


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Nanostructured molecular materials with tailored functionalities and competitive processing techniques are an area of intense interest in current material research. The monolayer and multilayer films on inorganic solids with specific interactions between the solid surfaces and the adsorbed organic species are of particular interest. The surface-based devices created by the organization of organic species on the covers of inorganic solids have been well investigated to use thin films as chemically modified electrodes. The surface of metal or metal oxide can be used for the direct deposition of layer-by-layer thin films from a solution by covalent or ionic bonds. This concept of absorption and reaction on the surface is universal and can assemble multi-component 2D or 3D film microstructures using an easy processing technique [1, 2, 3].
Preparation of modified electrodes. OTEs: optically transparent electrode, AG: anchoring, E: electrophilic building block, N: nucleophilic building block and EG: end group.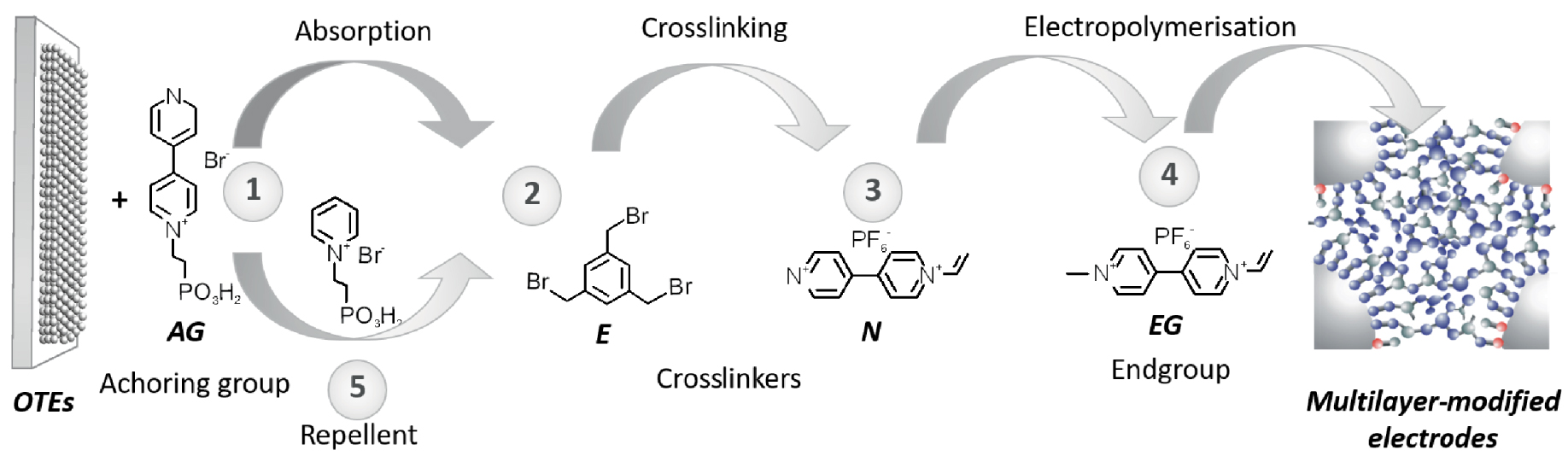
The molecularly controlled fabrication of nanostructured films has been dominated by Langmuir–Blodgett (LB) deposition for about 60 years [4]. In 1960 Kuhn et al. carried out controlled synthetic multi-composite films of organic molecules using the LB technique with donor and acceptor dyes [5]. The LB technique is limited because noncovalently attached films are not very robust; the molecules are often not firmly trapped and lost in solvents.
A LbL assembly technique with alternating physisorption of opposite charge at liquid/solid interfaces was first introduced by Decher et al. in 1991 [6, 7], giving access to multilayer films.
The multilayer film could be also formed using LbL assembly techniques including: electrostatic interaction π–π∗ stacking [8, 9, 10], hydrogen bonding [11], coordination bonding [12], charge transfer [13], molecular recognition [14, 15], adsorption/drying cycles [16], covalent bonds [17], etc. The deposition sequence of different materials defines the multilayer architecture and thus the device properties. Each deposited monolayer can enhance the deposition of the next layer. The same technique can be used with different components such as smart organic or inorganic molecules [18], colloids [19], macromolecules [20], and biological components [21].
Synthesis of the vinyl-viologen monomers, crosslinker, and repellent.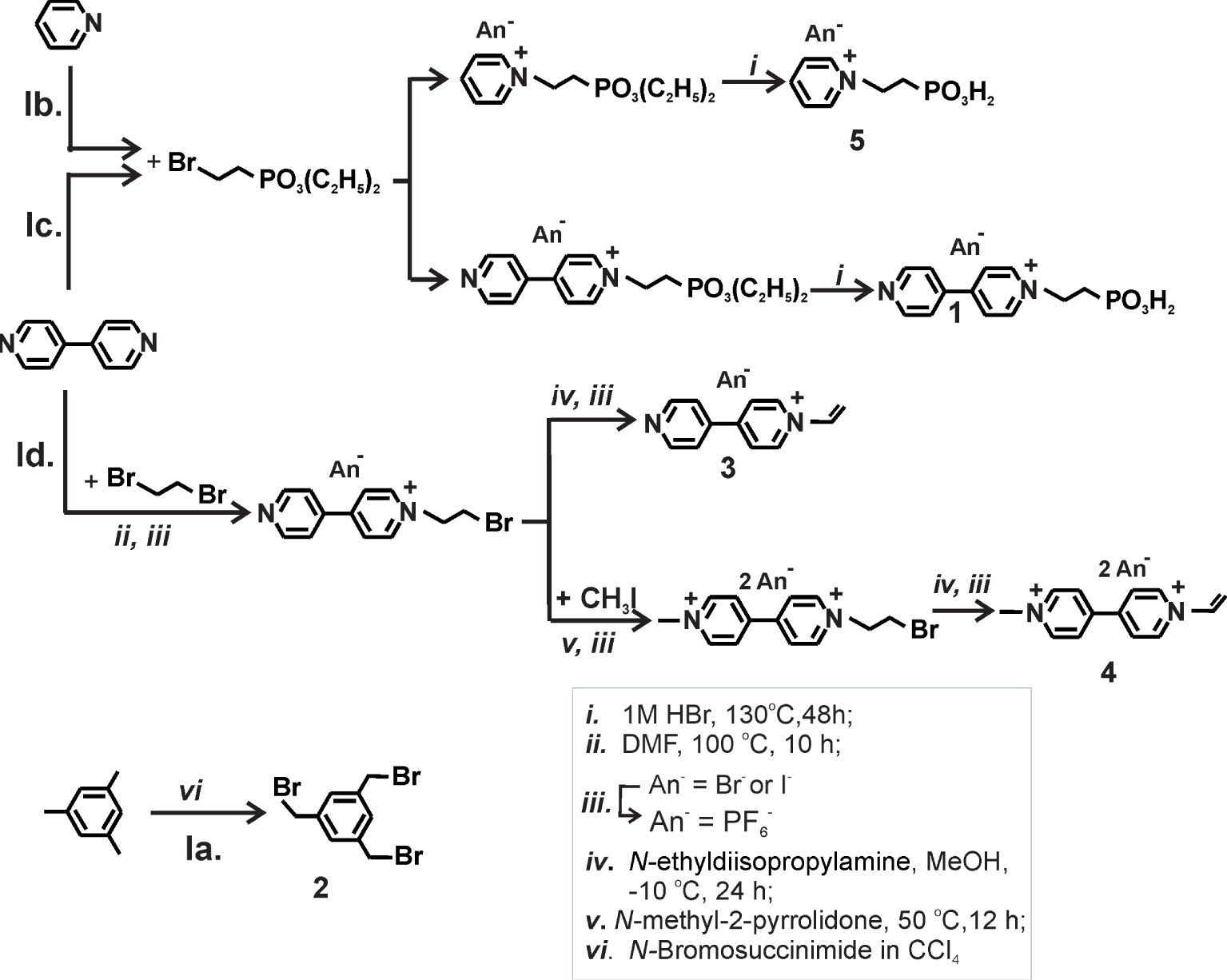
Suitable organic molecules widely utilized as electro-, thermo-, and photo-sensitive building blocks in numerous materials are 4,4′-bipyridine derivatives, known as viologens [22]. The viologens have excellent redox ability and can act as suitable electron acceptors because of Lewis acidity and π-deficiency to form charge-transfer (CT) complexes with electron-rich species [23]. The viologen (V) has three main oxidation states, namely V2+, V+⋅ and Vo. One-electron reduction yielding colorful radicals (V+⋅), i.e., cations that are stable in the absence of O2. The first reduction step is highly reversible and can be cycled many times without significant side reactions, while the second reduction state is less reversible. Change between the first reduction state (colored) and oxidation (colorless) through viologen-guest interaction or responsivity to electricity or light make viologens suitable for the fabrication of smart electrochromic devices [1, 24, 25], memory devices [26, 27, 28, 29], molecular machines [30], catalysts for hydrogen generation [31] and as dyes in the solar cell [32]. These materials have significant advantages such as a small switching voltage, a high contrast between their two-colored states, and high color purity in those states reversibility in colored or bleached.
