


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Enol ethers or vinyl ethers (VEs) are a class of chemical compounds that display ether and vinyl functional groups. These compounds have found numerous applications as key reagents in organic reactions such as the Diels–Alder reaction [1] and Claisen rearrangement [2], thanks to the remarkable reactivity of their electron-rich carbon–carbon double bond and the range of substituents they can bear [3, 4]. They have been extensively polymerized under cationic initiation to produce polymeric materials endowed with excellent properties used in various industrial applications such as adhesives, paints, coatings, and the synthesis of stimuli-sensitive materials [5, 6, 7, 8, 9, 10, 11, 12]. Despite their simple structure, no universal method has been found to carry out their synthesis. Nevertheless, many techniques have been developed to prepare VEs, particularly the catalyzed transetherification using transition metals. Winternheimer et al. reported the main available pathways to synthesize such VEs [2]. In the 1930’s, Reppe and Wolff developed a process to form VEs from acetylene and primary alcohols under rigorous pressure and temperature conditions and using a large basic catalyst amount [13, 14]. Then, four additional synthetic pathways have been developed for the chemical conversion of alcohols to VEs: (i) addition onto acetylene [13, 14], (ii) phase transfer catalysis based on Williamson reaction between an alkoxide and 2-chloroethyl vinyl ether [15], (iii) modification of vinyl acetate in the presence of an iridium complex [16, 17], and (iv) transetherification [18]. The latter strategy has been widely studied in the presence of either mercuric acetate [19, 20] or palladium acetate [21, 22, 23, 24] catalysts producing various VEs in fair to good yields. Complexes of palladium acetate and trifluoroacetate with bidentate ligands, such as 1,10-phenanthroline, have proven to efficiently catalyze most of the VEs preparations, and its catalysis mechanism was proposed by Handerson and Schlaf [23]. They successfully used (L–L) Pd(OAc)2 catalyst, where L–L stands for 2,2′-bipyridyl or 1,10-phenanthroline chelate, for the transfer vinylation of protected monosaccharides. The efficiency of palladium (II) complex in transfer vinylation has been initially described by McKeon and Fitton [25]. To the best of our knowledge, only two other examples of catalyzed transfer vinylation were reported in the literature: vinylation of steroids [22] and synthesis of glycidol vinyl ether [26]. In 2010, Pichavant et al. described the synthesis of a VE by transetherification catalyzed by a palladium (II) complex in the presence of ethyl vinyl ether (EVE) in dichloromethane [27]. The iridium-catalyzed reaction of vinyl acetate with alcohols later appeared as a complementary method to palladium-catalyzed transetherification [16, 17].
In 2013, our group published the trans- etherification between EVE and various alcohols leading to the corresponding original VEs in 40–84% yield in a single step synthesis [18, 28, 29, 30, 31, 32, 33]. A series of VEs monomers that bear diverse functional groups, such as halogenomethyl, amino, carbonate, oligo(ethylene oxide) side-groups, and oligo(fluoroether) chains, was prepared and characterized by NMR spectroscopy. The palladium complex has been found to accelerate the reaction upon using an excess of ethyl vinyl ether or alcohol.
In this work, a detailed study on the trans- etherification reaction of EVE was carried out. The aim was to prepare new vinyl ethers with various side functional groups (Scheme 1) for a wide use in the synthesis of polymeric materials and other applications. These VEs were synthesized from the corresponding alcohols through transetherification of EVE catalyzed by an organometallic complex generated in situ (Scheme 1). Several reaction parameters were studied: the initial EVE/alcohol molar ratio, the alcohol concentration, the nature of the solvent, the presence and nature of the ligand, the amount of the organometallic complex, and the choice of the metal precursor.
Synthesis of VEs bearing various functional groups by transetherification of the corresponding alcohol and EVE.
2. Results and discussion
In this study, liquid VEs bearing various side chains were prepared from four different alcohols (Scheme 1). Furfuryl alcohol was chosen to undergo a series of transetherification reactions to determine the optimal experimental conditions (Scheme 2).
Synthesis of 2-(vinyloxymethyl) furan by transetherification of the furfuryl alcohol and EVE.
