58] reported that in the absence of all transport limitations, the reaction rate correlates with sample weight and also the total number of the active sites of the catalyst.
Plots of −ln[1 − Yieldhigher ethers] vs Time at distinct temperature (30 °C–50 °C) over FeAlPO, ZrAlPO, CuAlPO and ZnAlPO catalysts are represented in Figure 10. The values of rate constant (first order) secured by the plotted graph’s slope as well as other important parameters like activation energy (Ea) and temperature coefficient (Tc) values were calculated by using equation (1) (Arrhenius equation) and equation (2) respectively (Table 5). In the present case, a faster kinetics was observed based on activation energy for the higher ethers yield (%). The activation energy of the catalysts for etherification was found to follow the order: ZrAlPO > ZnAlPO > FeAlPO > CuAlPO. Among the catalysts CuAlPO showed least energy of activation (Ea). This indicates that from kinetic measurements, CuAlPO is the best catalyst for etherification between GL and TBuA with high yield of higher ethers (%). The value of activation energy (Ea) was evaluated by making use of Arrhenius equation:
| (1) |
| (2) |
The plot of first order rate constant for the formation of higher ethers yield over FeAlPO, ZrAlPO, CuAlPO and ZnAlPO catalyst under MWV heating method.
3.6. Effect of reactant pre-adsorption
Pre-adsorption (reactant) study was executed successfully to find out the catalytic mechanism between GL with TBuA reactants. The simplest form of the mechanism occurred in etherification reaction over solid sample through the action of one (or) more surface-bonded type with respect to either a Langmuir–Hinshelwood (LH-type, two surface-bonded species) mechanism or Eley–Rideal (ER-type, one surface-bonded species) mechanism. In ER-type mechanism, one of the reactant (GL or TBuA) is adsorbed on the solid surface (catalyst) and the other reactant co-ordinates with the adsorbed species during final desorption of the resultant product [59]. However, LH-type mechanism is involved before the start of the reaction; initially two reactants (GL and TBuA) are adsorbed onto the surface of the catalyst [60]. Thus, the reactant pre-adsorption impact was examined under reaction conditions (optimized) i.e., 0.08 g of CuAlPO at 60 °C plus 1:4 molar ratio of GL:TBuA under MWV heat treatment for 30 min. Reactant pre-adsorption reaction was started by premixing the CuAlPO sample (0.08 g) with either GL or TBuA or both for one day at room temperature after the addition of remaining reactants. These experimental observations are shown in Figure 11 by plotting higher ethers yield (%) against time (min).
Effect of pre-adsorption studies on the etherification of GL with TBuA over CuAlPO solid acid catalyst under MWV heating method. (Reaction conditions: catalyst weight = 0.08 g, time = 15 min, temperature = 50 °C, molar ratio = 1:4 (GL:TBuA).)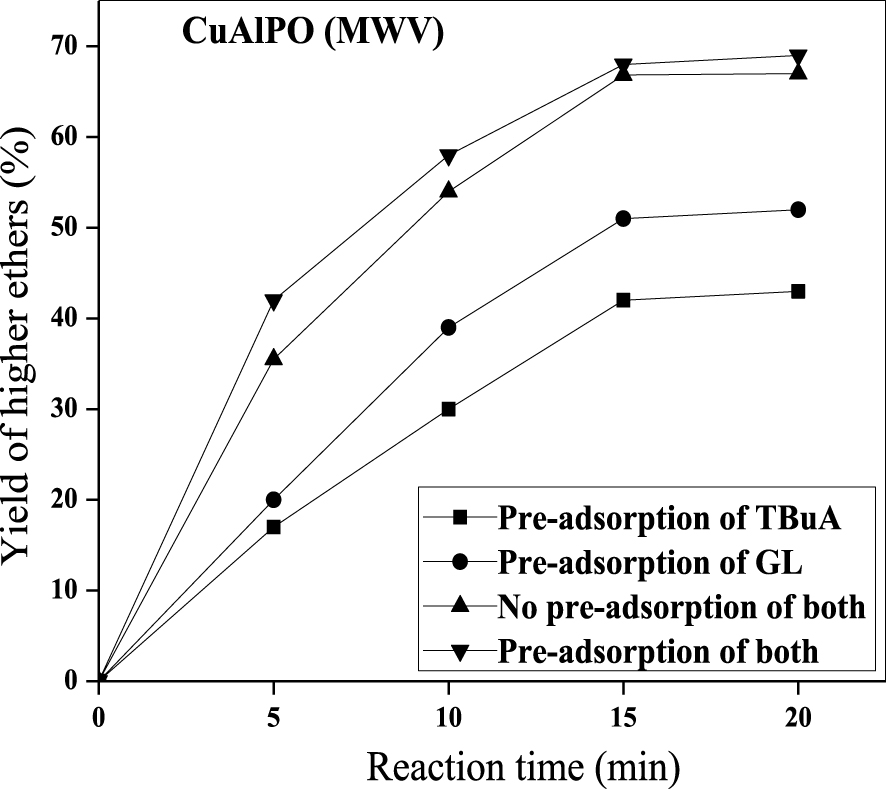
Premixing the catalyst with both the reactants produced highest higher ethers yield (%) whereas in the other cases lowest higher ethers yield (%) was attained (i.e., premixed the catalyst with only TBuA and without premixing). The order for pre-adsorption results was found to be: both reactants premixing > no premixing of both > premixing only with GL > premixing only with TBuA. Hence, the sample (CuAlPO) premixing with two reactants (GL and TBuA) showed highest yield (%) compared to without premixing. The lowest yield is obtained by premixing the catalyst with TBuA due to blockage of catalytically active sites by the adsorbed alcohol reactant without adding other reactant. From the literature also it is noticed that alcohol is chemisorbed strongly compared to other reactants [61]. This suggested that the etherification reaction involves LH-type mechanism because the reaction prefers chemisorption of both GL and TBuA.