This paper shows the principle and the application of a new approach to fabricating redox-active polymer film on mesoporous TiO2 electrodes. This new strategy involves the LbL self-assembly technique and crosslinking and “in situ” co-polymerization reactions. The first step consists of coordinating the bifunctional nucleophilic anchoring group (AG); the following steps consist of the sequential exposure of the electrodes to a counterpart solution containing electrophilic (E) or nucleophilic (N) building blocks, respectively. Finally, the co-polymerization occurs by applying a negative electrode potential in a cyclic voltammetry experiment with vinyl-monomers in solution according to Scheme 1.
The preliminary results showed growing redox species surface concentration and multilayer stability towards forced desorption in water. Further, the method was used to obtain electrochromic pictures by ink-jetting as the deposition technique of the monolayer anchoring group. The results obtained with picture-electrode occur by ink-jetted monolayer anchoring group, and crosslinking reactions confirm the efficiency and simplicity of the method for the in situ synthesis of redox polymers films fabrication. The polymers exhibit good switching in electrochromism response, attributed to their robust polymeric architecture and strong covalent linkage between electroactive film and TiO2 substrate. The method offers a practical, versatile way by choice and integration of well-matched monomers to manufacture nanostructured functional materials.
2. Experimental section
2.1. Materials
All chemicals were purchased from Merck (D-Hohenbrunn), Sigma-Aldrich, and Fluka. Solvents were of laboratory grade. UV Spectra: 8453 UV–Vis Spectrophotometer (Agilent, Germany) 𝜆max in nm (𝜀 in M−1⋅cm−1). NMR Spectra: Bruker AMX-500 spectrometer; 1H: 250 MH; chemical shifts 𝛿 are given in ppm relative to the solvent signal peaks as an internal standard for 1H.
Glass slides (69 mm × 69 mm), coated with ITO (In-doped SnO2, 20 Ω∕cm2) from BTE Bedampfungstechnik (Elsoff, Germany) or FTO glass (F-doped SnO2, 15 Ω∕cm2) from LOF were used as received.
2.2. Synthesis procedure
The monomeric 1 to 5 were synthesized according to Scheme 2 in Section 3.
2.2.1. Synthesis of 1-(2-phosphonoethyl)-4-pyridin-4-ylpyridinium bromide(1)
This synthesis follows a known procedure [33]. A suspension of 4 g (25.64 mmol) of 4,4′-bipyridyl with 14 g (57,14 mmol) diethyl 2-bromo-ethylphosphonate and in dibutyl ether (10 ml) was stirred at 50 °C for 23 h under reflux. After approximately 30 min, a white precipitate was obtained. The product was washed with dry diethyl ether and dried for 12 h in a high vacuum (HV) (14.08 mmol, 55% yields). The product, 1-[2-(Diethoxyphosphory)ethyl]-4-pyridin-4-ylpyridinium salt (5.65 g, 14.08 mmol) was heated with 170 ml of HBr (1 M, 72 h, 130 °C) under reflux. The cold mother liquor was evaporated, and the powder dried for 24 h in HV (13.33 mmol 94.7% yield). 1H-NMR: (250 MHz, D2O): 9.04 (m, 4H), 8.54 (m, 4H), 4.94 (d,3J[H,P] = 12.6 Hz, 3J[H,H] = 7.6 Hz, 2H), 2.50 (dt, 2J[P,H] = 12.9 Hz, 2J[H,H] = 7.6 2H).
2.2.2. Synthesis of 1,3,5-tris-(bromomethyl) benzene (2)
The synthesis follows a known procedure [33]. Mesitylene (20 g, 166 mmol) and 95 g (530 mmol) N-bromo succinimide (NBS) dissolved in 500 CCl4 (dried 24 h over CaCl2 and distilled) was irradiated with a 500 W-lamp for 24 h and stirred at 70 °C for 30 h under reflux. The mixture was kept at 6 °C for 24 h. The succinimide was filtered off, and the solvent evaporated. The residue was dissolved in chloroform and washed once with an aq. NaHCO3 solution and two times with H2O. The organic layer was then dried (Na2SO4), filtered, and the solvent evaporated. The oily residue was dried in HV, dissolved in CHCl3, and added petrol ether. Under these conditions, the crystallization starts. The mixture was kept for 3 days at 6 °C. Light yellow crystals were obtained, isolated, and dried for 24 h in HV (19 mmol, 12% yield). 1H-NMR: (250 MHz, CDCl3): 7.38 (s, 3H), 4.48 (s, 6H).
2.2.3. Synthesis of 4-pyridin-4-yl-1-vinylpyridinium bromide(3)
The synthesis was performed according to a literature procedure [33]. A solution of 3.2 g (20.5 mmol) of 4,4′-bipyridyl in 37.6 g (200 mmol) of 1,2-dibromoethane was stirred for 6 h at 100 °C. The resulting precipitate was filtered off and washed with ether. The yellow solid was isolated and dried for 24 h in HV (19.8 mmol, 97% yield). 1-Bromoethyl-4-pyridin-4-yl-pyridinium bromide (6.8 g, 19.8 mmol) was dissolved in MeOH (180 ml) and dropped to a solution of 2.5 g (19.4 mmol) of N-ethyl diisopropylamine in MeOH (20 ml) at − 10 °C. The reaction mixture was stirred at − 10 °C for 20 h. The pH value of the solution was controlled and kept below 10. The resulting solution was concentrated to about 50 ml and dropped into 250 ml of ether. The precipitate formed was filtered off and dissolved in 50 ml of MeOH. The precipitation procedure was repeated once to remove N-ethyl diisopropylamine hydrobromide salt. At last, the precipitate was filtered off, washed with ether, and dried in HV. The product was purified by chromatography (Sephadex LH-20, column: 35 × 3.5 cm, MeOH as eluent) to give a slightly colored product (3.7 g, 14.4 mmol, 73% yield). 1H-NMR (250 MHz, D2O): 9.00 (d, 3J[H,H] = 5.0 Hz, 2H), 8.67 (d, 3J[H,H] = 5.0 Hz, 2H), 8.37 (d, 3J[H,H] = 7.5 Hz, 2H), 7.83 (d, 3J[H,H] = 7.0 Hz, 2H), 7.46 (d,d, 3J[H] = 15 Hz, –CH =, 1H), 6.13 (d,d, 3J[H] = 15 Hz, =CH trans, 1H), 5.77 (d,d, 3J[H] = 7.5 Hz, =CH cis, 1H).
