


 CC-BY 4.0
CC-BY 4.0
1. Introduction
La famille des semi-conducteurs connue sous le nom d’halogènes pnictures de cadmium et de mercure suscite encore un grand intérêt dans la recherche des matériaux pour leurs caractéristiques structurelles particulières, et leurs propriétés électroniques distinctives.
Ainsi, des composés présentant de bonnes propriétés optiques et électriques sont répertoriés dans la littérature [1, 2, 3, 4, 5, 6, 7, 8, 9]. A ce jour, plus d’une trentaine de ces composés ternaires (MaZbXc) (voir Tableau S1 et Figure S1) et quaternaires (MaZb) (Ma′Xc) [M∕M′ = Hg, Cd; Z = P, As, Sb; X = Cl, Br, I] ont été découverts. Les données principales de leurs structures cristallines sont résumées dans trois revues principales [10, 11, 12].
La principale caractéristique de ces composés polyanioniques est la présence de liaisons anion–anion (Z–Z), la structure de base de ces composés est ainsi catégorisée selon les différents types de fragments des atomes pnictogène Z : (1) Anions Z3− séparés. (2) Anions binucléaires . (3) Bianions Z3− and . (4) Anions triangulaires. (5) Anions Z− unidimensionnels infinis.
La majorité des composés halogénopnictures de mercure contiennent des atomes pnictogène de type binucléaire dans leur charpente hôte, ceux qui contiennent les autres types sont relativement rares. Cet assemblage peut donner naissance à des charpentes de géométrie et de topologie différentes. En effet, les charpentes supramoléculaires qui sont typiques pour les atomes de mercure du fait de leur coordination linéaire sont rarement observées pour les atomes de cadmium [12]. Dans tous les cas, la charge positive de la charpente qui est composée de cations des métaux de transition et des atomes pnictogène comme des lieurs, est compensée par celle des anions hôtes.
TiO2 est le semi-conducteur le plus utilisé en photocatalyse pour la dégradation d’une large gamme de composés organiques, en raison de son faible coût, de son efficacité et de sa grande stabilité photochimique. Toutefois le vrai obstacle à son application étant sa large bande interdite (band gap) qui ne lui permet d’absorber que la lumière UV (𝜆 < 100 nm), qui ne présente qu’une faible portion (4%) du spectre solaire, contrairement à la lumière visible (43%). Dans ce but, de nombreuses recherches sont menées pour le développement de nouveaux photocatalyseurs ayant une grande activité dans le visible.
En plus de leurs structures cristallines avec des géométries architecturales diverses, les composés chalcogénures métalliques possèdent aussi des bandes interdites optiques étroites, les rendant ainsi susceptibles d’être utilisés comme photocatalyseurs pour la dégradation des polluants organiques à la lumière du visible [13].
La présente étude rapporte l’étude structurale, les propriétés optiques et électroniques ainsi que l’activité photocatalytique d’un nouvel halogénure d’antimoine et de mercure Hg12Sb6(Br5,186I6,814), en combinant une variété de techniques expérimentales (diffraction des rayons X, spectroscopie X à dispersion d’énergie, analyse par spectroscopie IR et mesure de l’activité photocatalytique sous la lumière du soleil) avec une étude théorique par la méthode DFT.
Notre composé est le deuxième halogène pnicture cristallisant dans le système triclinique après le composé Hg19As10Br18 [14]. Cependant, à notre connaissance, il s’agit du premier à présenter un mélange d’halogènes (I et Br).
2. Partie expérimentale
2.1. Synthèse des cristaux
Les cristaux de Hg12Sb6(Br5,186I6,814) ont été préparés par la méthode classique du transport en phase vapeur (CVT) à partir d’un mélange stœchiométrique des éléments : mercure (Hg), dihalogénures de bromure et de mercure (HgX2; X = Br et I), antimoine (Sb) et diiodide (I2).
Le mélange est broyé puis introduit dans un tube en quartz scellé sous vide ; des cristaux de couleur orange sont obtenus après un chauffage à 623–673 K durant une semaine.
2.2. Microscopie électronique et spectroscopie X à dispersion d’énergie (EDXS)
Dans le but de vérifier la nature des éléments présents dans le composé, une analyse chimique par spectroscopie à rayons X à dispersion d’énergie (EDX Oxford Instruments) sur MET de plusieurs cristaux a été effectuée. Les spectres obtenus confirment la présence des quatre éléments Hg, Sb, Br et I (Figure S2).
2.3. Propriétés optiques
L’analyse par spectroscopie IR à transformée de Fourier en mode réflexion totale atténuée (ATR) a été menée sur un spectromètre de type Perkin Elmer (Spectrum Two). Les acquisitions ont été réalisées en effectuant 64 balayages entre 4000 et 400 cm−1, avec une résolution de 2 cm−1. Le spectre de la réflectance diffuse a été enregistré avec un spectrophotomètre UV–visible de type Specord 200 Plus (190–1100 nm), équipé d’une sphère d’intégration PTFE (Polytétrafluoroéthylène). Cette mesure permet l’obtention de la réflectance (%R) en fonction de la longueur d’onde (𝜆) dans un domaine (300–700 nm).
Les travaux de Kubelka et Munk [15, 16] permettent de relier le coefficient d’absorption 𝛼 à la réflectance R par l’équation : (S étant le coefficient de diffusion)
| (1) |
L’énergie du gap d’un composé semi-conducteur peut être reliée au coefficient d’absorption par la relation de Tauc [17] :
| (2) |
où h est la constante de Planck, 𝜈 la fréquence de vibration, Eg le gap optique et A une constante. L’exposant n est une constante qui détermine le type de transition dans un semi-conducteur (n = 2 pour une transition optique directe et n = 1∕2 pour une transition optique indirecte).
2.4. Mesure de l’activité photocatalytique
L’activité photocatalytique de ce matériau a été évaluée par la dégradation du colorant organique la rhodamine (RhB) sous la lumière du soleil.
La Rhodamine B est un colorant Xanthène largement utilisé en tant que colorant dans les industries textiles et pigment dans des préparations pharmaceutiques. Les molécules de ce colorant sont difficilement biodégradables.
Ainsi, un mélange de 10 mg de photocatalyseur et 50 ml de solution de RhB (5 ppm), soumis à une agitation modérée est laissé dans l’obscurité pendant toute une nuit pour atteindre l’équilibre adsorption/désorption. Ensuite, le mélange est mis sous irradiation solaire directe pendant 3 h et des prélèvements de 4 ml sont effectués chaque 15 mn. La photolyse des solutions (en absence de photocatalyseur) a été réalisée en parallèle avec les expériences photocatalytiques afin d’évaluer la contribution de la photolyse directe sous la même intensité solaire. Enfin, la concentration de la RhB est mesurée par un spectrophotomètre UV–visible de type Specord 200 Plus (190–1100 nm). Le composé parent HgI2 (commercial), appliqué récemment comme photocatalyseur à la dégradation de la RhB sous la lumière du soleil [18], est utilisé comme référence dans les mêmes conditions.
2.5. Détermination et affinement de la structure
Les intensités ont été collectées à l’aide d’un diffractomètre Bruker AXS à détecteur bidimensionnel de type CCD. Le cristal étudié correspond à une macle non-mériédrique constituée de deux individus mis en évidence par le programme CELL-NOW (Bruker, 2006) avec une matrice de macle de : [−0,98 0,01 0,853 0,0025 −0,994 0,949 0,0025 0,01 0,993] obtenue par le programme PLATON/TwinRotMat.
Les réflexions des deux domaines ont été intégrées en utilisant le programme SAINT, résultant d’un total de 18 250 réflexions observées : 6423 et 6440 réflexions correspondent au premier et au deuxième domaine respectivement et 5387 réflexions communes.
Une solution préliminaire a été déterminée par méthodes directes, en utilisant le programme Superflip [19]. La structure finale a été affinée avec le programme Jana2006 [20] en utilisant un fichier de type HKL5 créée par le programme TWINABS (Bruker, 2006) contenant la contribution de ces deux individus.
