


 CC-BY 4.0
CC-BY 4.0
1. Introduction
In the recent years, humanity has registered an increase in consumption of medicines, other pharmaceuticals and personal care products (PPCP). Some of these species have evil effects if they reach the surface waters, therefore maximum acceptable contents of PPCPs need to be defined, in order to decide if that water source could be purified and turned potable. The maximum limits and nature of PPCS approved depend on the foreseen water use and differ for each country [1]. Clofibric acid (CA) is a micropollutant often found in wastewater, being a metabolite of clofibrate, a drug used since 1963 as a blood lipid and cholesterol lowering agent [2]. CA is a persistent pollutant that cannot be removed by conventional wastewater treatments [3]. In the wastewater treatment plants, CA has been detected at concentrations up to 41.4 mg/L [4]. Even very low concentrations of CA are a threat to human health and water systems. CA has high mobility, is toxic and can persist in waste water for approximately 21 days [5]. The waste water treatment procedures such as adsorption, coagulation, and biological methods could not remove CA completely [6]. In order to decrease even more the wastewater toxicity, methods allowing the complete mineralization of CA, such as the advanced oxidation processes are highly desirable. Several alternatives like photocatalytic degradation [7, 8, 9], ozonation [10], photo-Fenton processes [11], electrochemical oxidation [4], etc. are cited in the literature as advanced oxidation processes. A few papers dealing with the total removal of CA by photocatalytic reactions are available [12, 13].
Titanium dioxide (TiO2) is by far the most popular photocatalytic material, due to its photostability, lack of toxicity, low cost, and easy availability [14, 15]. The main three polymorphs of TiO2 are anatase, rutile and brookite, the first two being a lot more common [16]. Their band gap values are at 3.23 eV (anatase) and 3.02 eV (rutile). The electron promotion from the valence to the conduction band is possible by the UV irradiation and the electron–hole pair formation is a preamble for the generation of hydroxyl radicals HO∙, a key species in the mechanism of persistent organic compound degradation.
The major drawback of TiO2 is the high recombination rate of the electron–hole pairs, therefore new photocatalytic systems based on metal-doped titanium oxide were developed, as a strategy for reducing the recombination rate by a better charge carrier separation [17]. The use of noble metals in this respect is a preferred strategy [18].
In some previous works of our team, we have investigated the influence of TiO2 doping with metals as Pt, Pd, Au, Ag, etc., on the photodegradation of various organic compounds [15, 19, 20]. The mentioned noble metals increase the photocatalytic activity by slowing down the recombination of the electron–hole pair [21]. The noble metal–metal oxide interaction gives specific results for each support-metal couple; sometimes, the doping does not bring the expected improvement of the photocatalytic activity, as noted in a previous paper of ours for Pt/TiO2 photocatalyst [15].
The noble metal deposition on TiO2 particles is often performed by the impregnation from solutions. This method is simple and cheap and enables a proper dispersion of the noble metal on the oxide surface [22]. The incipient wet impregnation (IWI) method is an efficient version used for doping [23], having the advantage of realizing the uniform dispersion of small metal particles in the pores of the support. The doped solid often has a high stability in the photocatalytic reaction system [20]. When the proper choice of the doping agent can be made, the notable advantage of the absence of any waste during the photocatalyst preparation is achieved, if the impregnation salt of the noble metal is easily decomposed during the thermal activation.
In this study, the photocatalytic performance of palladium-doped TiO2 was investigated in comparison with that of several commercial titanium dioxide samples (Kronos 7000, Kronos 7500, PC500 and TiO2 from Across Chemicals). The catalyst doping was performed by applying the IWI method. This doped catalyst (TiO2_Pd) had been previously selected by Sescu et al. [20] for its possible photoactivity in the elimination of rhodamine B. As a continuation of this recent work, the main objective of this study was to investigate if the palladium-doped TiO2 catalyst can enhance the UV-A photodegradation of emerging persistent water pollutants such as CA. The improvement of the photocatalytic efficiency of the TiO2_Pd in comparison with those of raw titania was established probably due to the exhibition of the synergistic effects between the support and the palladium deposited on the titania. The original contribution of this study includes the investigation of the degradation kinetics of this persistent water micropollutant on TiO2_Pd suspensions and on the elucidation of the mineralization potential of this catalyst under UV-A irradiation conditions. To the best of the authors knowledge, this is the first work presenting an exhaustive evaluation of a Pd-doped TiO2 photocatalyst concerning the effect of different key operating parameters on the degradation and mineralization efficiency of this molecule including the type of catalyst and its load, the initial pollutant content, irradiation, water matrix and inorganic anions. In addition, the contribution of catalyst reusability on the degradation reaction was considered in this study.
2. Experimental part
2.1. Materials and reagents
The chemicals used in this study were of analytical purity and preliminary purification procedures were not necessary. Four different commercial titanium dioxide (TiO2) samples were tested as photocatalysts: TiO2 from Across Chemicals (CAS 13463-67-7) (powder containing 98%+ anatase, BET surface area = 9.99 m2∕g), Kronos vlp 7000 from Kronos Titan GmbH, Germany (anatase powder containing 95% TiO2, BET surface area = 225 m2∕g, Kronos uvlp 7500 (Kronos Titan GmbH, Germany; anatase powder containing 95% TiO2, BET surface area > 250 m2∕g) and Cristal activ PC500 (Cristal SAS, France; anatase 99%, BET surface area of 398 m2∕g).
Clofibric acid of analytical purity (>99%) was purchased from Across Organics (New Jersey, USA). Formic acid and acetonitrile employed in the HPLC analysis were obtained from Sigma-Aldrich (St. Louis, MO, USA). All stock and working solutions used for different analysis and photocatalytic runs were prepared in ultrapure water produced with a water purifier system (ELGA Option-Q DV 25, Veolia, High Wycombe, UK) having the following characteristics: 18.2 M𝛺⋅cm resistivity at 298 K, COT < 0.5 mg C/L; pH = 5.7. Additional experiments were also conducted in tap water (TW) collected from Rennes (France) and groundwater (coming from a well, WW) spiked with CA in order to evaluate the influence of the water matrix on the degradation kinetics of the target molecule. The main characteristics of the used real water samples were: TW: pH = 6.3, 6.4 mgC/L total organic carbon, 572 μS/cm conductivity, 8.76 mg/L nitrates, 6.73 mg/L sulfates, 38.8 mg/L chlorides; WW: pH = 5.9, 5.32 mgC/L, 448 μS/cm conductivity, 1.27 mg/l nitrates, 9.02 mg/L sulfates, 0.22 mg/L chlorides.