2.2.4. Synthesis of 1-methyl-1′-vinyl-4,4′- bipyridinium dihexafluorophosphate(4)
The synthesis was performed according to a literature procedure [33]. 1-Methyl-4-pyridin-4-yl-pyridinium iodide (6 g, 20 mmol) (also prepared according to the procedure already described) and 40 g (95 mmol) of 1,2 dibromoethane in DMF (80 ml) was heated for 10 h at 100 °C. The resulting precipitate was filtered off and washed with ether. After drying in vacuo (r.t., 24 h) 7.5 g (17.8 mmol, 89% yield) of 1-methyl-1′-(2-bromoethyl)-4-pyridin-4-yl-pyridinium bromodiodide was obtained. 1-Methyl-1′-(2-bromoethyl)-4-pyridin-4-yl-pyridinium bromidiodide (7.5 g, 17.8 mmol) was dissolved in MeOH (200 ml) and dropped slowly at − 10 °C to 1.5 ml of an aq. NaOH solution (10 M). After 13 h, the solution was adjusted to pH = 5 with 48% HBraq solution, and then the temperature was raised to 20 °C. The solvent was evaporated, and the crude product purified by chromatography (Sephadex LH-20, column: 35 × 3.5 cm, MeOH as eluent) to give 3.8 g (9 mmol, 50% yield) of 1-methyl-1′-vinyl-4,4′-bipyridinium salt as a yellow powder. 1-Methyl-1′-vinyl-4,4′-bipyridinium salt (3.8 g, 9 mmol) was dissolved in 20 ml of water, and 10 ml of a 10% NH4PF6 solution was added dropwise. The solid was filtered off, washed with cold water, and dried for 24 h in HV. 3.82 g (7.8 mmol, 44% yield) of 1-methyl-1′-vinyl-4,4′-bipyridinium hexafluorophosphate as the light yellow powder was obtained. 1H-NMR (250 MHz, CD3CN): 9.0 (d, 3J[H,H] = 7.0 Hz, 2H), 8.89 (d, 3J[H,H] = 7.0 Hz, 2H), 8.49 (d, 3J[H,H] = 7.0 Hz, 2H), 8.42 (d, 3J[H,H] = 7.0 Hz, 2H), 7.55 (dd, 3J[H] = 15 Hz, –CH =, 1H), 6.31 (d,d, 3J[H] = 15 Hz, =CH trans, 1H), 6.24 (dd, 3J[H] = 7.5 Hz, =CH cis, 1H), 4.43 (s, 3H).
2.2.5. Synthesis of 1-(2-phosphonoethyl)pyridinium bromide(5)
The synthesis was performed according to a literature procedure [33]. Pyridine (2 g, 25.3 mmol) was reacted with 7 g (28.6 mmol) diethyl-2-bromo-ethyl phosphonate at 50 °C for 23 h. The resulting precipitate (2 g, 25.3 mmol) was collected by filtration, washed with diethyl ether, and dried in vacuo at r.t. for 16 h yielding 5.6 g (17.3 mmol) of the corresponding diethyl ester as a white powder (68.3%). The total amount of the intermediate was subjected to hydrolysis in 180 ml of 1 M HBr at 130 °C for 48 h. After evaporation of the solvent, the resulting oil was dried for 48 h in vacuo. The crude product was dissolved in 100 ml MeOH and precipitated by the addition of diethyl ether. 1-(2-Phosphonoethyl) pyridinium bromide was isolated and dried in vacuo to yield 3.4 g (12.7 mmol, 73.4% yield). 1H-NMR (250 MHz, D2O): 8.83 (d, 3J[H,H] = 6.4 Hz, 2H), 8.49 (t, 3J[H,H] = 7.5 Hz, 1H), 8.01 (t, 3J[H,H] = 7.5 Hz, 2H), 4.81–4.73 (m, 2H), 2.43–2.30 (m, 2H).
“in situ” Synthesis of TiO2 electrodes with polymerizable viologen unit.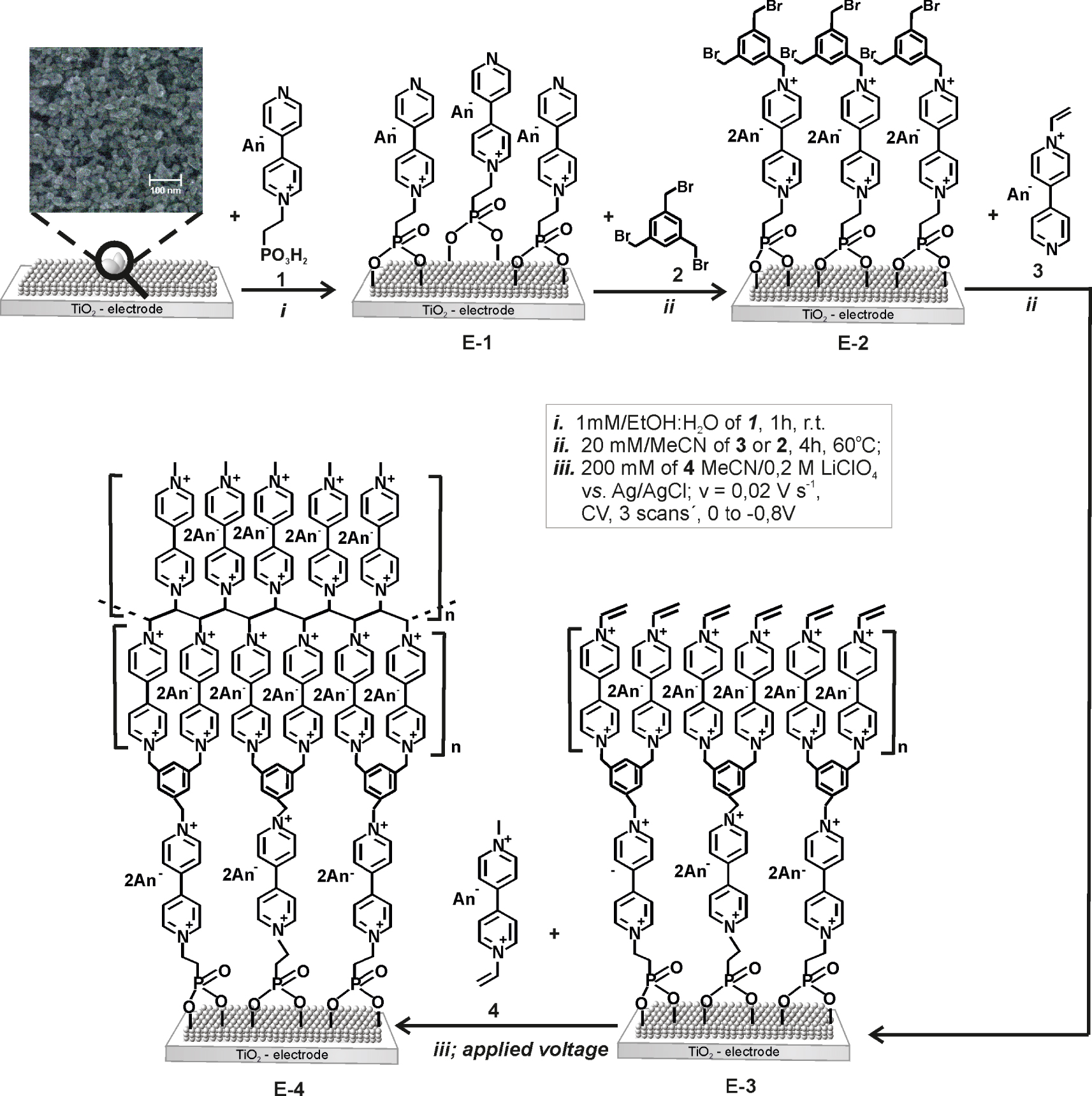
2.3. Preparation of the working electrode
2.3.1. In-situ synthesis of 1,1′-[(5-{[1′-(2- phosphonoethyl)-4,4′-bipyridinium-1-yl]methyl}-1,3-phenylene)bis(methylene)]bis(1′-vinyl-4,4′-bipyridinium) hexabromide (E-3)
Colloidal TiO2 [34] paste was coated on 10 mm × 10 mm FTO glass electrodes (F-doped SnO2, 15 Ω∕cm2) from LOF using the doctor-blade method to yield 5 μm thick films [32]. The glasses were cleaned according to the method reported by Hillebrandt et al. [35] before coating.