Dans cette structure il existe six (06) positions cristallographiques indépendantes pour le mercure, trois (03) pour l’antimoine et six (06) pour les halogènes (Br/I). L’affinement montre que les atomes d’halogènes I et Br ne sont pas ordonnés et occupent les six (06) positions de manière statistique (s.o.f : I > Br), comme déjà observés dans quelques composés sulfohalogénures de mercure [21, 22]. En fin d’affinement, la fraction en volume des composants est de : 0,519 (3) : 0,481 (3), et la carte de densité électronique est de : 𝜌max = 3,09 e ⋅Å−3 (localisée à 1,14 Å de X2) et 𝜌min = −2,77 e ⋅Å−3 (localisée à 1,68 Å de Hg6).
Les conditions de la collecte et le résultat de l’affinement sont rassemblés dans le Tableau 1.
Données cristallographiques, conditions de la collecte et résultats de l’affinement de Hg12Sb6(Br6,814I5,186)
| Formule chimique | Hg12Sb6(Br6,814I5,186) |
| Masse molaire (g ⋅ mol−1) | 4416,7 |
| Dimension du cristal (mm3) | 0,462 × 0,117 × 0,075 |
| Forme cristalline | Prismatique |
| Groupe d’espace | P −1 |
| Z | 1 |
| Paramètres de maille (Å, °) | a = 7,7911 (13) b = 11,680 (4) c = 12,404 (2) 𝛼 = 112,758 (16) 𝛽 = 99,988 (13) 𝛾 = 102,04 (3) |
| Volume (Å3) | 976,7(5) |
| Densité calculée (g ⋅ cm−3) | 7,4772 |
| Coefficient d’absorption (mm−1) | 61,7 |
| F(000) | 1808 |
| Intervalle angulaire T (°) | 3,52/30,2 |
| Intervalles d’index | −10<h<10 − 16<k<16 −17<l<17 |
| Réflexions enregistrées totales | 18 250 |
| Réflexions indépendantes | 3581 |
| Réflexions avec I > 3𝜎(I) | 2127 |
| Tmin∕Tmax | 0,0005/0,0322 |
| Nombre de paramètres affinés | 144 |
| GOF | 1,34 |
| (R∕Rw)obs (%) | 0,0774/0,0902 |
| (R∕Rw)all (%) | 0,1554/0,1024 |
| Δ𝜌maxΔ𝜌min (e−∕Å3) | 3,09/−2,77 |
Les coordonnées atomiques réduites, les facteurs d’agitation thermique anisotropes et les distances interatomiques sont donnés respectivement dans les Tableaux S2, S3 et S4.
2.6. Méthode de calculs théoriques
Tous les calculs du présent travail ont été effectués par la méthode du pseudopotentiel [23] et des ondes planes dans le cadre de la théorie fonctionnelle de la densité, telle qu’implémentée dans le code CASTEP (Cambridge Serial Total Energy Package [24].
Les pseudopotentiels de type ultra-soft de Vanderbilt [25] sont utilisés pour modéliser les interactions entre les électrons de valence Hg : 5d106s2, Sb : 5s25p3, Br : 4s24p5, I : 5s25p5 et les électrons de cœurs. La méthode d’approximation du gradient généralisée de Perdew–Burke–Ernzerhof (GGA-PBE) [26] a été utilisée pour traiter les interactions électroniques d’échange-corrélation. Les états de valence à un électron de Kohn–Sham ont été étendus sur un ensemble d’ondes planes avec une coupure d’énergie cinétique de 500 eV. Pour le calcul des paramètres structuraux, l’intégration sur la zone Brillouin (BZ) a été réalisée à l’aide d’un maillage 4 × 4 × 4 Monkhorst-Pack [27]. La relaxation des paramètres structuraux a été réalisée à l’aide de l’algorithme BFGS [28] avec les seuils suivants : la convergence d’énergie totale est de l’ordre de 10−6 eV/atome, la force sur chaque atome est inférieure à 5 × 10−3 eV/Å, à 10−2 GPa pour la pression maximale et enfin à 10−4 Å pour le déplacement maximal.
3. Résultats et discussion
3.1. Description de la structure
La principale caractéristique de ce composé est la présence de liaisons anion–anion (Sb–Sb). Les atomes d’antimoine entourés de trois atomes d’Hg et un atome de Sb, possèdent une coordination tétraédrique légèrement déformée. Des distances Sb–Sb [2,763(3)–2,815(3) Å] et Hg–Sb [2,626(3)–2,693(3) Å] légèrement inférieures à la somme des rayons covalents (2,82 et 2,90 Å) [29] indiquent de fortes interactions. Toutefois elles sont comparables à celles observées dans les composés halogénures d’antimoine et de mercure ternaires [Sb–Sb = 2,650–2,816 et Hg–Sb = 2,632(4)–2,686(4) Å] [30, 31, 32, 33] et quaternaires [Sb–Sb = 2,750(3)–2,856(4) et Hg–Sb = 2,624(1)–2,704(2) Å] [7, 9, 34, 35, 36, 37, 38]. Des angles Hg–Sb–Hg et Sb–Sb–Hg qui valent respectivement 105,75(9)–118,62(12)° et 99,00(8)–108,63(9)° indiquent des tétraèdres légèrement distordus. Les deux tétraèdres correspondant à une même liaison Sb–Sb forment un groupement octaédrique Sb2Hg6.
La charpente de cette structure est ainsi formée par des octaèdres Sb2Hg6 reliés par les sommets qui déterminent des files parallèles agencées en zigzag dans les trois directions adoptant une structure de type ReO3 distordue (Figure 1).