2.2. Doped photocatalyst preparation
A TiO2 Across sample doped with palladium was prepared by the IWI technique (1% Pd by weight ratio). Palladium (II) chloride and hydrochloric acid from Sigma-Aldrich were used in this respect. The IWI method occurs by mixing a thoroughly dried catalytic support with a small volume of salt solution containing the dopant, tempting to equalize the free pore volume of the solid [20]. The dopant solution (3 mL) was poured quickly on the TiO2 (2 g) heated for 2 h at 200 °C in a high and narrowly shaped crucible, then covered with a lid to prevent the powder spread-out. After cooling down to room temperature, the product was gently mixed in an agate mortar, then calcined at 400 °C for 4 h (1°/min heating rate). More details about the method used for the catalyst synthesis were described in our previous work [20]. Scanning electron microscopy (SEM) investigations and Energy Dispersive X-ray Analysis (EDAX) composition measurements were performed (JEOL JSM 7100 F EDS EBSD Oxford microscope; JEOL JSM-7100, Jeol Ltd., Tokyo, Japan) to characterize the samples.
2.3. Photocatalytic experiments and analytic methods
The photocatalytic performance of the prepared material was investigated via the photodegradation of CA under UV-A irradiation conditions. Clofibric acid was used here as a model molecule for photo-oxidation reactions of environmental refractory organic contaminants. The photocatalytic reactions were performed in a cylindrical glass photoreactor with a total volume of 1.5 L. The UV-A irradiation source (Philips PL-S lamp, 9 W, emission range: 350–400 nm, maximum at 365 nm) was placed in a quartz tube concentric within the photoreactor. The incident light intensity was measured with a radiometer (VLX-3W; Vilbert Lourmat, France). The experiments were run at an irradiation intensity of 6.2 mW∕cm2, excepting the series from the study concerning the influence of the irradiation intensity. They were conducted at room temperature (20 ±0.5 °C), under aeration, using 1 L of suspension, at the natural solution pH (pH = 6). During the photocatalytic reaction, the solution pH was not controlled. Before each photocatalytic run, the reaction media was continuously magnetically stirred in the dark to reach the adsorption–desorption equilibrium of CA at the surface of the solid. According to some previous studies, time duration of 60 min proved to be enough to achieve the complete equilibrium of adsorption. Afterwards, the UV-A lamp was turned on to start the photocatalytic reaction. Samples were taken from the reaction mass, filtered before each analysis through a H-PTFE syringe filter (pore size of 0.22 μm, Chromafil® Macherey-Nagel, Germany) to separate the solid and subjected to analysis for measuring the residual CA. All experiments were conducted in duplicate and for the results, a standard error ⩽5% was considered.
The residual CA concentration in the filtered samples was monitored using the high-performance liquid chromatography method. A reversed phase column (BEH C18, 250 mm × 4.6 mm, 5 μm particle size) was used for the separation. The HPLC instrument (WATERS®, Milford, MA, USA) was coupled with a photodiode array detector (PDA; Waters TM 996, Milford, MA, USA) for analyte detection. The selected value for the maximum absorption wavelength of CA was 227 nm. The considered mobile phase was composed of a mixture of acetonitrile/ultrapure water (60/40 containing 0.1% v/v of formic acid). The analysis were carried out under isocratic mode at a flow rate of 1 mL/min and the injection volume was 50 μL. Under the considered analytical conditions, the retention time for CA was about 6.6 min. The pollutant concentration was calculated by an external calibration procedure. The additional information about the calibration curve and the calculation of the detection and quantification limits were reported previously [24, 25, 26].
The assessment of the mineralization yield of CA and the corresponding reaction intermediates during the photocatalytic reaction was achieved by additional measurements of the total organic carbon content (TOC). These analyses were performed by using a TOC analyzer (TOC-5000-A, Shimadzu Corporation, Japan) coupled with a nondispersive IR detection. The instrument calibration was made following a methodology previously described in detail [27].
2.4. Reaction monitoring
The degradation yield of CA was calculated on the basis of the HPLC analysis data collected using the equation:
| (1) |
| (2) |
3. Results and discussions
3.1. Preliminary experiments for catalyst screening
A first evaluation of the influence of Pd doping on the photocatalytic activity was performed using a CA solution of 25 mg/L as a test analyte and a photocatalyst dose of 0.5 g/L. The photocatalytic performance of the sample TiO2_Pd was compared with these of four commercially available products: Kronos 7000, Kronos 7500, PC500 and the TiO2 support (Across Chemicals). The evolution of the CA decomposition (Figure 1) shows the fast conversion increase between the t-time and initial values. While the conversion values in the case of Kronos 7000, Kronos 7500, PC500 was about 75% after 110 min, almost 89% was reached on TiO2. In addition, the results presented in Figure 1 clearly confirm that the synthesized catalyst appears to be more photoactive than the other tested TiO2 samples for the oxidation of the target molecule leading to a complete pollutant elimination after 70 min of reaction.
Photocatalytic activity of various TiO2 photocatalysts (0.5 g/L) expressed as pollutant degradation efficiency (initial [CA] = 25 mg/L): 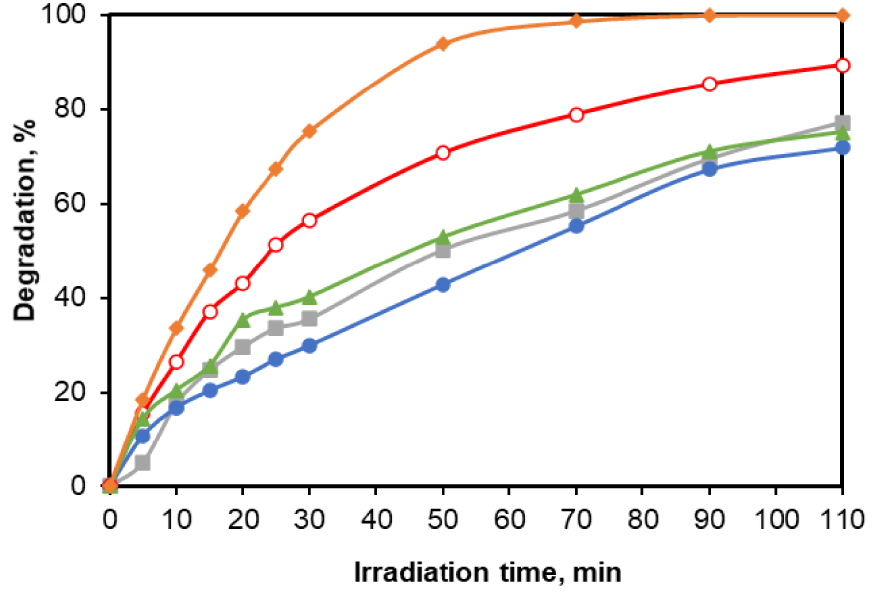
 K7500;
K7500;  PC500;
PC500;  TiO2;
TiO2;  K7000;
K7000;  TiO2_Pd.
TiO2_Pd.