Using our typical procedure, the following key parameters were investigated: initial EVE/alcohol molar ratio, alcohol concentration, nature of the solvent, presence and nature of the ligand, amount of catalyst, and the choice of metal precursor. Results are summarized in Table 1.
Transetherification of EVE and furfuryl alcohol in different reaction conditionsa
| Entry | M(OAc)2 (%) | Ligand (eq.) | Solvent | Initial EVE/alcohol molar ratio | [Alcohol] (mol/L) | Conv.b (%) |
|---|---|---|---|---|---|---|
| 1 | Pd(OAc)2 (2%) | — | CH2Cl2 | 12 | 2.2 | 0 |
| 2 | Pd(OAc)2 (2%) | 1-phenyl-1,2-ethanediol (1.5) | CH2Cl2 | 12 | 2.2 | 0 |
| 3 | Pd(OAc)2 (2%) | Triphenylphosphine (2) | CH2Cl2 | 12 | 2.2 | 0 |
| 4 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1.5) | CH2Cl2 | 12 | 2.2 | 69 |
| 5 | Zn(OAc)2 (2%) | 1,10-phenanthroline (1.5) | CH2Cl2 | 12 | 2.2 | 0 |
| 6 | Co(OAc)2 (2%) | 1,10-phenanthroline (1.5) | CH2Cl2 | 12 | 2.2 | 0 |
| 7 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1.5) | DMF | 12 | 2.2 | 8 |
| 8 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1.5) | THF | 12 | 2.2 | 16 |
| 9 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1.5) | Dioxane | 12 | 2.2 | 50 |
| 10 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1.5) | Toluene | 12 | 2.2 | 57 |
| 11 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1.5) | — | 12 | 2.2 | 66 |
| 12 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1.5) | CH2Cl2 | 6 | 2.2 | 60 |
| 13 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1.5) | CH2Cl2 | 24 | 2.2 | 57 |
| 14 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1.5) | CH2Cl2 | 12 | 4.4 | 65 |
| 15 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1.5) | CH2Cl2 | 12 | 0.5 | 37 |
| 16 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1.5) | CH2Cl2 | 12 | 1 | 52 |
| 17 | Pd(OAc)2 (2%) | 1,10-phenanthroline (1) | CH2Cl2 | 12 | 2.2 | 65 |
| 18 | Pd(OAc)2 (2%) | 1,10-phenanthroline (2) | CH2Cl2 | 12 | 2.2 | 63 |
| 19 | Pd(OAc)2 (2%) | 1,10-phenanthroline (3) | CH2Cl2 | 12 | 2.2 | 59 |
| 20 | Pd(OAc)2 (1%) | 1,10-phenanthroline (1.5) | CH2Cl2 | 12 | 2.2 | 52 |
| 21 | Pd(OAc)2 (5%) | 1,10-phenanthroline (1.5) | CH2Cl2 | 12 | 2.2 | 69 |
| 22 | Pd(OAc)2 (10%) | 1,10-phenanthroline (1.5) | CH2Cl2 | 12 | 2.2 | 66 |
aReaction conditions: Solvent or bulk (entry 11), 24 h, 20 °C, M(OAc)2(%), ligand (eq./Pd(OAc)2). bAlcohol conversion was assessed by 1H NMR spectroscopy; Conv. = (∫4.71 CH2O(VE)∕2)∕(∫4.71 CH2O(VE)∕2 + ∫4.61 CH2OH alcohol∕2) × 100, where ∫i represent the integral of the signal centered at i ppm in the 1H NMR spectrum.