(a) Projection de la structure Hg12Sb6(Br5,186I6,814) dans le plan (b,c), montrant l’agencement en zigzag des octaèdres Sb2Hg6 délimitant les cavités occupées par les atomes d’halogènes, (b) Agrandissement de la partie en encadrée (en rouge), (c) La liaison anionique Sb–Sb dans les octaèdres Sb2Hg6.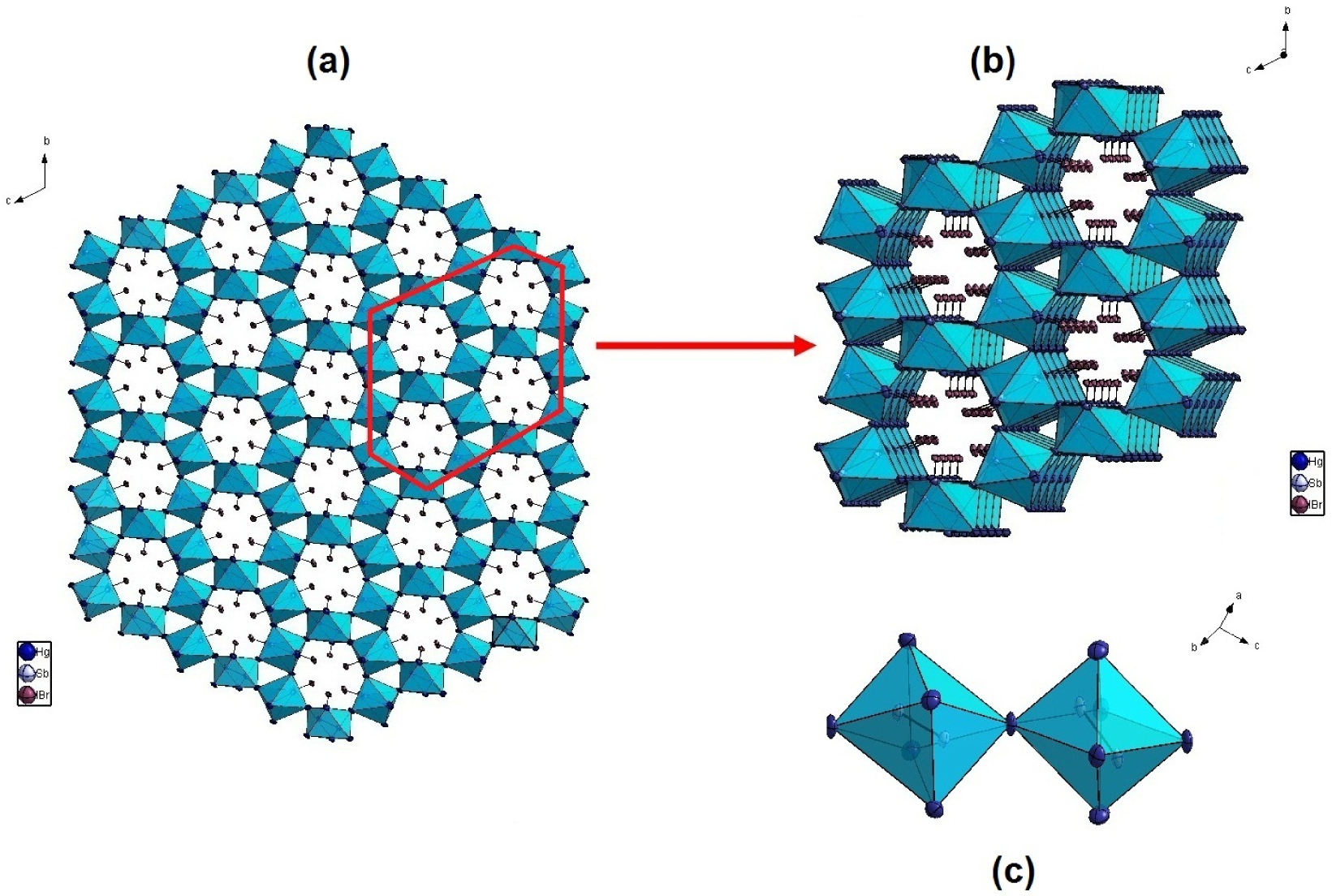
Ces files parallèles agencées en zigzag déterminent des cavités occupées par les atomes d’halogène X (X = Br et I). Ces derniers assurent ainsi la cohésion de la structure par l’intermédiaire de liaisons Hg–X (X = Br et I) en reliant de part et d’autre les groupements octaédriques Sb2Hg6.
Comme pour la plupart des composés halogénopnictures de mercure, les atomes Hg peuvent adopter des coordinations variées : linéaire, tétraédrique ou octaédrique.
Dans cette structure les atomes d’Hg occupent six (06) positions cristallographiques indépendantes. La moitié des atomes de mercure (Hg1, Hg4 et Hg5) est entourée de deux (02) atomes de Sb avec des angles de liaisons Sb–Hg–Sb qui dévient de la linéarité [158,24(8)–171,69(10)°]; ceci est typique des composés chalcogénides au mercure Hg2+ [39]. L’autre moitié des atomes de mercure (Hg2, Hg3 et Hg6) est entourée d’un seul atome de Sb et de trois (03) atomes d’halogène X dans une configuration tétraédrique légèrement déformée (Figure S3).
Des distances Hg–X (X = I, Br) varient sur un large intervalle entre 2,662(3) et 3,455(4) Å (distances < à 3,5 Å) (Tableau S3) selon le nombre d’atomes d’halogène voisins. Les courtes distances Hg–X (Br/I) correspondent aux atomes d’Hg (Hg2, Hg3 et Hg6) adoptant une coordination tétraédrique. Ces distances sont proches de celles observées dans les halogénures de mercure correspondant HgX2 (X = I, Br) (Hg–I = 2,63–2,80 Å et Hg–Br = 2,72 Å) [40, 41, 42, 43], et Hg2X2 (Hg–I = 2,72661(17) Å et Hg–Br = 2,71 Å) [44, 45], indiquant de fortes liaisons covalentes. Quant aux plus grandes distances Hg–X (Br/I) elles correspondent aux atomes d’Hg (Hg1, Hg4 et Hg5), et concernent les atomes d’halogènes complétant la sphère de coordination de ces atomes d’Hg. Ces distances qui correspondent aux interactions secondaires dans les halogénures de mercure correspondant HgX2, restent toutefois proches de la somme des rayons de Van der Waals [Hg = 1,7 Å, Br = 1,91 Å et I = 2,15 Å donc Hg–I = 3,85 Å et Hg–Br = 3,61 Å] [29], indiquant le caractère ionique de ces liaisons. Le nombre de ces liaisons semble jouer un rôle important pour la formation et la stabilisation des composés halogènes pnictures [12].
Dans cette structure, la moitié des atomes d’Hg (Hg1, Hg4 et Hg5) alterne avec les atomes Sb selon des liaisons Hg–Sb–Hg en forme ‘d’arc’ pour constituer une branche polymère 1D le long de l’axe a. Ces branches sont pontées par des liaisons anioniques Sb–Sb conduisant à la formation de couches 2D selon le plan (ac) (Figure 2). Enfin, la superposition de ces couches 2D le long de l’axe b via des liaisons Hg–Sb qui concernent l’autre moitié des atomes d’Hg (Hg2, Hg3 et Hg6), permet de concevoir une charpente 3D.
(a) Branches polymères 2D dans le plan (a,c), résultat de l’interconnexion des liaisons Hg–Sb–Hg (en forme d’arc) via des liaisons Sb–Sb, (b) Agrandissement de la partie en encadrée (en rouge).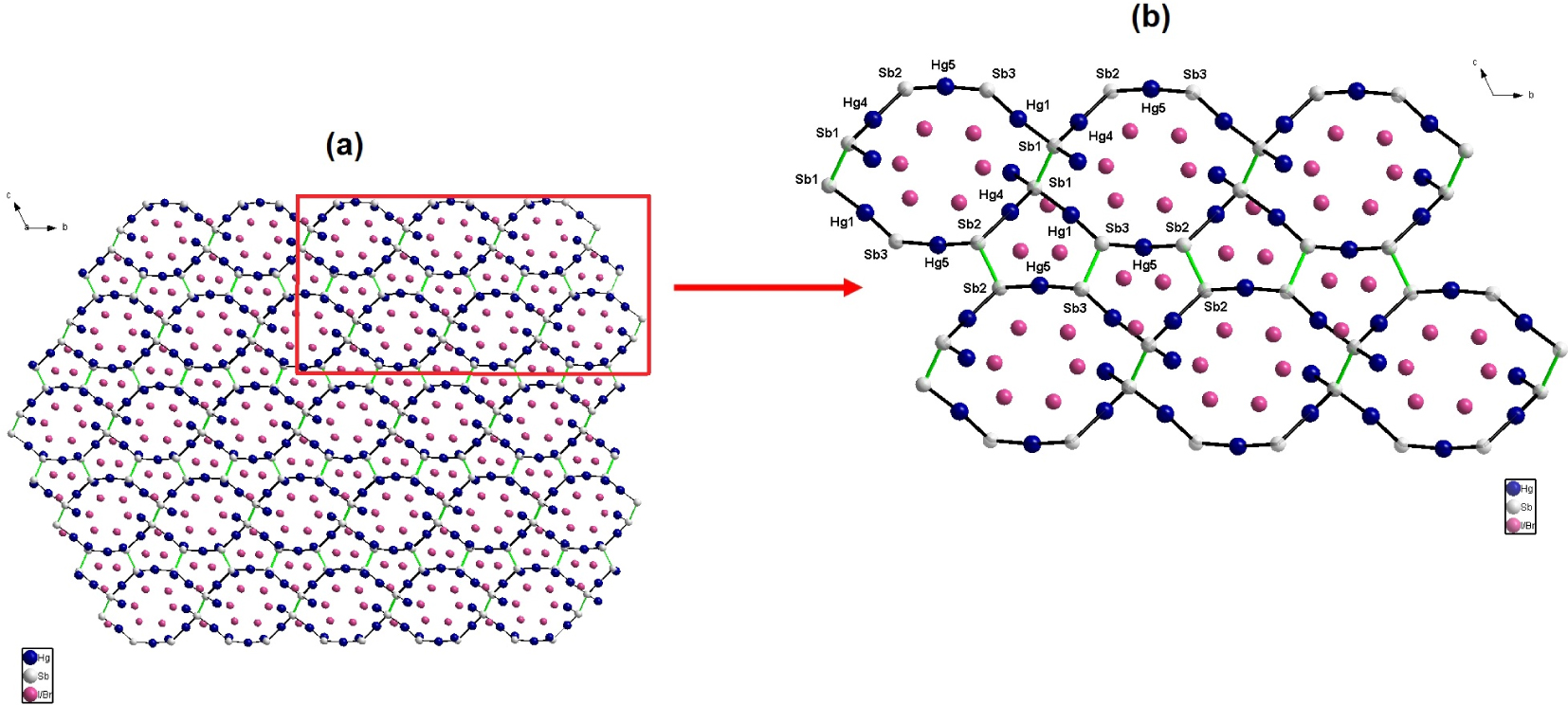
Ainsi, dans cette charpente 3D, les files parallèles des octaèdres Sb2Hg6 agencées en zigzag délimitent deux types de cavités de forme triangulaire et hexagonale de taille plus importante. Le diamètre interne de la cavité hexagonale qui est occupée par les atomes d’halogènes est autour de 6,7 Å (il est défini à partir des plus courtes distances entre les atomes de mercure opposés). L’autre cavité triangulaire vide possède un diamètre autour de 3,8 Å (Figure 1b).