3.7. Reaction mechanism
Probable reaction mechanism for an acid catalysed etherification of GL with TBuA over Brønsted acidic sites of the catalyst.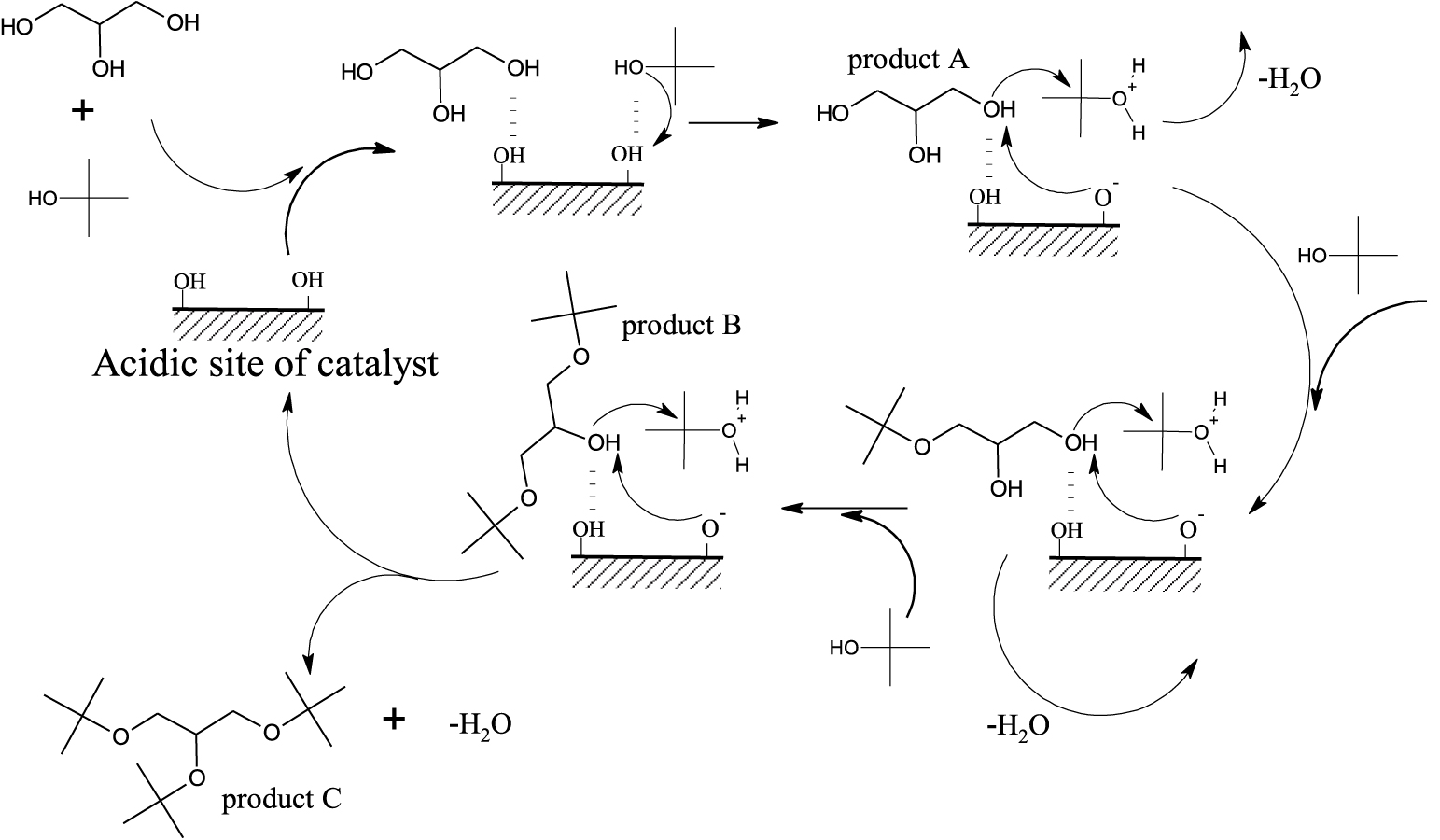
The majority of the strength of catalytic action of various acid samples for the present reaction has been attributed to Brønsted acid sites present on the catalyst [48, 50, 15]. The probable reaction mechanism for the GL conversion with maximum selectivity towards higher ethers over CuAlPO solid acid catalyst under MWV heating is shown in Scheme 2. In etherification reaction, steps include the adsorption of reactant’s (GL or TBuA) OH group onto the Brønsted acidic sites of catalyst. The lone pair electrons present on the oxygen atom of the TBuA strongly co-ordinate with Brønsted acidic sites of the catalyst as well as attract the acidic proton from the acidic sites, which generates a protonated tertiary intermediate alcohol that is unstable. This unstable protonated tertiary intermediate alcohol loses a water molecule by attacking the lone pair electrons of an oxygen atom of the glycerol i.e., nucleophilic attack of glycerol through SN2 mechanism [62] and it leads to the formation of product A. This adsorbed m-TBGe molecule again reacts with another adsorbed TBuA reactant by the same mechanism, which result in the formation of product B and similarly product C is formed by product B with GL. This mechanism clearly reveals that the successive transformation of GL to higher ethers is achieved by the CuAlPO solid acid catalyst due to the regeneration of the acidic sites. In the present work, the highest selectivity (68%) towards higher ethers shows that the used catalyst has no effect on water formed during the reaction which is in accordance with earlier reports [48].