Step 1: TiO2 plates were exposed to the anchoring electrophilic 1-[3,5-bis(bromomethyl)benzyl]-1′-(2-phosphonoethyl)-4,4′-bipyridinium dibromide (1) 1 mM in ethanol/water (1:1 vol.%)] for 1 h at 20 °C (E-1).
Step 2: The electrode was then exposed to compound 2 (20 mM in MeCN) solution 4 h at 60 °C (E-2).
Step 3: Further the electrodes was treated with 4-pyridin-4-yl-1-vinylpyridinium bromide 3 (20 mM in MeCN) for 4 h at 60 °C, to produce the surface-confined compound 1,1′-[(5-{[1′-(2-phosphonoethyl)-4,4′-bipyridinium-1-yl] methyl}-1,3-phenylene) bis (methylene)] bis (1′-vinyl-4,4′-bipyridinium) hexabromide (E-3).
Step 4: The vinyl group layer on the electrode surface was co-polymerized with 200 mM solution of 4 in MeCN/0.2 M LiClO4 by applying a negative electrode potential between 0 to −0.8 V vs. AgCl using three scans in cyclic voltammetry to yield the E-4 according to Scheme 3.
The electrochromic picture was prepared by inkjet printing of 0.1 M compound 1 (electrochromic anchoring group) on a TiO2 coated ITO-electrode according to the method described by Möller and Asaftei [36]. To protect the “white regions”, the printed electrodes were exposed to a 1 mM solution of repellent compound 5 in isopropanol, 1 h at r.t. to yield E-1 as a picture. After deposition of the anchoring monolayer, the electrode was prepared in the same way as described above (see steps 2–4).
2.4. Electrochemical characterization
Spectroelectrochemical measurement, cyclic voltammetry, and square voltage excitations experiments were performed under argon in a three electrode electrochemical cell with the potentiostat PGSTAT 302N from AUTOLAB controlled by a P.C. running under GPES from Windows, version 4.9 (ECO Chemie B.V.). TiO2 modified electrodes with an electrochemically active surface area of A = 1 to 4.9 cm2 were used as a working electrode. The reference electrode was Ag/AgCl (3 M, KCl in water) and Ag wire, respectively. Counter electrodes were Pt wire and ITO–glass plate (In-doped SnO2, 20 Ω∕cm2) from Bedampfungstechnik (Elsoff, Germany).
3. Results and discussion
The compounds 1 and 5, respectively, have been prepared from 4,4′-bipyridine and 2-bromoethane-diethyl phosphonate via an SN 2 mechanism in a Menschutkin-type reaction. Under these conditions, double alkylation was excluded. Solvolysis of diethyl phosphonate derivatives was achieved by refluxing 48 h in 1 M HBr to yield compounds 1 and 5 as hygroscopic bromides.
The electrophilic 3-fold crosslinker 2 was synthesized by a radical allylic bromination from mesitylene with N-bromosuccinimide and light as a catalyst in CCl4 (low yield 11%) [37]. The monomers 3 and 4, used further in the electropolymerization reaction, were obtained in two reactions steps. The first step consisted of the quaternization of 4,4′ bipyridine with an excess of 1,2-dibromethane. The elimination of HBr was occurred under base conditions by N-ethyl diisopropylamine/methanol to form the N-vinyl bipyridinium monocation 3 according to the procedure described in the literature [37, 38]. The synthesis ways are presented in Scheme 2.
The synthesis of the anchoring group with alkyl phosphonic acids on one side and the vinyl group on the underside of the viologen unit in solution was unsuccessful. The phosphonic acids group provide acts as a catalyst and promotes the polymerization of vinyl groups during the final eliminations step of the synthesis.
The way to obtain the 1,1′-[(5-{[1′-(2-phosphonoethyl)-4,4′-bipyridinium-1-yl]methyl}-1,3-phenylene)bis(methylene)]bis(1′-vinyl-4,4′-bipyridinium) hexabromide E-3 and E-4 respectively were the in situ synthesis directly at the electrode surface by LBL technique and crosslinking reaction (Scheme 3).
In the last synthesis step, co-polymerization is achieved using galvanostatic (constant current), galvanodynamic (pulsed current), potentiostatic (constant potential), or potentiodynamic (cyclic voltammetry (CV) or pulsed potential) methods. For the co-polymerization was chosen the potentiodynamic cyclic voltammetry experiment with compound 4 in MeCN/0.2 M LiClO4. The three-electrode system comprising working E-3, counter Pt wire, and reference Ag/AgCl was applied negative electrode potential between 0 to −0.8 V. After three scans are, the electropolymerization finished because no significant increase of peak currents reduction was observed on successive cycles (Figure 1).
Cyclic voltammograms of E3 with 3 mM of monomer 4 in 0.2 M TBABr/MeCN, scan rate v = 0.02 V⋅s−1.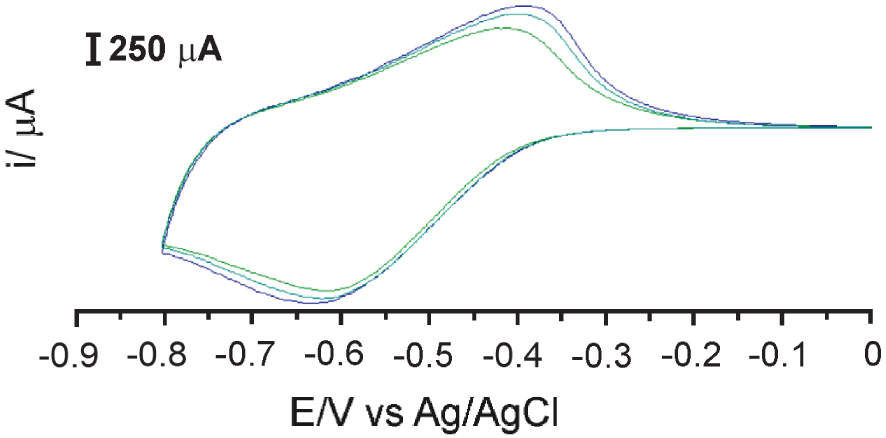
Electrochemical characterization of electrodes E1 to E-4 was performed to regard the influence of the crosslinking steps on surfaces concentration of redox species, stability, and switching in electrochromism response. The electrodes were checked for their quality by absorbance, switching time, and stability.
Figure 2 shows the typical cyclic voltammogram of 3 mM of monomer 4 in 0.2 M tetrabutylammonium hexafluorophosphate (TBAPF6) in acetonitrile obtained on a glassy carbon electrode after the first scan. Two pairs of redox couples appeared at ca. −0.3 V and −0.65 V, respectively. They are ascribable to the well-known reversible one-electron transfer reaction of viologen.