The data collected from the photocatalytic experiments performed in the catalyst screening step are fitted by using the pseudo-first-order reaction model:
| (3) |
The suitability of the model fit with the obtained experimental data was evaluated by the value of the linear regression coefficient R2 corresponding to (3) and ka values were determined from the slope of the computed straight lines for the first 35 min of irradiation. The net superiority of the catalytic activity of TiO2_Pd sample in comparison with the commercial products could also be highlighted from the apparent degradation rates given in Figure 2. The ka values determined from the slopes of the linear regression are: 0.0448; 0.0286; 0.0192; 0.0162 and 0.0131 min−1 for the samples TiO2_Pd, TiO2 (undoped), Kronos vlp 7000, Kronos uvlp 7500 and PC500, respectively. These data clearly indicate that the reaction rate is almost double after the TiO2 doping with Pd, while raw TiO2 Across is still roughly twice more active than Kronos and PC500. The morphology of the used TiO2_Pd samples is presented in Figure S1 while its surface composition was confirmed by the results related to the spatial distribution of elements (mapping) (EDAX), attached to SEM analysis (Figure S2). More information concerning the characterization of the samples are reported in our previous work [20]. Furthermore, the high values obtained for all calculated correlation coefficients (over 95%) confirms that for all solids the photodegradation of the target compound is well described by the pseudo-first-order kinetic model.
Pseudo-first-order kinetics deduced for the CA aqueous solution (25 mg/L) photodegradation in the presence of various photocatalysts (0.5 g/L dose). Regression equations: 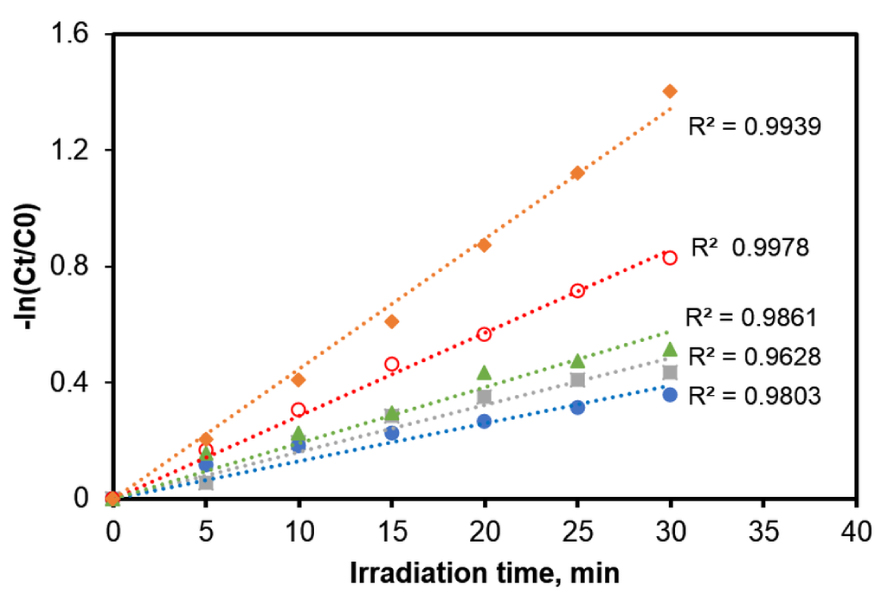
 K7500 (y = 0.0162x);
K7500 (y = 0.0162x);  PC500 (y = 0.0131x);
PC500 (y = 0.0131x);  TiO2 (y = 0.0521x);
TiO2 (y = 0.0521x);  K7000 (y = 0.0192x);
K7000 (y = 0.0192x);  TiO2_Pd (y = 0.0448x).
TiO2_Pd (y = 0.0448x).
We should point out that all these data revealed that TiO2 doping with Pd significantly enhanced the photocatalytic activity of TiO2 for the CA degradation in aqueous media. The higher photocatalytic activity of TiO2_Pd than these of the other investigated TiO2 samples could be expected, since different studies reported that noble metals (Ag, Au, Pt, and Pd) are efficient dopants improving the photocatalytic activity of TiO2 catalysts [28]. It was highlighted that the Fermi levels for the noble metals are lower than that of TiO2, improving the transfer of the photogenerated electrons from the conduction band of the semiconductor towards the particles of noble metal. Such electron trapping mechanism highly reduces the recombination rate between the photoinduced electrons and holes, significantly enhancing the catalyst photoactivity [28]. Based on the results obtained for the preliminary screening tests, all further photocatalytic runs were conducted in the presence of TiO2_Pd.
3.2. Influence of the light intensity
Since the preliminary experiments indicate the necessity of the irradiation in the system together with the photocatalyst use, the effect of the irradiation intensity on the reaction efficiency was tested, because this parameter affects the degradation efficiency. The experiments were performed at a quite low concentration of CA (5 ppm), in the presence of a lower TiO2_Pd dose (0.2 g/L), in order to highlight better the role of the light intensity on the elimination efficiency of the molecule. The results are displayed in Figure 3, indicating the degradation evolution in time, the initial reaction rates and the mineralization extent after 190 min of reaction.