3.8. Reusability check in etherification reaction
Effect of catalytic reusability of the CuAlPO solid acid catalyst on GL etherification with TBuA under MWV heating method. (Reaction condition: molar ratio = 1:4 (GL:TBuA), catalyst weight = 0.08 g, reaction time = 15 min, reaction temperature = 50 °C.)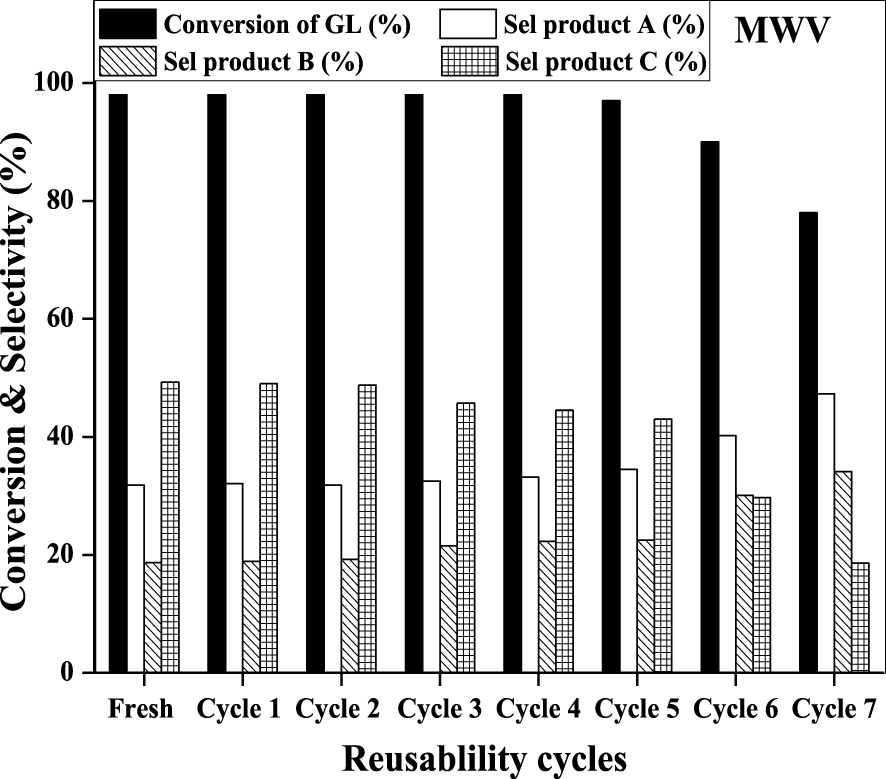
The reusability check was executed in order to evaluate recyclability and stability of catalyst on etherification reaction over CuAlPO sample. After each cycle completion, the solid sample was isolated by centrifugation and filtration. The separated catalyst was washed by C2H5OH, dried at 120 °C (5 h) and ultimately calcined at 550 °C (2h) to remove adsorbed organic species. The calcined catalyst was weighed and changes in weight were observed. Thus, the catalyst was reused for further cycles in the same experimental setup. As demonstrated in Figure 12, CuAlPO catalyst performed constant catalytic activity up to five cycles. There was a slight decrease in percentage of GL conversion and also selectivity for higher ethers after the 5th cycle due to chemisorption of alcohol on the catalytic acid sites, which may have blocked the active acidic sites [63].
4. Conclusions
The important results of this study can be summarized as follows:
- The etherification of GL with TBuA effectively takes place over CuAlPO solid acid catalyst under microwave heating method.
- Microwave heating method was superior to ultrasonic and conventional heating with respect to % selectivity towards higher ethers.
- Presence of large amount of mesoporosity and strong acid sites in CuAlPO catalyst provides an excellent platform to transform GL into value-added products to be used as oxygenated fuel additives (d-TBGe + t-TBGe) blended with diesel fuels.
- The CuAlPO catalyst showed highest GL conversion (98%) as well as selectivity (68%) towards higher ethers (d-TBGe + t-TBGe) in the microwave-irradiated etherification reaction.
- Hot leaching test proves that CuAlPO catalyst is truly heterogeneous in nature.
- Pre-adsorption study suggested that the etherification reaction followed the Langmuir–Hinshelwood mechanism.
- Kinetic measurements confirmed that the CuAlPO is the best catalyst in the present reaction due to less energy of activation (Ea) compared to other catalysts.
- The catalyst showed excellent catalytic reusability with constant activity.
- Etherification reaction under catalysed microwave-assisted method offers many advantages like eco-friendliness, short reaction time, low energy consumption, simple protocol, high yield and a simple technique for the application of green chemistry.