Cyclic voltammograms of 3 mM solution of monomer 4 in 0.2 M TBAPF6/MeCN at glassy carbon electrode (aria ca. 0.0680 cm2), scan rates v = 0.1 V⋅s−1 (a) and one-electron redox process of viologen (b).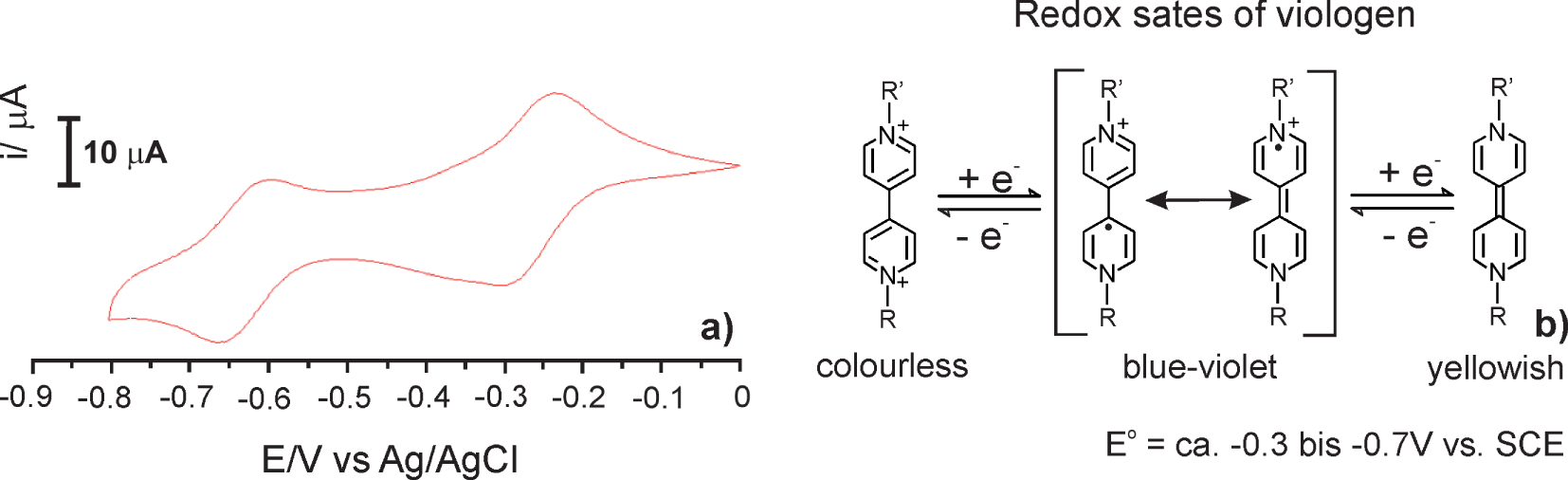
The electropolymerisation of 3 mM of compound 4 on GC and TiO2 electrodes was checked by cyclic voltammograms between 0 to −0.9 V in 0.2 TBABr/MeCN at 20 °C with 0.1 V⋅s−1 scan rate. After polymer deposition, the electrodes were rinsed and immersed in the fresh electrolyte, and a cyclic voltammogram was taken. Figures 3a and c show the viologen-film formation on GC and TiO2 electrode, respectively, and Figures 3b and d show the first redox peak of the formed polymer into the pure electrolyte.
Cyclic voltammograms: (a) 3 mM of 4 in 0.2 M LiClO4/MeCN at GC electrode, (b) in pure electrolyte after polymer deposition, (c) 3 mM of 4 in 0.2 M in 0.2 M TBABr/MeCN at TiO2 electrode (aria ca. 1 cm2), (d) in pure electrolyte after polymer deposition. Scan rates for (a) and (c) v = 0.1 V⋅s−1 and for (b) and (d) v = 0.02 V⋅s−1.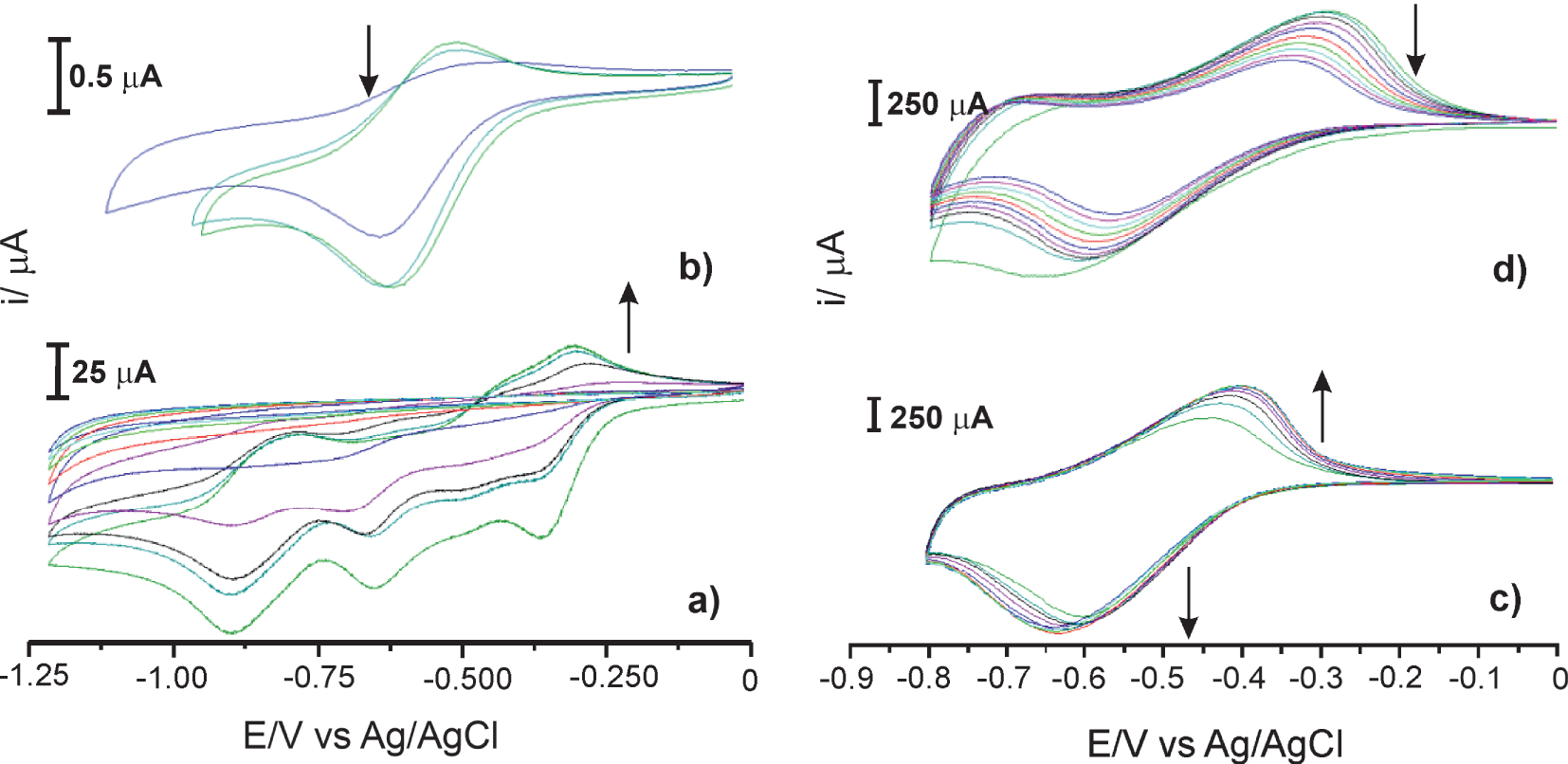
The gradual increase in the cathodic peak currents after each cycle in the CV polymerization curves demonstrates that polymer films are successfully electrodeposited on both electrodes. The second peak appears on the volumetric curve (Figure 3a), corresponding to a two-electron reduction of viologen sites. This deeper reduction of viologen film causes a rapid decrease of the current peak in subsequent polarization cycles after ca. five scans. The inactivation of the electrode finally leads to the I–E curve corresponding to the residual current on an unmodified electrode. These results indicate that the desorption of viologen film does not cause the inactivation of the electrode (see Figure 3b). Two-electron reduction of viologen leads to loss of the reversibility of redox reactions of the polymer viologen groups. For this reason, the electropolymerization experiments were only carried out in the first redox state of the viologen monomer. The current peak of polymer film on GC and TiO2 electrodes measured in pure electrolyte, decreases with the number of scans because it is not attached to the surface.