(a) Degradation yield dependence with irradiation intensity; (b) pseudo-first-order reaction kinetics fit; (c) mineralization yield (standard deviation from duplicate experiment by error bars). ([CA] = 25 mg/L, 0.5 g/L photocatalyst dose). Regression equations: 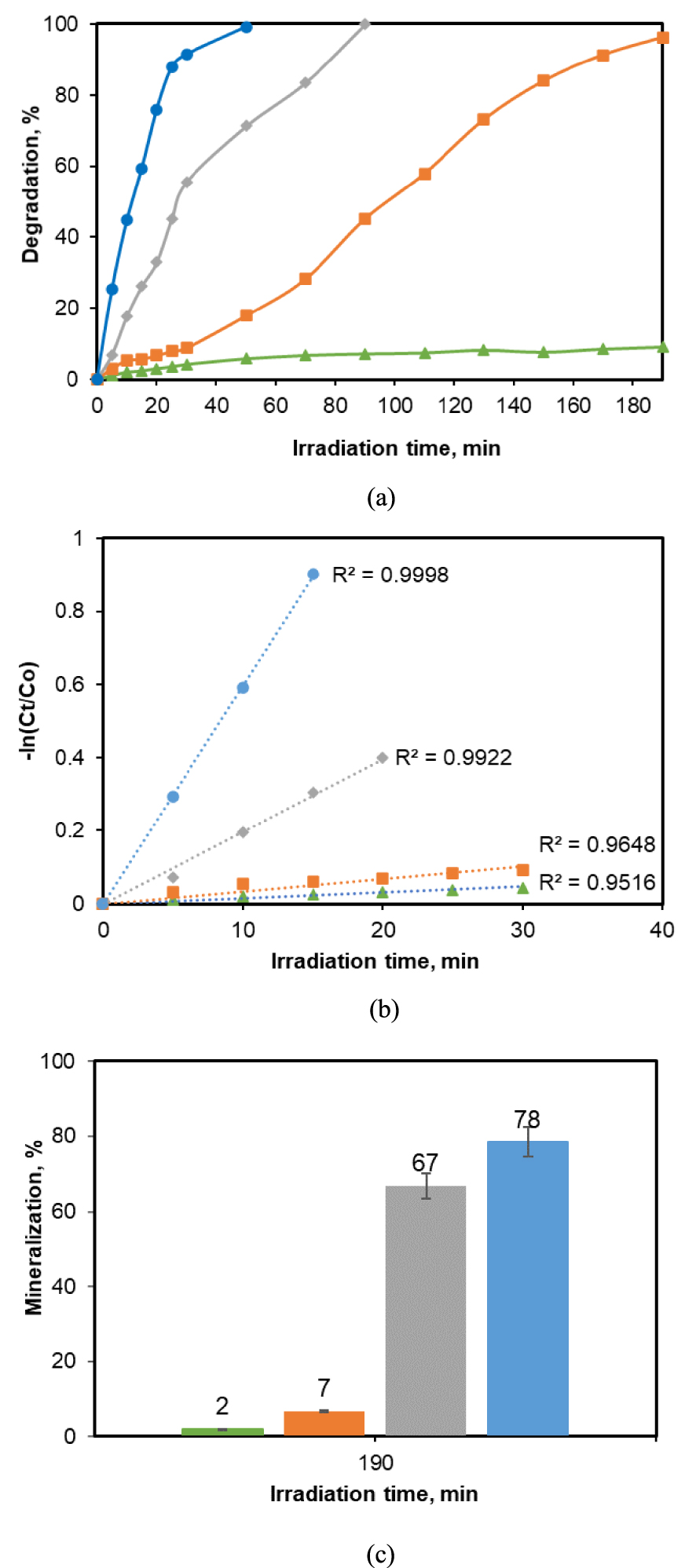
 0.075 mW∕cm2 (y = 0.0015x);
0.075 mW∕cm2 (y = 0.0015x);  0.76 mW∕cm2 (y = 0.0034x);
0.76 mW∕cm2 (y = 0.0034x);  2.45 mW∕cm2 (y = 0.0198x);
2.45 mW∕cm2 (y = 0.0198x);  6.2 mW∕cm2 (y = 0.0597x).
6.2 mW∕cm2 (y = 0.0597x).
The results show that the radiation intensity has utmost influence on the efficiency of the CA decomposition (Figure 3). The lowest value (0.075 mW∕cm2) does not have a significant effect on initiating the photocatalytic degradation (Figure 3a): after 180 min of reaction, only about 10% of the analyte is decomposed, this value being just a little bit higher than that obtained in the blank experiment investigating the role of irradiation only. A quite high degradation yield of 96% was found at an irradiation intensity of 0.67 mW∕cm2, but the reaction time to reach this value was quite long (190 min). When a radiation intensity of about 2.5 mW∕cm2 was applied, the total decomposition of CA occurred after 90 min. The increase of the light intensity to 6.2 mW∕cm2 made possible the total reaction after only 50 min. In conclusion, applying the highest intensity is beneficial on the reaction promotion. The beneficial role of the increase in light intensity to enhance the elimination efficiency was previously reported in other studies using other catalysts [29, 30, 31].
For all the irradiation power levels employed, it was found that the reaction kinetics fit excellently the pseudo-first-order model (Figure 3b). However, the ratios between the rate constant of the lowest and of the highest irradiation intensities varied by a factor of almost 50 (the equations are given together with the symbols under the legend of Figure 3).
The clear superiority of the exposure reaction conditions, under the highest intensity, results especially from the values of the mineralization yield of the organic molecule during the photocatalytic transformation. The decomposition yield determined by HPLC shows only the incipient transformations of the target molecule, i.e., its partial split in various fragments. Sometimes this step leads to the transformation of the initial, stable pollutant, in some smaller, biodegradable molecules, but cases are cited [32, 33] when these fragments are still nonbiodegradable and even more toxic than the initial molecule. The total mineralization indicated a process cancelling totally the presence of toxic organic compounds in water. The comparison between the mineralization yield under the four different light intensities reveals that the highest level gave a yield of 78% mineralization after 190 min of photocatalytic reaction. A relatively good value of 67% was obtained also at an intensity of 2.45 mW∕cm2. By contrast, the low irradiation intensity values develop only some percent of mineralization. The importance of the intense irradiation is well highlighted by the case of using the value of 0.76 mW∕cm2: the CA molecule decomposition is very high (96%), but the mineralization was of only about 7%. According to literature, the first step in the photocatalytic degradation of aromatic molecules is the hydroxylation of free sites, then the cycle opening leading to smaller molecules (such as organic acids). If the irradiation of solution continues, the fragments could finally be transformed in CO2 and H2O [16]. The higher mineralization yields achieved in our work certainly certify the complete mineralization of the target molecule under high irradiation conditions.
3.3. Influence of the photocatalyst dose
Since the photocatalytic transformations assume the reactions occurring between the active oxidizing species generated on the photocatalyst surface as a consequence of the electron–hole effects on the water and dissolved oxygen, on the one part, and the organic molecule adsorbed on the solid, on the other part, the photocatalyst dose is an important factor influencing the reaction progress. A high dose of photocatalyst certainly brings more opportunities for the adsorption of the molecule which ought to be transformed on the solid’s surface. In the meantime, a too high dose will result in light absorption and scattering due to the interaction with the particles [34, 35] and thus, the light energy conversion for the promotion of the electron from the valence band to the conduction band will decrease a lot. An optimal equilibrium between delivering adsorption opportunities and establishing a number of solid particles per unit volume so as to favour the oxidation of the target molecule has to be considered. The photocatalyst doses were varied between 0.05–0.5 g/L at an initial pollutant concentration of 25 mg/L. In the meantime, blank experiments were also performed in order to assess the role of the UV irradiation in the degradation of the aqueous solution of CA and the contribution of the pollutant adsorption on the solid without irradiation, to the overall removal of the organic pollutant. The results are illustrated in Figure 4, displaying the degradation yield (Figure 4a), the kinetic parameters (Figure 4b) and the mineralization yields (Figure 4c).