Transetherification of primary alcohols that bear various side groups with ethyl vinyl ethera
| Entry | ROH | VE | Pd(OAc)2 (%) | Conv.b (%) | Yieldc (%) |
|---|---|---|---|---|---|
| 1 | 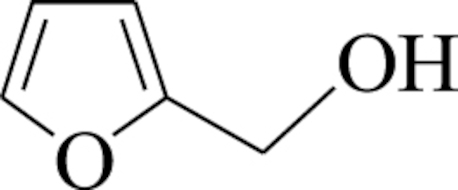 | 2-(vinyloxymethyl) furan (1) | 2 | 69 | 59 |
| 2 |  | 1,2,3 trimethoxy-5-(vinyloxymethyl) benzene (2) | 2 | 82 | 75 |
| 3 | 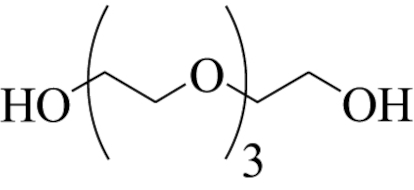 | 2-(2-(vinyloxy)ethoxy) ethanol (mono-3) | 2 | 75 | 50 |
| (2-(2-(vinyloxy)ethoxy)ethoxy) ethene (bis-3′) | 5 | 61 | 40 | ||
| 4d |  | 6-(vinyloxy)-hexahydrofuro[3,2-b]furan-3-ol (4) | 2 | 50 | 42 |
aReaction conditions: 20 °C, 24 h, Pd(OAc)2 (2%) generated in situ, 1,10-phenanthroline (1.5 eq./Pd(OAc)2). bConversion was assessed by 1H NMR spectroscopy; Conv. = (∫4.71 CH2O(VE)∕2)∕(∫4.71 CH2O(VE)∕2 + ∫4.61 CH2OH alcohol∕2) × 100. cIsolated yield. dReaction carried out in THF.
The presence of the ligand played an essential role in the transetherification reaction. This was proven in entry 1 that was carried out without any ligand, for which no transformation was observed. Using 1-phenyl-1,2-ethanediol (entry 2) and triphenylphosphine (entry 3) ligands did not yield the desired product. However, the 1,10-phenanthroline ligand showed a remarkable catalytic activity of Pd(OAc)2 with 69% conversion, obtained after 24 h at 20 °C (entry 4).
In order to assess the impact of the metal present in the catalyst, use of Zn(OAc)2 or Co(OAc)2 complex catalysts did not result in the formation of the expected VE (entries 5–6). However, using Pd(OAc)2 led to the formation of the desired VE in 69% conversion. These results confirm Boutevin et al.’s studies. They demonstrated that the 1,10-phenanthroline-palladium (II) acetate complex led to VEs by transetherification of EVE in fair yields [15].
When studying the effect of solvent nature, moderate conversions were observed with 1,4-dioxane or toluene (entries 9–10). On the other hand, poor conversions were obtained using THF and DMF (entries 7–8). Dichloromethane was noted to be the best solvent for this reaction, leading to the highest conversion (entry 4). This variation in the catalytic activity can be related to the solubility of the catalyst, which differs from one solvent to another depending on the solvent polarity. A slight decrease in the catalytic activity (66% conversion) was observed when the reaction was conducted without any solvent (entry 11).
It was noted that the increase in the initial EVE/alcohol molar ratio from 6 to 12 eq. shifted the equilibrium in favor of the desired VE product (entries 4 and 12). Furthermore, an increase in the EVE equivalents above 12 did not benefit the conversion (entry 13). This may be due to the dilution effect. Thus, 12 eq. of EVE was sufficient to promote this reaction.
The effect of the initial alcohol concentration was examined as well. 69% conversion was obtained with a concentration of 2.2 mol/L (entry 4). A similar conversion was obtained when the alcohol concentration was raised to 4.4 mol/L (entry 14), showing that the maximum possible conversion was already reached with a concentration of 2.2 mol/L. Dropping the alcohol concentration to 0.5 mol/L (entry 15), lessened the conversion to 37%. Thus, medium concentration ( ≈2.2 mol/L) was good enough to be used for the catalyst to be effective and give high conversions.
Since the presence of the ligand was crucial, the variation of the number of equivalents of 1,10-phenanthroline was also studied. When the number of the 1,10-phenanthroline ligand equivalents was increased from 1.0 to 1.5, the conversion was improved from 65% to 69% (entries 4 and 17). In entry 18, a slightly lower conversion was obtained with 2 ligand equivalents (63%). Furthermore, adding 3 equivalents of the ligand resulted in an apparent reduction of the conversion (entry 19).
A significant increase in the conversion was noted by raising the catalyst amount from 1 to 2% (entries 4 and 20). 5% catalyst (entry 21) did not show any notable advantage. However, 10% catalyst (entry 22) caused a slight drop in the conversion, indicating that a 2% catalyst is sufficient to reach the highest conversion in this transetherification reaction.