The stability of polymer in the mesoporous electrode can be achieved by LbL cross-linking reaction and co-polymerization. The preparation of homogeneously coloring working electrodes with TiO2-coordinating groups such as phosphonic, salicylic, or benzoate acid functionality was described by Campus et al. [32]. It was not clear if electrochromic materials can be deposited without reversible diffusion into the solution. This phenomenon reduces the concentration of redox species on the electrode surface. A molecular cross-linking reaction can be used to prevent the diffusion of viologen monolayer in the solution. The crosslinking takes place in a singular or multi-step reaction. The process i.e., involves a cascade reaction and takes place in a solvent, where the migration of anchoring material is suppressed. This procedure has the following advantages: limited desorption of monolayer and amplified the concentration of redox species on the surfaces; fixation of the compound applied on the surface; formation of 3D branched structure or hybrid materials.
The effect of the cascade cycles on electrochemistry is shown in Figure 4. The electrochemical response of the redox species in polymer film on the surface grows linearly with the number of cascade cycles. The current peak in CV increases almost by ten times with each reaction step.
Cyclic voltammograms of E1 to E4 TiO2 electrodes. Inset: blanc TiO2 electrode. The electrodes were measured in 0.2 LiClO4/MeCN vs. Ag/AgCl at v = 0.02 V⋅s−1.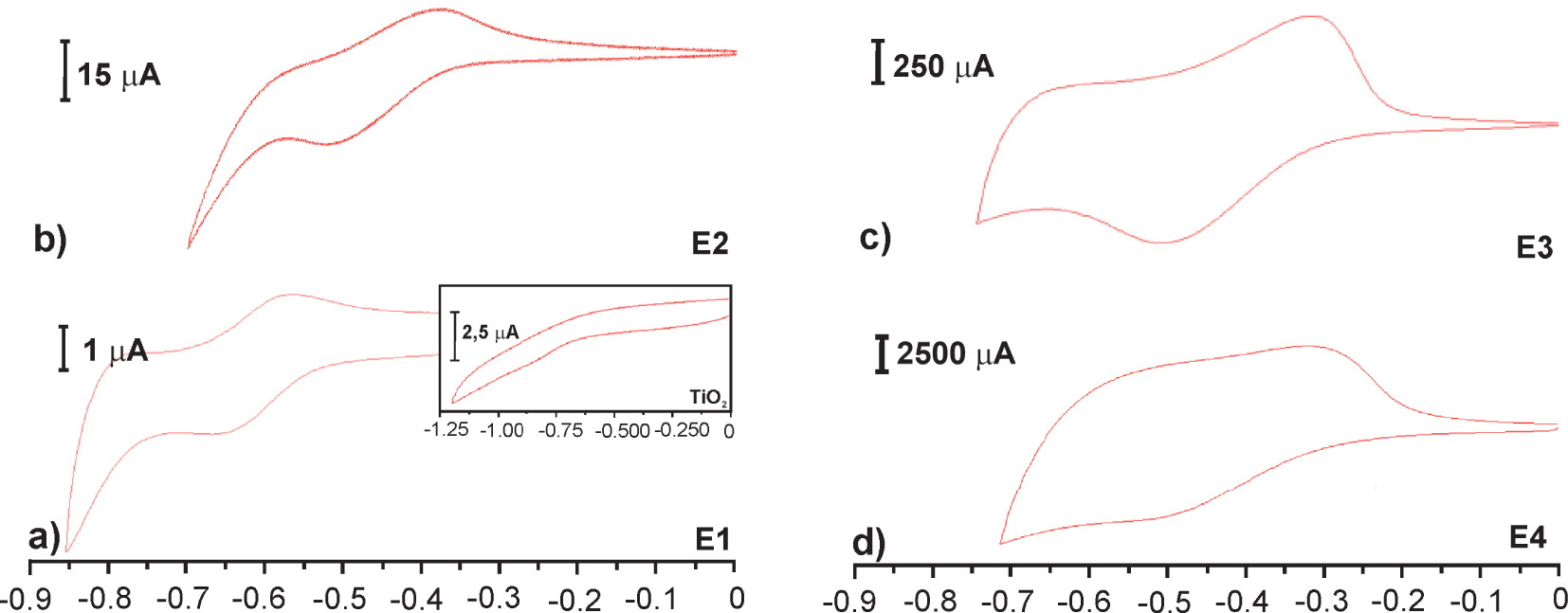
The UV–VIS results of modified electrodes (E-1 to E-4) in the reduced state show that the viologen monolayer absorption is weak because the bipyridine units are not closed. The crosslinking steps increase absorbance intensities by up to 6-fold at 720 nm. After the third reaction step, the absorbance intensities curve flattens out, and a maxim of the concentration of redox species on the electrode is reached (Figure 5).