Dans cette structure une forte agitation thermique (ADP’s) des atomes Hg est observée selon la direction U11 pour les atomes de mercure Hg1, Hg4 et Hg5 et selon la direction U22 pour les atomes Hg2, Hg3 et Hg6 (Tableau S2). Pour les atomes d’halogènes elle est observée pour les atomes X2, X5 et X4 selon respectivement les mêmes directions. Cette agitation semble orientée vers l’espace vacant formé par l’agencement des octaèdres Sb2Hg6.
Le calcul des différentes valences de liaison (BVS), utilisant la formule empirique de Brown and Altermatt [46, 47, 48] conduit aux valeurs moyennes des charges des ions :
Hg (2,66), Sb (2,29) et I/Br (0,98) (Tableau 2) et confirme le degré d’oxydation attendu pour les différents ions dans la structure Hg2+, Sb2− et (I/Br)−. L’introduction des distances interatomiques relativement longues (> à3 Å) et correspondant à la deuxième sphère de coordination peut expliquer les valences un peu élevées des atomes d’Hg. L’équilibre des charges dans ce composé peut ainsi s’écrire : .
Analyse de la population de Mulliken
| s | P | d | Totale | Charge | |
|---|---|---|---|---|---|
| Hg | 1,20–1,30 | 0,83–0,94 | 9,94–9,95 | 12,03–12,15 | +0,20/+0,23 |
| Sb | 1,71–1,75 | 3,36–3,40 | 0,00 | 5,09–5,12 | −0,02/−0,03 |
| X | 1,57–1,65 | 5,18–5,34 | 0,00 | 6,64–6,95 | −0,15/−0,27 |
| Liaisons | Hg–Sb | Sb–Sb | Hg–X | ||
| Populations | 0,81–0,92 | 0,85–0.89 | −0,15/+0,35 | ||
| dCalc (Å) | 2,616–2,693 | 2,714–2,759 | 2,699–2,930 | ||
| dExp (Å) | 2,622(3)–2,693(3) | 2,763(3)–2,815(3) | 2,662(3)–2,958(5) | ||
| Ionicité f | 0,209–0,08 | 0,161–0,116 | 0,843–≈1 |
X = Br/I; dCalc par défaut < à 3 Å.
3.2. Mesure des propriétés optiques
3.2.1. Analyse par spectroscopie ATR-FTIR et UV–visible
L’analyse par spectroscopie IR à transformée de Fourier en mode réflexion totale atténuée (ATR) de l’échantillon cristallin est représentée dans la Figure S4. Le spectre ATR-FTIR, ne montre pas d’absorption importante dans le moyen IR entre 4000 et 400 cm−1 (2,5–25 μm), ce qui permet d’envisager ce type de matériau comme candidat pour des applications dans le domaine des télécommunications et multimédias [49, 50]. La caractérisation par spectrophotométrie en réflectance diffuse UV–visible de cet halogène pnicture révèle la présence d’une bande d’absorption optique dans le domaine du visible autour de 570 nm qui correspond à la couleur orange des cristaux, et d’un gap optique de l’ordre de 2,1 eV, qui suggère un caractère semi-conducteur de ce composé dont la transition est indirecte (Figure 3). La valeur de l’énergie de ce gap est inférieure à celle des binaires Hg2I2 (2,4 eV), Hg2Br2 (2,6 eV) et HgBr2 (3,65 eV) [51, 52]; mais supérieure à celle mesurées dans les composés halogénopnictures ternaires et quaternaires (1,23–2,05 eV) [6, 7, 13, 9, 36, 53]. Enfin, il convient de noter que la valeur de ce gap est très proche de celle de HgI2 (2,1 eV) [18].
Détermination de l’énergie du gap du composé Hg12Sb6(Br5,186I6,814). (a) Cas d’une transition directe, (b) Cas d’une transition indirecte.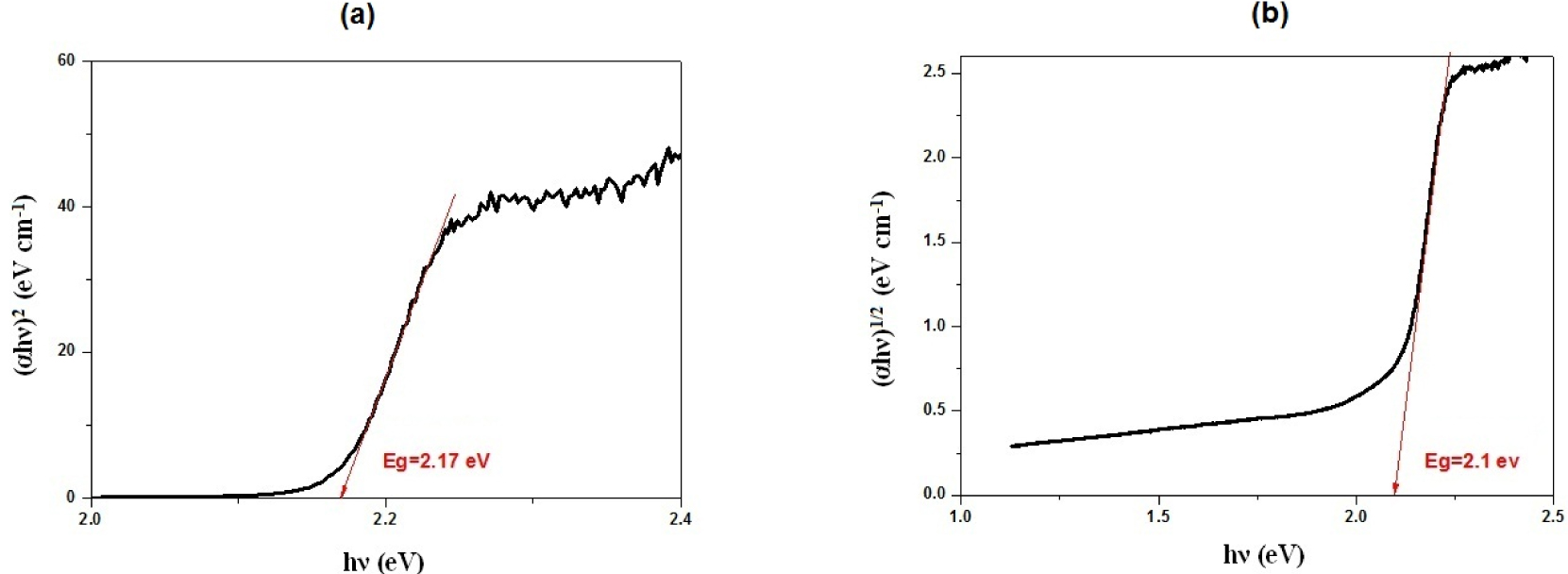
3.2.2. Energie des bandes de valence et de conduction
Le potentiel des bords des bandes de valence (EVB) et de conduction (ECB) peuvent se calculer comme suit [54, 55] :
| (3) |
| (4) |
| (5) |
EC est l’énergie potentielle des électrons libres à l’électrode à hydrogène standard ( ∼4,5 eV) et Eg est l’énergie de gap de l’halogène pnicture.