Influence of the TiO2_Pd dose on the evolution of the photocatalytic reaction. (a) Decomposition yield/HPLC; (b) pseudo-first-order kinetic data fit; (c) mineralization yields (error bars represent standard deviation from duplicate experiment). ([CA] = 25 ppm). Regression equations: 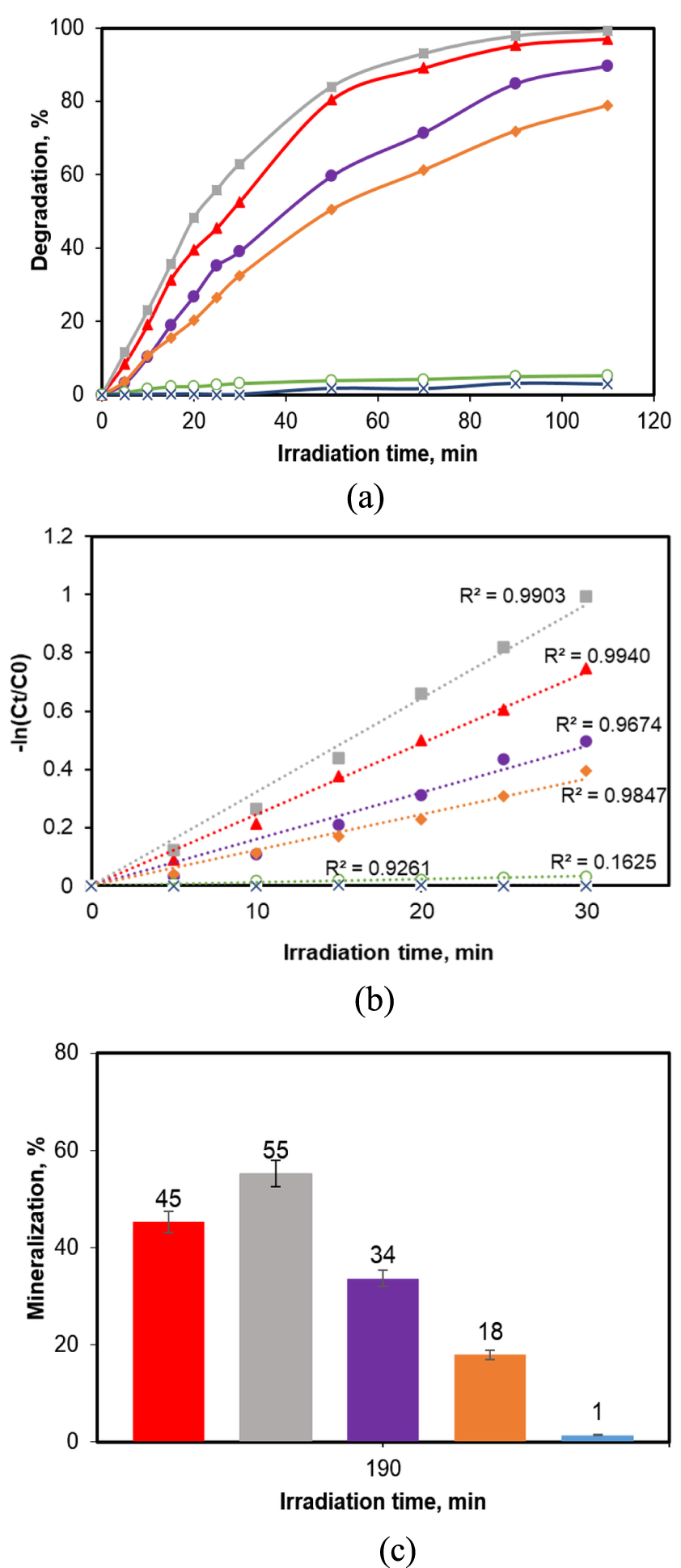
 0.5 g/L (y = 0.0245x);
0.5 g/L (y = 0.0245x);  0.2 g/L (y = 0.0322x);
0.2 g/L (y = 0.0322x);  0.1 g/L (y = 0.0116x);
0.1 g/L (y = 0.0116x);  0.05 g/L (y = 0.0123x);
0.05 g/L (y = 0.0123x);  Photolysis (y = 5E-05x) × Adsorption.
Photolysis (y = 5E-05x) × Adsorption.
The blank experiments clearly indicate that the presence of the photocatalyst is absolutely necessary for the removal of the CA. Indeed, the adsorption blank shows that only 3% of the initial CA was retained on the solid surface. In the photolysis test (UV irradiation but no photocatalyst), the CA removal was of 5%. These results are in line with the ones found in other studies carried out on the same pollutant [24, 36]. So, the data prove that the CA removal was enhanced significantly when both irradiation and photocatalyst were applied in the reaction media. The data from Figures 4a and b show that even a very low photocatalyst dose of 0.05 g/L is able to produce a CA conversion of almost 79% after 110 min of reaction, while the dose increase to 0.2 g/L leads to the total degradation of the test molecule. On the contrary, the extra addition of photocatalyst up to a dose of 0.5 g/L brings a slight decrease of the decomposition yield. It should be noted that the value of the catalyst dose beyond which the effect of this parameter became less pronounced depends on several parameters such as pollutant concentration, geometry of the reactor, light source (mainly its intensity and wavelength) [35]. In terms of reaction rate and mineralization yield, the much better effect of the dose of 0.2 g/L in comparison with both higher (0.5 g/L) and lower (0.1 g/L) doses is even more evident: the reaction rates are substantially higher (values of the slopes of the corresponding equations of the kinetics fits in Figure 4b), while the mineralization yield of 55% (at 0.2 g/L) is much superior to the values of 45% and 34% found, respectively, at doses of 0.5 and 0.1 g/L. As expected, the obtained mineralization yields are lower than the yields of CA removal, because the pollutant mineralization involves a sequence of chemical reactions for the transformation of the reaction intermediates to carbon dioxide and water. Therefore, based on the results of this parametric study, the subsequent photocatalytic runs were conducted at 0.2 g/L of catalyst.
3.4. Influence of CA concentration
Another parametric study usually approached for evaluating the photocatalysts is the investigation of the influence of the initial concentration of the pollutant species on the evolution of the reaction. The CA concentration range used in our investigations was from 5 to 25 ppm.
The data concerning the degradation yield of CA (Figure 5a) indicate that, as expected, the pollutant concentration has an important influence on the photocatalytic process. The general trend of the degradation is the same for all CA concentrations values, but there are obvious delays in reaching the total conversion of the analyte. The 100% yield in CA transformation needed only 50 min for the initial concentration of 5 ppm, about 70 min for 10 and 15 ppm and 110 min for 25 ppm, respectively. As also noted in the other described parametric studies [30, 37] the kinetic data collected in this work fitted very well to the pseudo-first-order reaction model (Figure 6b). The computed values of the reaction rate constants have quite similar close values for the concentrations of 10, 15 and 25 ppm, while the transformation of the most diluted sample (5 ppm) occurred at an almost double rate. The mineralization yield (Figure 6c) reached, after 190 min of photocatalytic reaction, had values situated between 55% (25 ppm) to 78% (5 ppm CA). The effectiveness of the tested reaction system, with 78% mineralization and total pollutant molecule split, makes the tested systems noteworthy for practical applications.