Influence of the cascade reaction numbers on the absorbance at 740 nm in a three-electrode system, 1 M LiClO4/MeCN vs. Ag/AgCl, Pt-wire, at −0.7 V.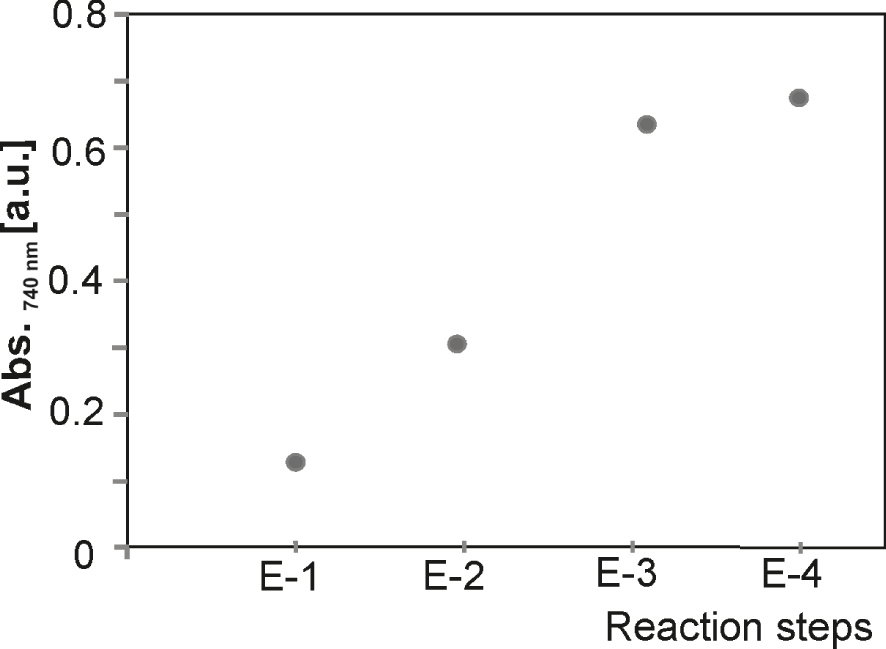
Absorption of viologens modified electrodes E1 to E4 after synthesis and after the accelerated aging process at 740 nm in a three-electrode system in 1 M LiClO4/MeCN vs. Ag/AgCl, Pt-wire counter electrode at −0.7 V.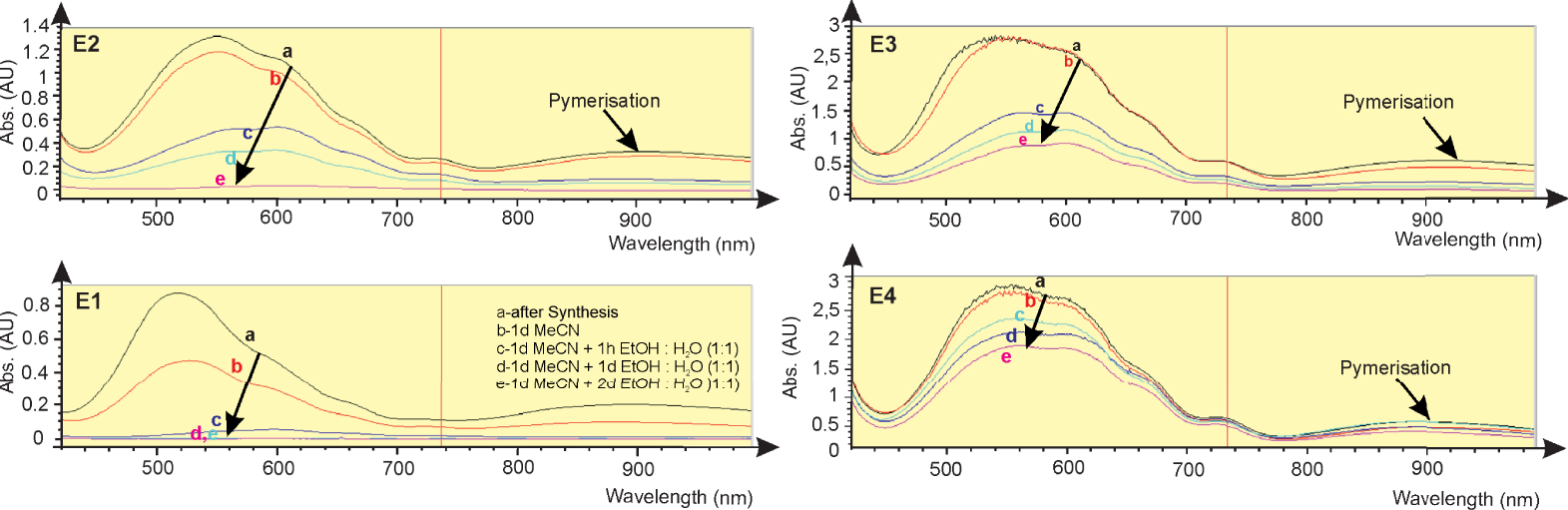
Influence of the cascade reaction numbers on the relative stability towards forced desorption.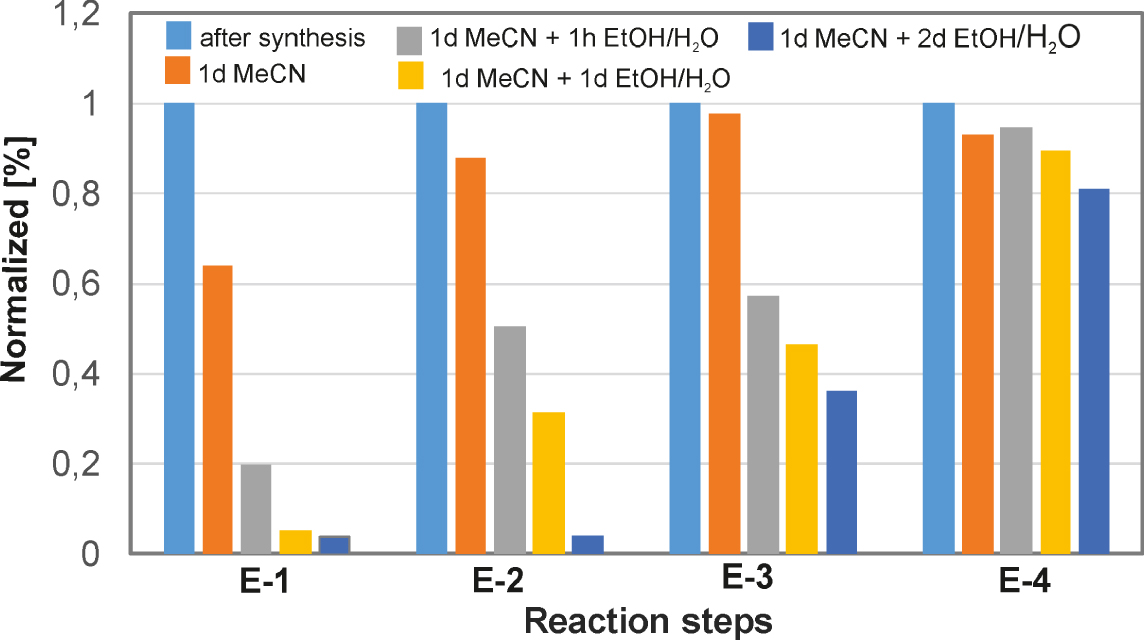
Electrode (a) after synthesis and (b) after aging process one day in H2O.
Absorption in the three electrodes system after equilibration.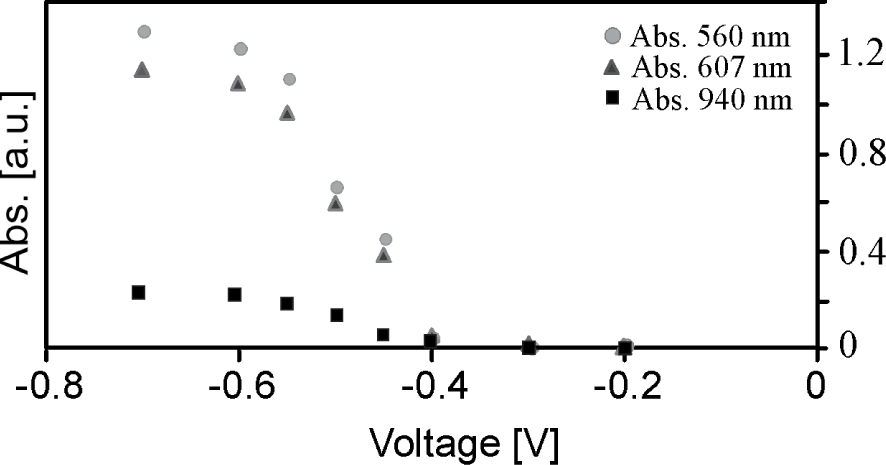
The effect of the number of the crosslinking reaction steps on the surface concentration 𝛤 is shown in Table 1.
Surface concentration (mol⋅cm−2) of redox layers on TiO2
| 𝛤 × 10−7 (mol⋅cm−2) at 𝜆 = 740 nm, 𝜀 = 3200 | |||
|---|---|---|---|
| E-1 | E-2 | E-3 | E-4 |
| 0.40 | 0.86 | 1.98 | 2.1 |
The values are obtained using 𝛤 = abs.740 nm∕𝜀 ⋅ 1000 with abs. = absorbance at 740 nm and 𝜀 = extinction coefficient (dm3⋅mol−1⋅cm−1). The concentration of redox species increases minimally with the last reaction step, which shows that the saturation degree of redox species was reached after three reaction steps. But the polymerization step is important for the stability of electrodes.
In another set of experiments, the same electrodes were subjected to an aging test to check their stability using UV–Vis spectroelectroscopy (Figure 6).