3.3. Propriétés photocatalytique
Comparé à HgI2, dont le taux de dégradation de la RhB après 30 mn d’exposition au soleil est de l’ordre de 30%, un taux de plus de 50% est observé avec notre composé pour le même temps d’exposition, et une décoloration presque totale après une durée de 180 mn d’exposition au soleil (Figure 4).
Cinétique de la photodégradation de la RhB sous irradiation solaire par les photocatalyseurs : Hg12Sb6(Br5,186I6,814) et HgI2.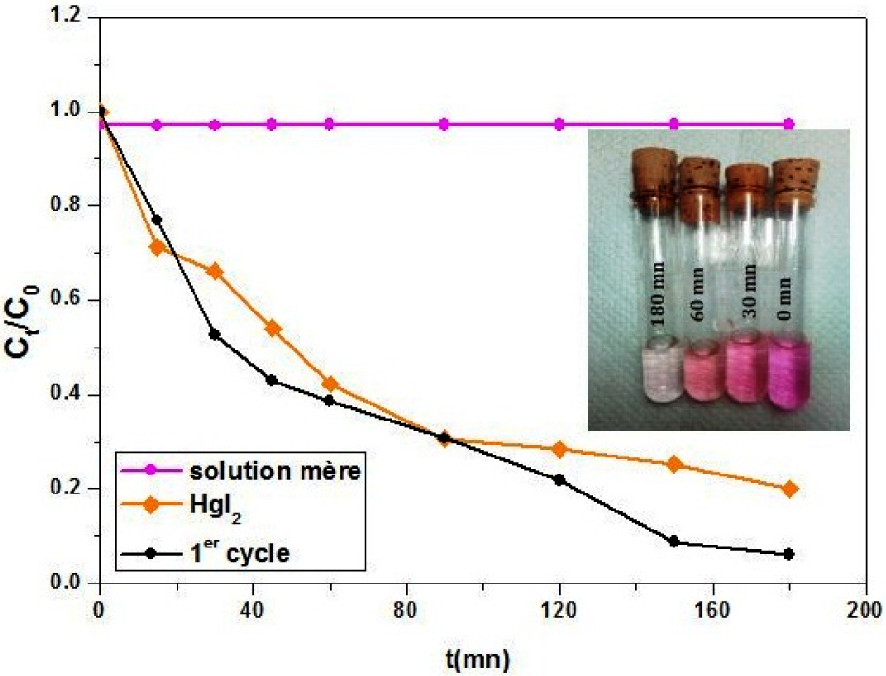
Le modèle de Langmuir–Hinshelwood a été utilisé pour le suivi de la cinétique de la réaction de photodégradation :
| (6) |
Celle-ci suit un modèle de pseudo-premier ordre C et Co étant respectivement les concentrations finales et initiales.
L’activité photocatalytique du composé Hg12Sb6(Br5,186I6,814) est meilleure que celle de HgI2, avec des constantes apparentes de dégradation respectives de 0,882 et 0,546 h−1 (Figure S5).
Trois cycles successifs de dégradation ont été effectués avec l’halogène pnicture afin de mesurer la stabilité et l’efficacité du photocatalyseur. Pour le deuxième cycle cette efficacité est restée identique, mais elle a légèrement chuté pour le troisième cycle donnant un taux de dégradation de l’ordre de 70% (180 mn) (Figure 5).
Représentation des trois cycles de la photodégradation de la RhB par le semi-conducteur Hg12Sb6(Br5,186I6,814).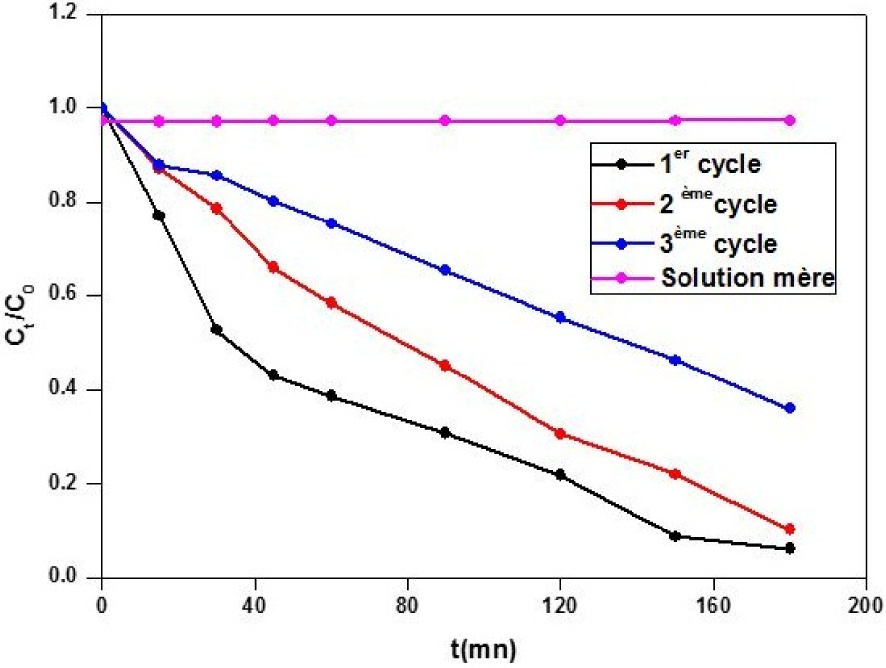
3.3.1. Mécanisme de la photodégradation
La décomposition de la RhB suit un mécanisme très connu. Ainsi lors de l’irradiation par la lumière du soleil d’un semi-conducteur les paires électrons/trous (e−∕h+) générées sont utilisées dans le processus d’oxydation et de réduction de la RhB.
L’énergie des niveaux ELUMO et EHOMO de la RhB sont respectivement de − 1,10 eV et 1,14 eV. Dans Hg12Sb6(Br5,186I6,814), le bord de la bande de valence (VB) située à environ 0,095 eV vs ENH, n’est pas plus négatif que le potentiel redox de l’ (−0,33 V), alors que le bord de la bande de conduction (CB) située à environ 2,265 eV vs ENH, est plus positif que le potentiel redox de (1,99 V). L’électron dans la CB ne peut donc pas réagir avec les molécules oxygène pour produire les radicaux anions superoxyde , et par conséquent le processus de réduction ne peut avoir lieu. D’autre part les trous photogénérés (h+) peuvent à la fois oxyder le colorant et réagir avec H2O et OH− pour former un radical hydroxyle () qui possède un fort potentiel oxydant pour la dégradation d’un tel colorant toxique. Toutefois une bonne partie des paires électrons/trous (e−∕h+) se recombine, ce qui diminue le rendement de la réaction.