Influence of the pollutant concentration on the reaction performance. (a) Degradation yield; (b) pseudo-first-order kinetics; (c) mineralization yield (error bars represent standard deviation from duplicate experiment). (Photocatalyst dose 0.2 g/L) Regression equations: 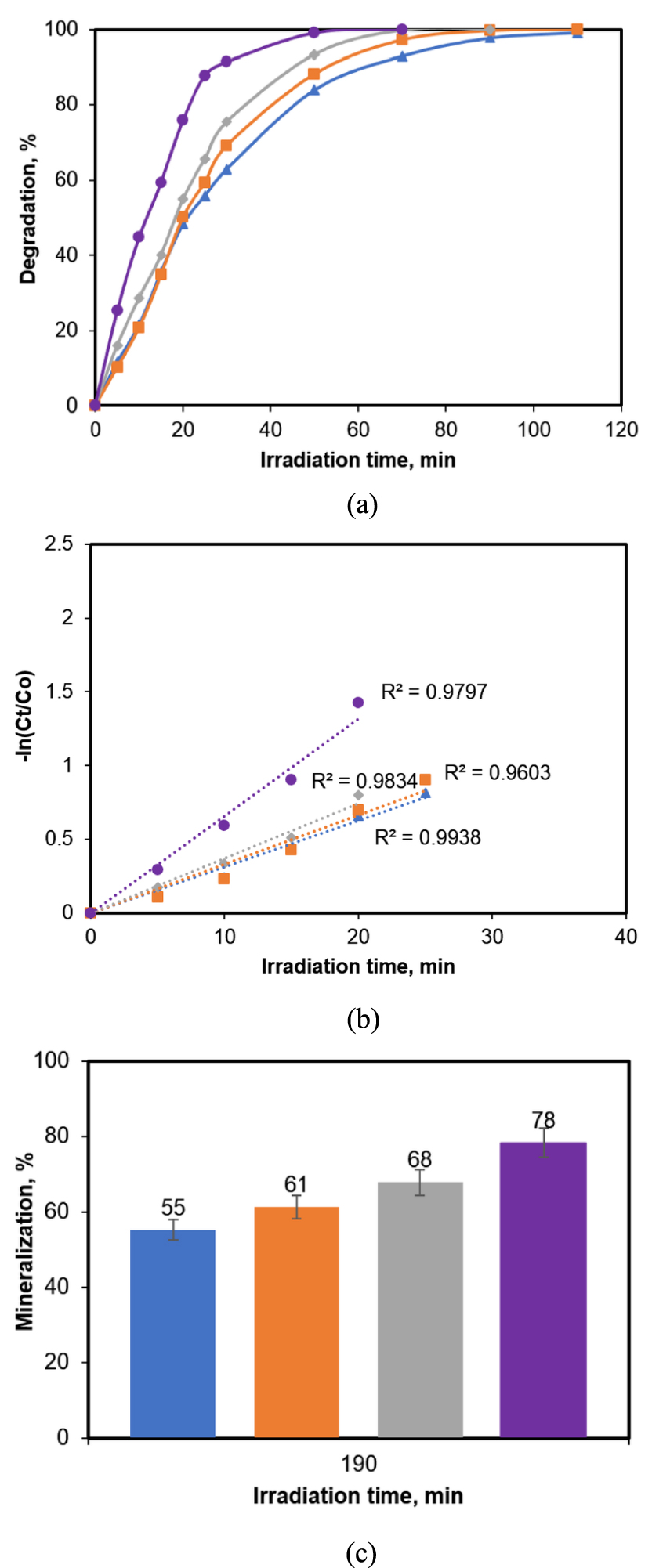
 25 mg/L (y = 0.0345x);
25 mg/L (y = 0.0345x);  15 mg/L (y = 0.0333x);
15 mg/L (y = 0.0333x);  10 mg/L (y = 0.0372x);
10 mg/L (y = 0.0372x);  5 mg/L (y = 0.0659x).
5 mg/L (y = 0.0659x).
3.5. Evaluation of the effect of water matrix
All the photocatalytic results discussed so far were performed in aqueous solutions prepared by using ultrapure water. For a potential practical application however, it is crucial to evaluate the viability of the photocatalytic treatment over the TiO2_Pd under experimental conditions closer to real ones. Hence, we have also examined the degradation of CA in TW and WW samples spiked with CA at 5 mg/L. For these investigations, 0.2 g/L of catalyst was used. The collected results for the degradation efficiency in the considered water matrices as well as the one obtained in ultrapure water (UPW) are illustrated in Figure 6. Accordingly, the highest degradation efficiency was achieved in UPW (99%) after 50 min of reaction, while only 47% and 36% of elimination was obtained for the same reaction time in WW and TW, respectively. In their study, Pourakbar et al. [38] reported a lower degradation for amoxicillin (AMX) in TW than in distilled water [38]. In addition, a similar trend was reported for the case of a secondary effluent spiked in AMX subjected to photocatalysis [35].