From this optimization, a series of VEs (1–4) was then synthesized by transetherification catalyzed by 2% palladium (II) acetate generated in situ. The reactions were carried out in dichloromethane in the presence of 1.5 equivalents of 1,10-phenanthroline and excess of EVE (12 eq.). The reaction was stirred at room temperature for 24 h. The results are summarized in Table 2.
2-(Vinyloxymethyl) furan (1) was prepared under the optimal conditions by transetherification catalyzed by a palladium (II) complex generated in situ; 69% conversion and 59% yield were obtained (entry 1). 1,2,3-trimethoxy-5-(vinyloxymethyl) benzene (2) was synthesized under the same conditions with 82% conversion and 75% yield (entry 2). The transetherification of tetraethylene glycol and EVE (Scheme 3) led to mono-vinyl ether (3) with 75% conversion (calculation based on the 1H NMR analysis of the reaction mixture). Surprisingly, in a random experimental trial with the amount of the catalyst raised to 5%, telechelic bis vinyl-ether (3′) was formed as a major product while the yield dropped from 50 to 40% (entry 3). Finally, the transetherification of dianhydro-D-glucitol was exceptionally performed in THF/CH2Cl2 due to its low solubility in dichloromethane. The desired VE (4) was obtained with 50% conversion and 42% yield (entry 4).
Transetherification of bis(2-2(hydroxyethoxy)ethyl)ether and EVE leading to either mono-functional VE (3) or telechelic VE (3′).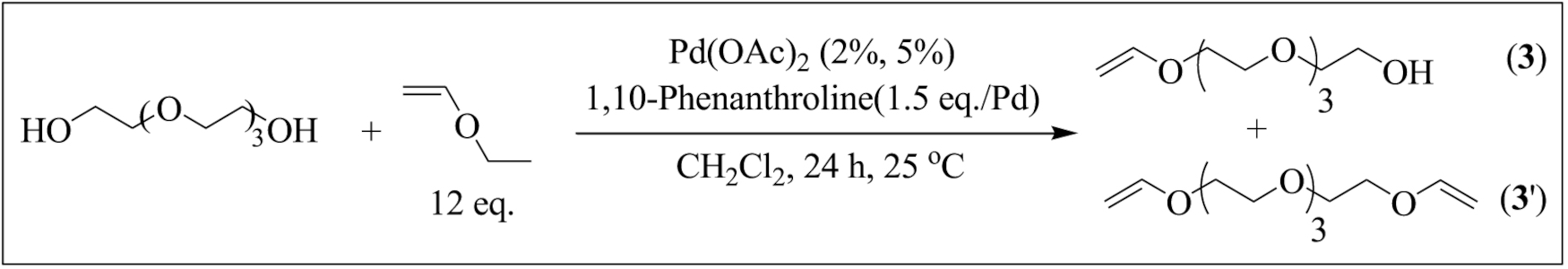
All synthesized VEs (1–4) were characterized by 1H and 13C NMR spectroscopy. In the following section, the 1H and 13C NMR spectra of 2-(vinyloxymethyl) furan (1) are provided as a representative sample. The 1H NMR spectrum (Figure 1) of VE (1) shows both nonequivalent protons (H2C=) of the vinyl group as two doublets of doublets centered at 4.11 and 4.34 ppm (3 JHHgem = 2.21 Hz, 3 JHHcis = 6.78 Hz, 3 JHHtrans = 14.26 Hz). Furthermore, the signal assigned to the vinyl proton (=CH−) is located at 6.53 ppm as a doublet of doublets. Similar signals with identical multiplicities, integrations, and close chemical shifts have been observed for the other synthesized VEs (2–4), confirming the success of the transetherification process. For VE (1), the methylenic protons in CH2–O exhibit very close signals at around 4.7 ppm. Another singlet can be observed at 4.6 ppm corresponding to the methylenic protons of the alcohol precursor that was not totally removed after purification.