La décomposition de la RhB (Figure 6) peut ainsi être décrite selon le mécanisme qui suit :
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
Schéma du principe général de la photocatalyse hétérogène de la RhB par le semi-conducteur Hg12Sb6(Br5,186I6,814).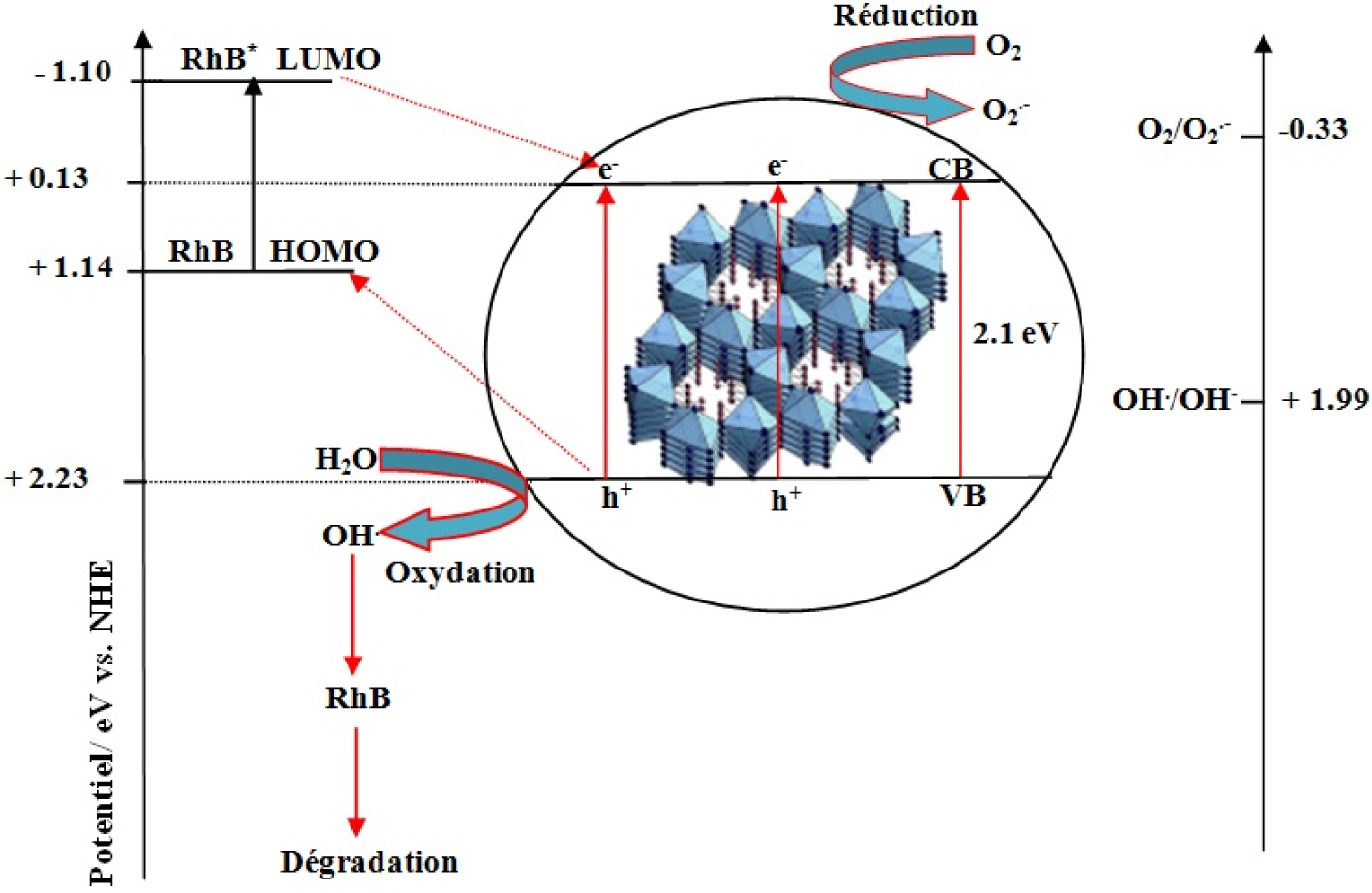
Pnicture∗ représente l’état excité du composé pnicture; électrons photoexcités dans la CB; trous photogènes ; Radical hydroxyde et ion hydroxyde adsorbée.
3.4. Propriétés électroniques
La structure des bandes d’énergie, les densités totales et partielles d’états ainsi que l’analyse de la population de Mulliken peuvent fournir des informations microscopiques sur diverses propriétés des matériaux et ainsi mettre en évidence leurs éventuelles applications potentielles.
3.4.1. Densités d’états (DOS et PDOS)
Pour plus d’informations sur la nature de l’impulsion orbitale des états électroniques dans le spectre d’énergie et un aperçu du caractère des liaisons chimiques interatomiques, nous avons calculé la densité totale d’états (TDOS) et la densité atomique (PDOS) projetée en Figure S6 pour le composé étudié dans la gamme énergétique de −20 à 15 eV.
L’approximation du gradient généralisé GGA-PBE sous-estime le gap autour de 1,70 eV (Figure 7), comparée à la valeur expérimentale de 2,1 eV. La bande de conduction (BC) et celle de valence (BV) ont des formes plutôt aplaties, ce qui est très fréquent dans ce genre de composés impliquant de faibles interactions entre la sous-structure hôte et les atomes d’halogènes invités. On note aussi que le bas de la BC est plus dispersé que le haut de la BV.
(a) Structure de bandes, (b) densité d’états totale DOS, (c) Première zone de Brillouin du composé Hg12Sb6(Br5,186I6,814).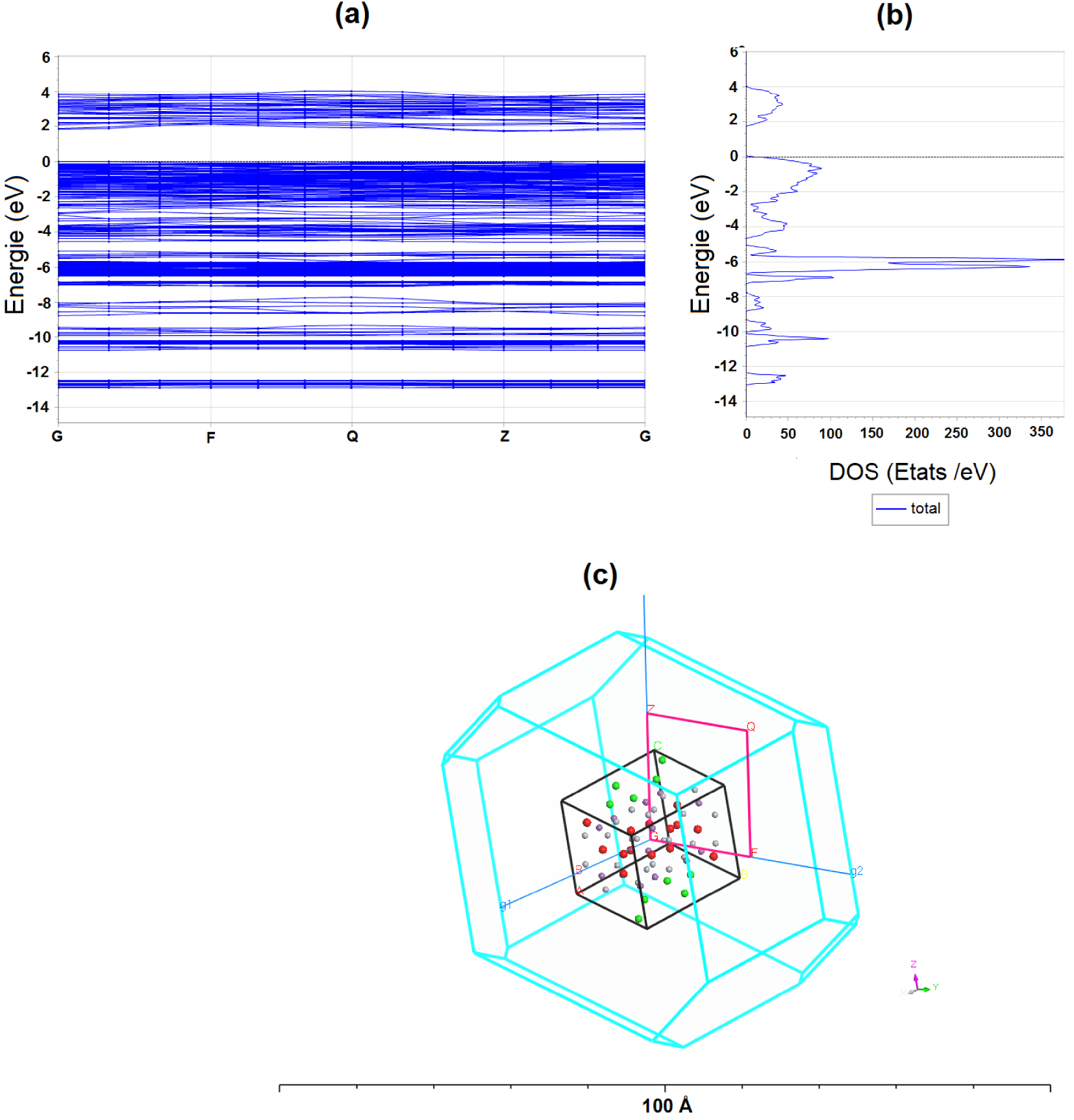
La BC dans le domaine 2,00 et 5,00 eV, est formée essentiellement par les états électroniques Hg-6s, avec une faible contribution des orbitales ‘s’ et ‘p’ des atomes Sb et X (Br/I). La BV entre −15,00 et −7,00 eV, est composée par les états Sb-5s ainsi que des orbitales ‘p’ des atomes d’halogènes, alors qu’entre −7,00 et −5,00 eV, elle est composée par la contribution des états Hg-5d et Sb-5p. La partie supérieure à partir de −5 eV jusqu’au niveau de Fermi quant à elle contient les états Sb-5p ainsi que des orbitales ‘p’ des atomes X (Br/I), avec une petite contribution des états Hg-6s. On peut ainsi en conclure que l’absorption optique est principalement attribuée aux transitions de charge des états Sb-5p ainsi que des orbitales ‘p’ des atomes X (Br/I) vers les états Hg-6s.