Effect of water matrix on CA degradation by TiO2_Pd in the UV-A photocatalytic process. (a) Degradation efficiency; (b) analysis of the experimental data for CA degradation according to pseudo-first-order kinetic model; (c) mineralization (error bars represent standard deviation from duplicate experiment). ([CA] = 5 mg/L, 0.2 g/L photocatalyst). Regression equations: 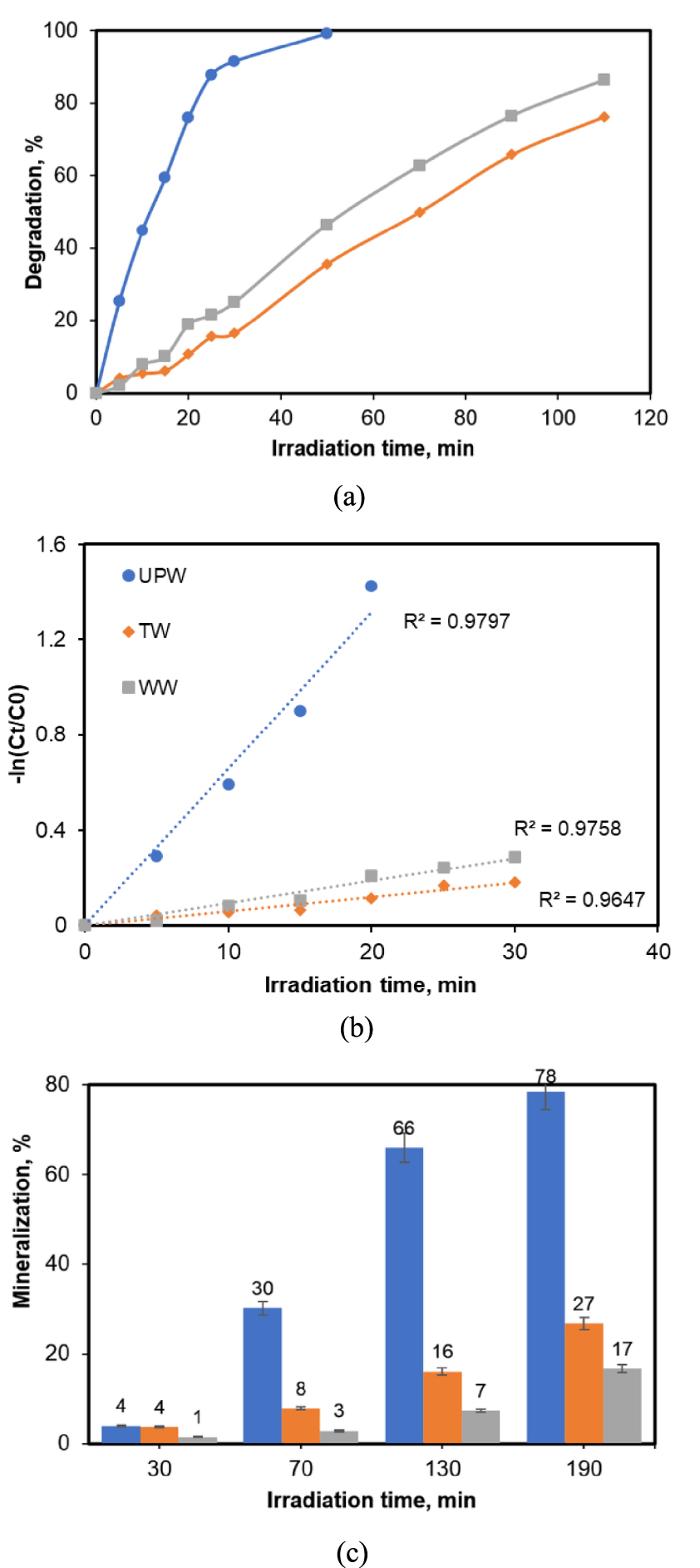
 UPW (y = 0.0659x);
UPW (y = 0.0659x);  TW (y = 0.006x);
TW (y = 0.006x);  WW (y = 0.0094x).
WW (y = 0.0094x).
Also, the degradation kinetics of CA in real water samples over the UV-A photocatalytic process were analysed using the equation (3) and the results are depicted in Figure 6b. For all investigated conditions, the experimental data are well described by the pseudo-first kinetic model. As shown in Figure 6b, the calculated apparent rate constant decreases as follows: UP > WW > TW, confirming the results of the extent of pollutant elimination presented above. Furthermore, Figure 6c clearly shows the effect of water matrix on the mineralization of the target molecule after different reaction durations. As expected, from this figure a reduction of the mineralization degree was observed in both investigated real water samples compared to that achieved in ultrapure water.
A possible explanation of the observed variations in the photocatalyst performance in the case of the real water samples is that the organic matter naturally present in these samples competes with the pollutant molecule for the adsorption active sites present on the catalyst surface, thus reducing the photocatalytic activity [35, 39]. Other authors have also reported that the recalcitrant organic matter present in natural waters has a negative effect on the degradation rate, because this source of organic carbon competes with the molecules of pollutant for the hydroxyl radicals or other oxidative species [40]. These oxidative species are recognized to have a high activity in oxidizing various organic pollutants enabling their photodegradation [16].
3.6. Effect of anions
Since real water samples often contain inorganic anions, the evaluation of their influence on the photocatalytic degradation of CA is of particular importance. Generally, anions are reported to affect potentially the efficiency of treatments using reactive oxygen species (ROS) or the electromagnetic spectrum. Hence, in this study we focused our attention on the evaluation of the effect of some anions (, ) on the performance of the photocatalytic process. Figure 7 depicts the results for the role of some anions on the photodegradation of the target molecule by the TiO2_Pd system. The experiments were conducted individually in aqueous solution for each anion at a concentration of 1 mM. The collected results for the CA degradation are presented in Figure 7a. It was found that the presence of anions in aqueous solution leads to a decrease of the pollutant degradation efficiency. Under these conditions, CA is almost totally removed after 100 min of UV-A light exposure. In the un-spiked solution, the pollutant elimination rate is much higher and the complete removal was achieved after 50 min of irradiation. It should be noted that the greatest reduction in the CA removal was reached in the presence of phosphate ions which is in line with previous studies [38, 41].
| (4) |
(a) Effect of inorganic ions on the CA degradation efficiency; (b) analysis of the experimental data for CA degradation according to pseudo-first-order kinetic model. ([CA] = 5 mg/L, 0.2 g/L photocatalyst). Regression equations: 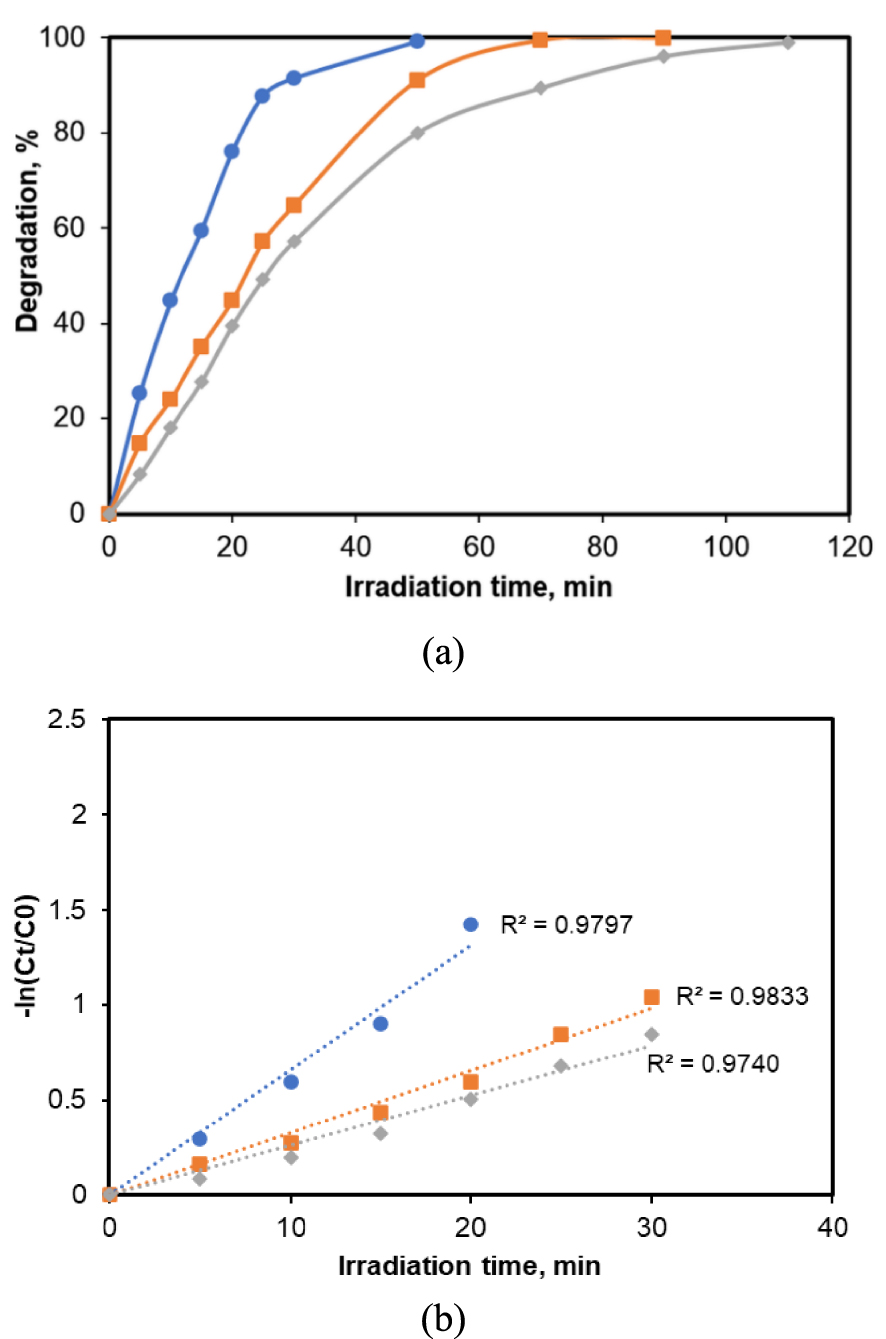
 UPW (y = 0.0659x);
UPW (y = 0.0659x);  Na2SO4 (y = 0.0327x);
Na2SO4 (y = 0.0327x);  Na3PO4 (y = 0.0263x).