The electrodes have been sequentially rinsed in acetonitrile and water–ethanol (1:1 vol.%) for several hours up to days. Such a treatment releases most of the dye if it is only present as an anchored monolayer.
In this case, normalized surface concentrations of redox species were used on electrodes. The sample anchored modifier decays to approximately 2 percent of its initial surface concentration during this treatment. Still, the cross-linked electrodes E-3 and E-4 withstand the accelerated aging, generally showing more than 50% activity at the end of the experiment. The persistence is much better in the cross-linked state. The results are shown in Figure 7.
The new concept of in situ syntheses of multilayer electrochromic film on mesoporous TiO2 combined with co-polymerization was studied on picture ink-jetted electrodes.
The picture on the TiO2 surface was prepared with ink-solution containing anchoring compound 1 according to the method described by Möller and Asaftei [36]. Ink-jetted TiO2 electrodes were treated with a solution of a repellent 5 before each was crosslinked with 2, 3, and electropolymerization steps to suppress the absorption of viologen monomers in “white areas”. A good repellent is, i.e., pyridinium ethyl phosphonic acid 5. The aging tests showed that crosslinking reaction leads to the formation of anchored multilayers in the TiO2 electrode, thereby increasing stability for both approaches. The picture in Figure 8 is still present after accelerated aging one day in the water.
Figure 9 shows the absorption of the working electrode in the 3-electrode system of the printed electrode presented in Figure 8 using an ITO-glass as a counter electrode and a silver wire as a reference electrode as a function of the applied potential. The working electrode was prepared according to the crosslinking procedure described in Scheme 3.
Absorbance responses of a three-electrode system with (a) five, (b) ten, and (c) twenty seconds square voltage steps (0 to −0.7 V) and (d–f) reduction–oxidation hysteresis.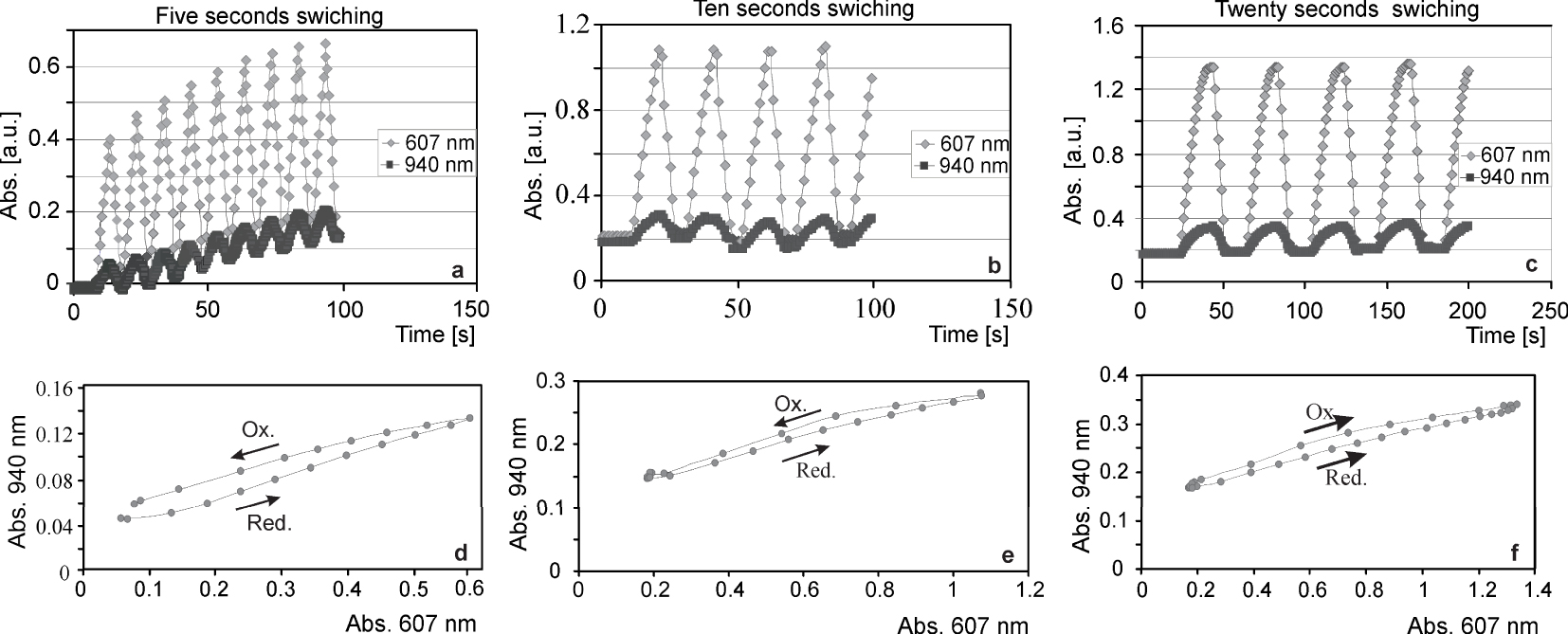
The absorbance vs. voltage shows a typical Nernstian behavior and a maximum absorbance at 560 nm of ca. 1.35, i.e., an excellent dynamic behavior. The absorbance at 607 nm shows a minor contribution of pymerization; the coupling of two radicals in a reduction state. A typically pymerization peak is present in UV–Vis spectra at 940 nm.
Square voltage excitation experiment (Figure 10) shows the absorbance responses of the three-electrode system with 5, 10, and 20-second square voltage steps. The absorbance vs. time plots shows that in the system with 10-s excitation, the absorbance changes are almost doubled compared to the 5-s excitation. With a 10-s excitation (Figure 10b) the absorbance changes grow to approx. 1.1 at 607 nm and with 20-s excitation (Figure 10c) an absorbance change of ca. 1.3 was reached.
It is interesting to observe a time-dependent change in the pymerization that results in a hysteresis when the absorbance at 607 nm (slightly influenced by pymerization) is plotted vs. the absorbance at 940 nm (exclusively pymerization). In practice, this results in a fast change to blue when the electrode is reduced, followed by a slow build-up of the violet tint due to pymerization. When oxidized, first, the blue tint disappears, and later the violet tint disappears. The counter electrode does not influence the kinetics. However, it is not sure if it got the best results regarding the electrolyte/solvent system.
It can be concluded that working electrodes with excellent stability and high concentration of redox species using in situ synthesis and electropolymerization have been achieved. A microscope check of the electrode shows no lateral diffusion of redox species as in the monolayer printed electrode.
4. Conclusions
The starting point of this work was the fabrication of redox-active polymers film on mesoporous TiO2 electrodes by a new approach. The strategy involves the top-down path (adsorption) combined with bottom-up layer-by-layer (LbL) crosslinking and co-polymerization reactions in situ. Derivative consists 4,4′-bipyridinium salts, so-called viologen with an anchoring phosphono ethyl acid and the vinyl group as building blocks molecules, and 1,3,5-tris-(bromomethyl) benzene as crosslinker were synthesized and characterized. Molecular entrapping of the oligomeric layer and co-polymerization prevents loss of sharpness due to entropy-driven broadening of the absorbed redox species. The effect of crosslinking reaction in situ and co-polymerizations steps lead to amplification of redox species on the surface concentration 𝛤. The preliminary aging tests indicate good long-term stability. The results obtained with picture-electrode occur by ink-jetted monolayer anchoring group, and crosslinking reactions confirm the efficiency and simplicity of the method for the in situ synthesis of redox polymers films fabrication. The polymers exhibit good switching in electrochromism response, attributed to their robust polymeric architecture and strong covalent linkage between electroactive film and TiO2 substrate. The method offers a practical, versatile way by choice and integration of well-matched monomers to manufacture nanostructured functional materials.
Conflicts of interest
The author has no conflict of interest to declare.