3.4.2. Analyse de population de Mulliken
L’analyse de population de Mulliken (PMA) [56] est un outil important couramment utilisé pour caractériser la distribution de la charge électronique dans une molécule et pour déterminer la nature de la liaison interatomique d’une paire d’atomes spécifique. En particulier, la population de liaisons (chevauchement), qui est l’un des paramètres essentiels tirés de cette technique, est un grand critère quantitatif pour étudier la force et la nature de la liaison entre une paire d’atomes voisins dans les matériaux. Selon Segall et al. [57], une valeur positive (négative) de la population de chevauchement indique des états de liaison (anti-bonding). Une valeur de zéro de la population de liaisons implique une liaison ionique idéale et une valeur supérieure à zéro indique la présence d’un degré de covalence.
En effet, les valeurs de populations de Mulliken calculées via le code de CASTEP, sont résumées dans le Tableau 2. À partir de ces résultats, il est clair que les liaisons Hg–Sb et Sb–Sb possèdent un caractère fortement covalent, comparées aux faibles interactions des liaisons Hg–X (Br/I), selon l’ordre Hg–Sb > Sb–Sb > Hg–X (Br/I). Ceci est en bon accord avec les résultats expérimentaux.
Cette population de recouvrement (PMA) peut aussi refléter le degré ou la tendance d’ionicité des liaisons, cette dernière peut être calculée comme suit [58] :
| (12) |
Des valeurs de fh = 0 ou 1, indiquent des natures de liaisons purement covalentes ou ioniques.
Selon le Tableau 2, le fort degré d’ionicité indique de faibles interactions Hg–X (Br/I) dont les liaisons semblent totalement ioniques, contrairement aux fortes interactions existantes entre Hg–Sb et Sb–Sb qui se traduisent ainsi par un faible degré d’ionicité et un degré élevé de covalence. Ceci confirme la nature mixte covalente–ionique des liaisons qui caractérisent ce composé halogène pnicture.
3.4.3. Propriétés optiques
Les propriétés optiques sont calculées dans la gamme de 0 à 20 eV, en utilisant une correction du scissor (l’opérateur ciseau) de 0,466 eV implanté dans le code CASTEP. Il s’agit d’une correction empirique appliquée aux énergies de gap sous-estimées par l’approximation DFT du Gradient Généralisé (GGA-PBE), pour porter la valeur du gap vers la valeur expérimentale par un décalage rigide de la bande de conduction.
Les paramètres optiques de ce composé à savoir le coefficient d’absorption, l’indice de réfraction, le coefficient de réflectivité, etc., peuvent être calculés à partir de la fonction diélectrique dépendante de la fréquence et étroitement liée à la structure de la bande électronique.
| (13) |
La partie imaginaire 𝜀2(𝜔) de la fonction diélectrique qui est exprimée comme les éléments de la matrice de momentum entre les états électroniques occupés et inoccupés, et la partie réelle 𝜀1(𝜔) qui se dérive de la partie imaginaire par les relations de Kramers–Kronig, peuvent être obtenues directement à l’aide du manuel CASTEP de Materials Studio.
La courbe de la partie imaginaire 𝜀2(𝜔) (Figure 8a) atteint une valeur maximale de 3,99 eV, résultat du transfert des électrons de valences extrêmes des états Sb-5p ainsi que des orbitales ‘p’ des atomes X (Br/I) à la première bande de conduction des états Hg-6s. Dans la zone à haute énergie, la valeur de la partie imaginaire 𝜀2(𝜔) est très faible.
Propriétés optiques du composé Hg12Sb6(Br5,186I6,814). (a) La partie imaginaire et réelle de la fonction diélectrique, (b) L’indice de réfraction et coefficient d’extinction, (c) Spectre d’absorption, (d) La réflectivité, (e) Fonction de perte d’énergie.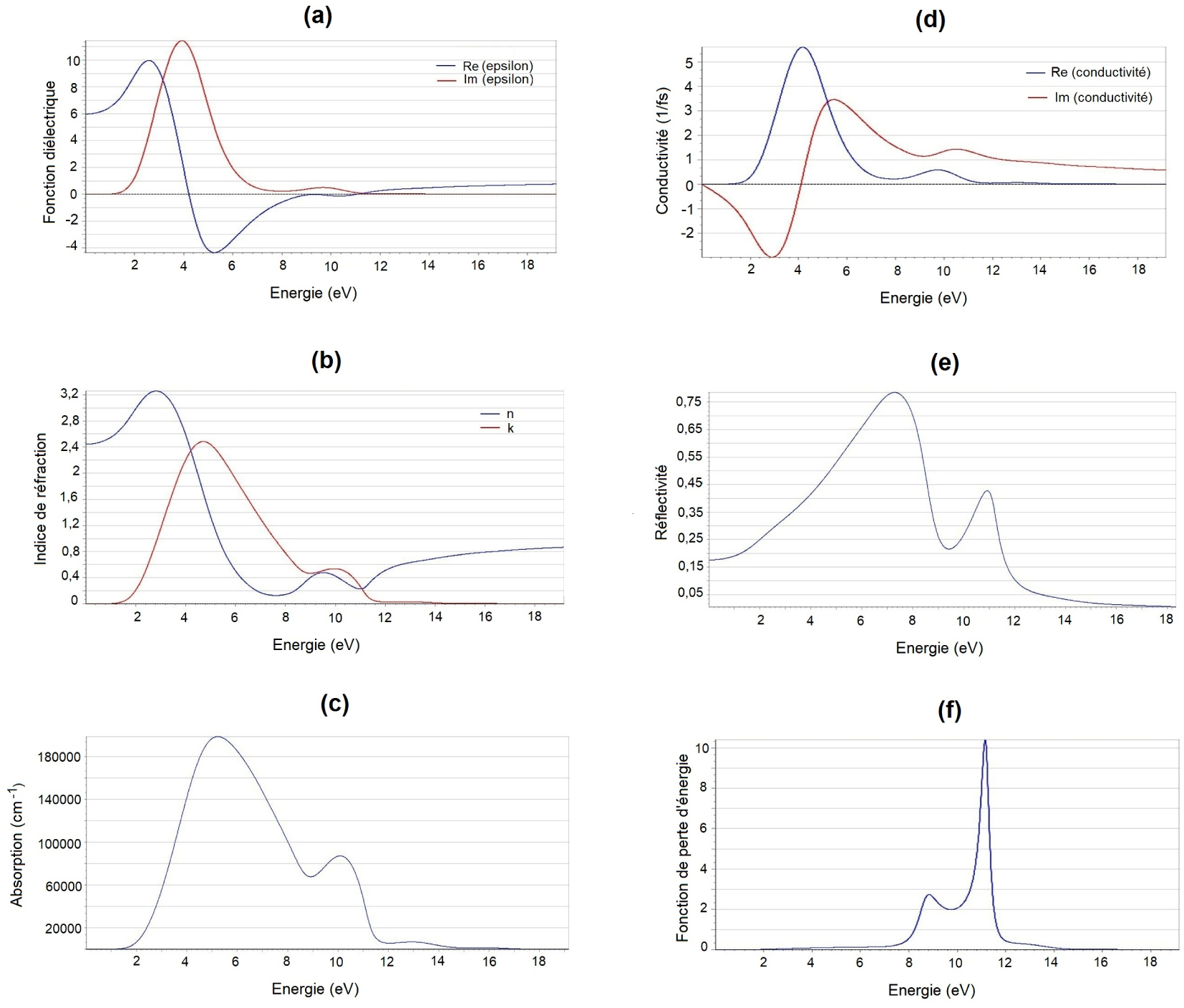
La courbe de la partie réelle 𝜀1(𝜔) décroît rapidement à partir de 2,60 eV, puis devient négative pour atteindre un minimum à 5,23 eV. Une croissance de cette courbe est ensuite observée au-delà du niveau zéro, mais comme pour 𝜀2(𝜔), les valeurs dans la zone à haute énergie restent faibles. 𝜀1(𝜔) et 𝜀2(𝜔) approchent des valeurs nulles autour de 11,5 eV, indiquant la limite de la présence de l’effet diélectrique après cette valeur dans la région de l’ultraviolet. Ceci correspond à une énergie pour laquelle une chute brutale de l’absorption et de la réflectivité (Figure 8c et e) est observée, ainsi que l’apparition du premier pic de la fonction perte d’énergie (Figure 8f). La constante diélectrique statique 𝜀1(𝜔) est obtenue comme la limite de la partie réelle de la fonction diélectrique près des fréquences nulles, vaut 6,00. L’indice de réfraction et le coefficient d’extinction sont représentés sur la Figure 8b. En général, la partie réelle n est liée à la dispersion, alors que la partie imaginaire k est directement liée au coefficient d’absorption. L’indice de réfraction statique n(0) est de 2,42, ce dernier est calculé à partir de la fonction diélectrique 𝜀(0) par la relation.