The 13C NMR spectrum of 2-(vinyloxymethyl) furan (1) (Figure 2) displays the characteristic signals of both carbon atoms in the OCH=CH2 double bond, at 151.14 ppm and 87.67 ppm. These signals appear in opposite phases as the former is tertiary while the latter is secondary. In the same manner, the carbon atom in methylene exhibits a negative signal at 62.34 ppm. Finally, the positive signals of the carbon atoms in the furan ring appear at 107.77, 110.35, and 142.58 ppm, confirming the structure of the product. Similar results have been obtained for the other VEs (2–4).
1H NMR spectrum of 2-(vinyloxymethyl) furan (1) (CDCl3, 20 °C, 300 MHz), ∗represents the signal of remaining traces of the alcohol precursor.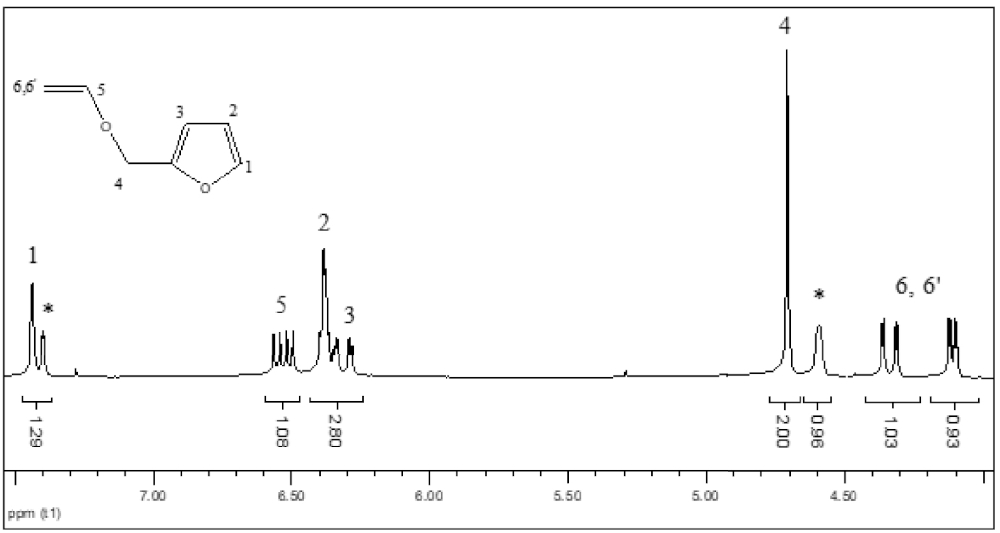
13C NMR spectrum of 2-(vinyloxymethyl) furan (1) (CDCl3, 20 °C, 300 MHz).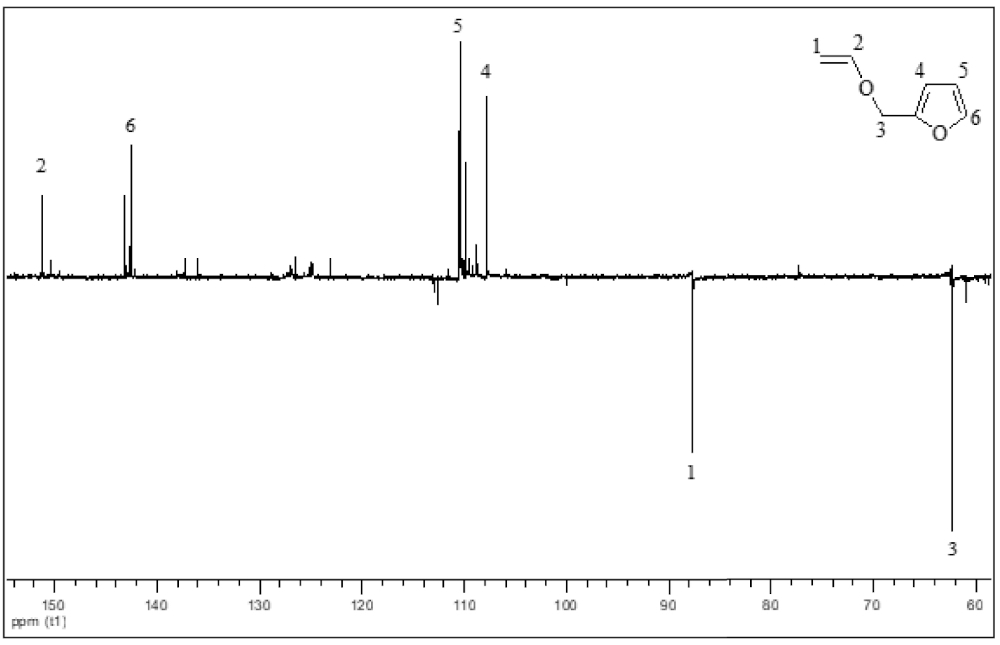
3. Experimental section
3.1. Materials
Ethyl vinyl ether, cobalt (II) acetate tetrahydrate, zinc (II) acetate, palladium (II) acetate, 1,10-phenanthroline, 1,2-phenylethanediol, triphenylphosphine, 2-(hydroxymethyl) furan, 3,4,5-trimethoxybenzylalcohol, tetraethylene glycol, dianhydro-D-glucitol, dichloromethane (99.9%), THF (anhydrous, 99.9%), sodium sulfate, were purchased from Sigma Aldrich. Deuterated chloroform (99.96 atom% D), 1.4-dioxane (anhydrous, 99.8% purity), DMF (anhydrous, 99.8% purity), and toluene (anhydrous, 99.8%) were purchased from Euriso-top, JTBaker, Riedel de Haen and Merck, respectively. THF was dried over sodium/benzophenone and distilled before use.
3.2. Characterization by nuclear magnetic resonance
The NMR spectra were recorded at room temperature on a Bruker AC 300 instrument using either d-chloroform or d-acetone as solvents and tetramethylsilane as reference. Coupling constant and chemical shifts (δ) are given in hertz (Hz) and part per million (ppm), respectively. The experimental conditions for recording 1H and 13C NMR spectra were as following: flip angle 90° (or 30°), acquisition time 4.5 s (or 0.7 s), pulse delay 2 s (or 2 s), and a number of scans 128 (or 512).