Na3PO4 (y = 0.0263x).
Figure 7b shows the kinetic results achieved for the degradation of CA for both anions and in ultrapure water. It can be observed that the experimental data are well fitted by the pseudo-first-order kinetic model (R2 > 0.96). Also, it was found that the pollutant degradation rate decreases for the solutions spiked in anions which is in agreement with the results illustrated in Figure 7a.
3.7. Reusability
The catalysts recyclability and recovery are important criteria in the evaluation of the possibilities of using a photocatalyst in practical applications of the photocatalysis as an advanced oxidation process.
To address these issues, the photodegradation efficiency of CA in the presence of TiO2_Pd was investigated in five successive runs. In order to prove the reusability, the photocatalytic experiments were performed in the presence of the material retrieved from a photocatalytic run. After the first run consisting of 50 min of reaction under maximal irradiation, the photocatalyst was collected by filtration, washed twice with ultrapure water, dried under mild conditions and then reused in a new photocatalytic test. Figure 8 shows the results in terms of pollutant removal efficiency. Accordingly, no significant loss in the photocatalytic activity of TiO2_Pd was found after five successive cycles. For the considered series of tests, the elimination yields are as follows: 99.11, 98.91, 98.89, 97.11, 95.74, respectively. Our findings are in agreement with previous studies by Sescu et al. [20], evaluating the photodegradation of 2,4-dinitrophenol in the same catalytic system.
Performance of TiO2_Pd photocatalyst over five consecutive working cycles in the photocatalytic degradation of CA ([CA] = 5 mg/L, 0.2 g/L photocatalyst).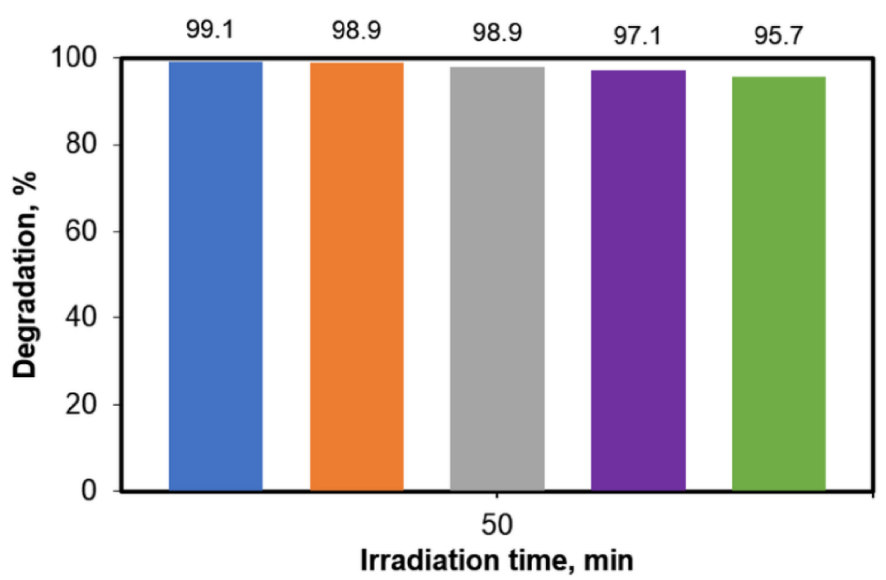
These results clearly demonstrate that TiO2_Pd catalyst shows high chemical stability in reaction and good recyclability. Hence, it is important to highlight that such characteristics qualify this material as significant in future practical water/wastewater applications.
4. Conclusions
The advanced removal of CA from aqueous solutions was investigated in the presence of several titanium dioxide commercially available photocatalysts, as well as by using a titanium dioxide sample doped with 1% palladium by the IWI technique. The TiO2_Pd sample exhibited an excellent activity at a quite low dose (0.2 g/L) and at an irradiation intensity of 6.2 mW∕cm2, i.e. the total decomposition of CA molecule after a reaction time of 50 min and a mineralization yield of 78% after a reaction time of 190 min, respectively, much better than the commercial titanium dioxide products Kronos 7000, Kronos 7500, PC500 and TiO2 from Across Chemicals.
The parametric studies performed on TiO2_Pd sample from this work highlighted the influences of the irradiation intensity, photocatalyst dose, CA initial concentration, as well as the influence of the water quality (pure water, TW and WW) and that of two salts (sodium sulfate and sodium phosphate) spiked in the reaction medium.
The degradation yields and mineralization extents decrease drastically when complex real water samples were used for the preparation of the working solutions, while the presence of phosphate and sulfate led to the decrease of the degradation reaction rate. The tests of photocatalyst reuse revealed that the values of the CA degradation remained over 95% even after five cycles of photocatalytic reaction during 50 min each.
The pseudo-first reaction order was proved to describe very well all the investigated reactions, although the rate constants vary quite much, especially depending on the irradiation intensity.
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgement
This work was supported by the publications grant of the TUIASI, project number GI/P14/2021.