| (14) |
Celui déduit à partir de la courbe de l’indice de réfraction en fonction de l’énergie est autour de 2,44, cette valeur augmente pour atteindre sa valeur maximale d’environ 3,26 dans la région du visible à 2,85 eV. Le spectre de n(𝜔) décroît fortement avec la croissance de l’énergie du photon pour atteindre une valeur minimale de 0,128. La valeur du maximum du coefficient d’extinction k(𝜔) quant à elle est localisée à l’énergie 4,72 eV. L’absorption de la lumière par un milieu optique est quantifiée par son coefficient d’absorption 𝛼 (𝜔). Le spectre d’absorption linéaire calculé 𝛼 (𝜔) (Figure 8c), montre qu’il y a absorption dans l’UV et le visible. Le seuil d’absorption commence à partir d’environ 2 eV, et correspond aux transitions des états électroniques (p) de Sb et X (Br/I) aux états (s) de Hg.
On observe deux pics, un premier à 5,22 eV et un deuxième moins important à 10,1 eV, ce composé montre plutôt un bon coefficient absorption dans la région 4–7 eV.
Ce seuil d’absorption diminue dans la région de haute énergie où l’absorption est très faible pour l’énergie des photons supérieure à 14 eV, caractère typique des semi-conducteurs.
Les parties réelle et imaginaire de la conductivité optique sont représentées par la Figure 8d. Lorsque l’énergie du photon est inférieure à 1 eV la conductivité est nulle, puis elle augmente pour atteindre une valeur maximale de 5,60 f−1 ⋅ s−1, pour une énergie de 4,21 eV, pour diminuer et enfin s’annuler autour de 11 eV.
La réflectivité (Figure 8e) calculée est inférieure à 20% à plus basse énergie, elle augmente avec l’énergie du photon pour atteindre une valeur maximale (supérieure à 70%) égale à 78,4% à 7,36 eV dans la région de l’ultraviolet. La fonction électronique de perte d’énergie L(𝜔) (Figure 8f) décrit la perte d’énergie d’un électron traversant dans un matériau. Le pic principal qui est autour de 11,1 eV correspond à la fréquence de plasma.
4. Conclusions
En conclusion, l’étude structurale par diffraction des rayons X du nouvel halogénure d’antimoine et de mercure Hg12Sb6(Br5,186I6,814), montre une charpente formée par des octaèdres Sb2Hg6 en files parallèles agencées en zigzag adoptant une structure de type ReO3 distordue, et déterminant des cavités occupées par les atomes d’halogène I/Br.
La spectrophotométrie en réflectance diffuse UV–visible révèle la présence d’une bande d’absorption optique dans le domaine du visible autour de 570 nm, et d’un gap optique de l’ordre de 2,1 eV qui suggère un caractère semi-conducteur de ce composé.
L’activité photocatalytique par la dégradation du colorant organique la rhodamine (RhB) sous la lumière du soleil, montre une décoloration presque totale après une exposition de 180 mn, et une efficacité pour un troisième cycle de l’ordre de 70%.
Par ailleurs, l’étude des densités d’états (DOS) confirme le caractère semi-conducteur de ce composé et permet de conclure que l’absorption optique est principalement attribuée aux transitions de charge des états Sb-5p ainsi que des orbitales ‘p’ des atomes X (Br/I) vers les états Hg-6s.
L’analyse de population de Mulliken (PMA) montre la nature mixte covalente–ionique des liaisons qui caractérisent ce composé halogène pnicture.
L’étude des propriétés optiques indique que la partie imaginaire 𝜀2(𝜔) de la fonction diélectrique atteint une valeur maximale de 3,99 eV, alors que la constante diélectrique statique 𝜀1(𝜔) vaut 6. L’indice de réfraction statique n(0) augmente pour atteindre une valeur maximale d’environ 3,26 dans la région du visible à 2,85 eV, et la valeur du maximum du coefficient d’extinction k(𝜔) quant à elle est localisée à l’énergie 4,72 eV. Le spectre d’absorption linéaire calculé 𝛼(𝜔) montre qu’il y a absorption dans l’UV et le visible avec un bon coefficient d’absorption dans la région 4–7 eV. La conductivité atteint une valeur maximale de 5,60 f−1 ⋅ s−1, pour une énergie de 4,21 eV, pour diminuer et enfin s’annuler autour de 11 eV.
La réflectivité augmente avec l’énergie du photon pour atteindre une valeur maximale égale à 78,4% à 7,36 eV dans la région de l’ultraviolet, et le pic principal de la fonction électronique correspondant à la fréquence plasma de perte d’énergie L(𝜔) est autour de 11,1 eV.
Conflit d’intérêt
Les auteurs n’ont aucun conflit d’intérêt à déclarer.
Remerciements
Nos remerciements vont au Professeur L. Carlos Otéro-Diaz du Département de chimie Inorganique de l’Université Complutense de Madrid pour son aide très précieuse.