3.3. Synthetic procedures
3.3.1. Synthesis of 2-(vinyloxymethyl) furan (1)
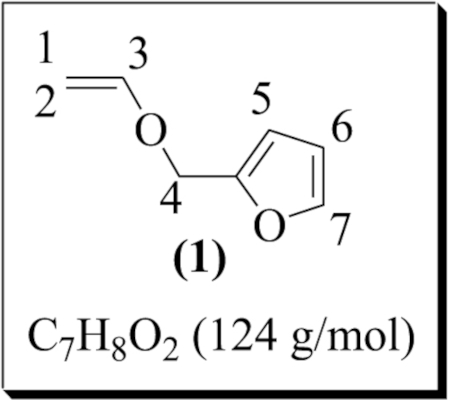
The catalyst solution was initially prepared by dissolving palladium (II) acetate (91.00 mg, 4.00 × 10−4 mol) in 2 mL of dichloromethane. A solution of 1,10-phenanthroline (110.20 mg, 6.00 × 10−4 mol) in dichloromethane (2 mL) was then added drop-wise. The reaction medium was stirred at room temperature for 30 min to generate the palladium catalyst in situ. Then, a solution of 2-(hydroxymethyl) furan (2.00 g, 0.02 mol) and ethyl vinyl ether (17.30 g, 0.24 mol) in dichloromethane (5 mL) was added to the catalytic solution. The reaction mixture was stirred at room temperature for 24 h. The conversion was tracked by withdrawing a 0.2 mL sample and drying it under vacuum to eliminate the EVE residue. The conversion was 69%. The purification of the synthesized EV was conducted by adding 50 mL of distilled water; the solution was extracted with dichloromethane (2 × 50 mL). The organic phase was then dried over Na2SO4, filtered, and the solvent was evaporated under vacuum to give the desired product (1) in the form of a brown liquid (1.46 g, 59% yield). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.40 (d, –CH–O, 1H, H7), 6.53 (dd, –CH=CH2, 1H, H3, 3 Jtrans = 14.27 Hz, 3 jcis = 6.78 Hz), 6.34 (m, –CH=CH–O, 1H, H6), 6.29 (d, –CH=C–O, 1H, H5, 3 J = 2.27 Hz), 4.70 (s, –CH2–O–, 2H, H4), 4.34 (dd, CHaHb=CH (E), 1H, H1, 3 Jcis = 6.78 Hz, 2 Jgem = 2.23 Hz), 4.11 (dd, CHaHb=CH (Z), 1H, H2, 3 Jtrans = 14.26 Hz, 2 Jgem = 2.21 Hz). 13C NMR (300 MHz, CDCl3) δ (ppm): 151.14 (–CH=CH2, 1C, C3), 142.58 (–CH–O, 1C, C7), 110.35 (–CH=CH–O, 1C, C6), 107.77 (–CH=C–O, 1C, C5), 87.67 (–CH2=CH, 1C, C1), 62.34 (–CH2–O–, 1C, C4).
3.3.2. Synthesis of 1,2,3-trimethoxy-5- (vinyloxymethyl) benzene (2)
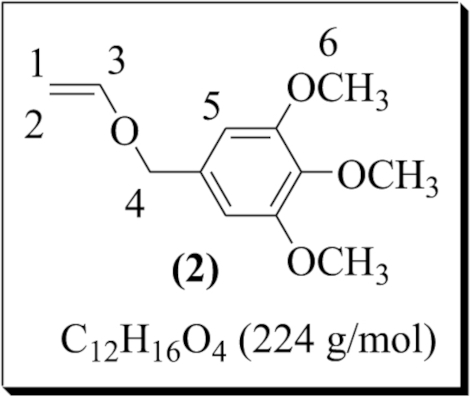
The same procedure used for (1) was applied to 3,4,5-trimethoxybenzylalcohol (2.00 g, 0.01 mol) and ethyl vinyl ether (8.65 g, 0.12 mol) which were dissolved in 2.5 mL of dichloromethane. The solution was then added to the catalytic solution prepared by dissolving palladium (II) acetate (112.50 mg, 5.00 × 10−4 mol) and 1,10-phenanthroline (135.15 mg, 7.50 × 10−4 mol) in 2 mL of dichloromethane. The 1H NMR analysis of an aliquot in CDCl3 showed 82% conversion. The product was then purified in the same manner as described for (1). 1,2,3-trimethoxy-5-(vinyloxymethyl) benzene (2) was obtained as an orange–yellow liquid (2.67 g, 75% yield). 1H NMR (300 MHz, CDCl3) δ (ppm): 6.52 (s, CH of benzene, 2H, H5), 6.45 (dd, –CH=CH2, 1H, H3, 3 Jtrans = 14.25 Hz, 3 Jcis = 6.80 Hz), 4.52 (s, CH2–O, 2H, H4), 4.25 (dd, CHaHb=CH, 1H, H1, 3 Jtrans = 14.3 Hz, 2 Jgem = 2.16 Hz), 4.00 (dd, CHaHb=CH, 1H, H2, 3 Jcis = 6.8 Hz, 2 Jgem = 2.16 Hz), 3.78 (s, CH3–O, 9H, H6). 13C NMR (300 MHz, CDCl3) δ (ppm): 151.50 (–CH=CH2, 1C, C3), 104.54 (C of benzene, 2C, C5), 87.72 (CH2=CH, 1C, C1), 70.23 (CH2–O, 1C, C4), 60.80–56.01 (CH3–O, 3C, C6).
3.3.3. Synthesis of 2-(2-(vinyloxy)ethoxy)ethanol (3)
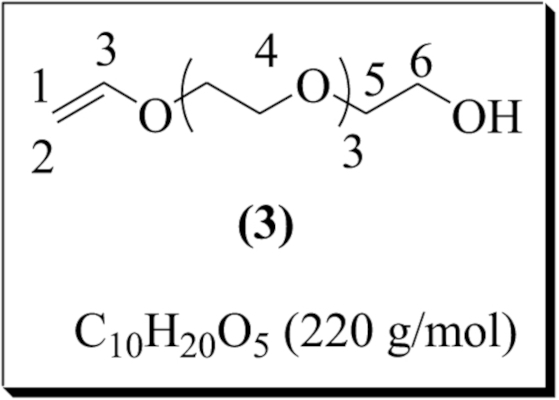
The same procedure used for (1) was applied to tetraethylene glycol (2.00 g, 0.01 mol) and ethyl vinyl ether (8.65 g, 0.12 mol) which were dissolved in 2.5 mL of dichloromethane. The solution was then added to the catalytic solution prepared by dissolving palladium (II) acetate (44.90 mg, 2.00 × 10−4 mol) and 1,10-phenanthroline (54.06 mg, 3.00 × 10−4 mol) in 2 mL of dichloromethane. The 1H NMR analysis of an aliquot in CDCl3 showed 75% conversion. The product was then purified in the same manner as described for (1). 2-(2-(vinyloxy)ethoxy)ethanol was obtained as a yellow liquid (0.60 g, 50% yield). 1H NMR (300 MHz, CDCl3) δ (ppm): 6.40 (dd, –CH=CH2, 1H, H3, 3 Jtrans = 14.34 Hz, 3 Jcis = 6.79 Hz), 4.10 (dd, CHaHb=CH, 1H, H1, 3 Jtrans = 14.33 Hz, 2 Jgem = 1.66 Hz), 3.90 (dd, CHaHb=CH, 1H, H2, 3 Jcis = 6.77 Hz, 2 Jgem = 1.06 Hz), 3.80–3.50 (m, CH2 on the terminal chain, 16H, H4–6). 13C NMR (300 MHz, CDCl3) δ (ppm): 151.63 (–CH=CH2, 1C, C3), 86.55 (CH2=CH, 1C, C1), 72.90 (1C, C5), 70.50–67.19 (6C, C4), 61.55 (–CH2–OH, 1C, C6).
3.3.4. Synthesis of 6-(vinyloxy)-hexahydrofuro[3,2-b]furan-3-ol (4)

The same procedure used for (1) was applied to dianhydro-D-glucitol (0.50 g, 3.42 × 10−3 mol) and ethyl vinyl ether (2.92 g, 0.041 mol) which were dissolved in 2.5 mL of THF. The solution was then added to the catalytic solution prepared by dissolving palladium (II) acetate (0.038 mg, 1.70 × 10−4 mol) and 1,10-phenanthroline (0.045 mg, 2.55 × 10−4 mol) in 2 mL of CH2Cl2. 1H NMR analysis of an aliquot in acetone-d6 showed 50% conversion. The product was then purified in the same manner as described for (1). 6-(Vinyloxy)-hexahydrofuro[3,2-b]furan-3-ol (4) was obtained as an orange–yellow liquid (0.25 g, 42% yield). 1H NMR (300 MHz, acetone-d6) δ (ppm): 6.49 (dd, –CH=CH2, 1H, H3, 3 Jtrans = 14.37 Hz, 3 Jcis = 6.87 Hz), 4.10 (dd, CHaHb=CH, 1H, H1, 3 Jcis = 6.76 Hz, 2 Jgem = 2.03 Hz), 3.90 (dd, CHaHb=CH, 1H, H2), 4.50–3.50 (cyclic CH2 and CH, 8H). 13C NMR (300 MHz, CDCl3) δ (ppm): 151.00 (–CH=CH2, 1C, C2), 88.90 (cyclic CH, 2C, C4 and C7), 78.00 (CH–O–CH=, 1C, C3), 72.00 (–CH–OH, 1C, C6), 82.50 (CH=CH2, 1C, C1), 76.50 and 73.10 (cyclic CH2, 2C, C8 and C5).
4. Conclusion
Several functionalized vinyl ethers were successfully synthesized by transetherification catalyzed by a palladium complex generated in situ with conversions between 50 and 82% and in yields ranging from 40 to 75%. Different experimental reaction parameters were optimized with a series of reactions carried out at room temperature for 24 h using a large excess of EVE. All VEs were characterized by 1H and 13C NMR spectroscopy. The newly synthesized VE can bear various functional groups which can potentially induce remarkable properties and different polymerization applications.
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgements
The authors thank Ecole Nat. Sup. de Chimie de Montpellier (ENSCM) and Ecole doctorale à l’université Libanaise (EDST). The authors thank Université Libanaise-programme de recherche for the financial support. The authors also thank Pr. Bernard Boutevin (ENSCM), and Dr. Ali Hachem and Dr. Iyad Karameh (Université Libanaise).