


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Developments in research in pharmaceutical and biomedical fields have led to extended life expectancies and dramatic changes in people’s general health care systems. In the past two decades, there have been many interesting discoveries related to nanomaterials for drug delivery application, especially in achieving controlled release behavior [1]. Controlling drug release has been introduced as a method to deliver the correct drug dose in the targeted area at the required period to improve the efficacy of therapy [2]. The encapsulation of drug in nanocarriers could enhance the drug’s efficacy and reduce the lateral effect [3]. Inorganic nanoparticles have shown versatility in shape, size, structure, and geometry to be used as nanocarriers [4]. Calcium phosphate-based materials have been successfully used in gene and drug delivery, while quantum dots as unique nanoparticles with luminescence, which is suitable for in-vitro imaging, have shown potential for in-vivo diagnostics [4].
As the most stable form of calcium phosphate, hydroxyapatite (HAp) has been highly utilized as biocompatible and bioactive nanoparticles in drug delivery, tissue engineering, and implant application [5]. The tailorable structure and size, longer biodegradation time, stability, and high active surface of HAp make it favorable as a drug delivery carrier [6]. The synthesis of HAp, with rod or needle shape, usually produced through sol–gel method improves the chemical homogeneity of pure HAp [6]. Moreover, the combination of HAp and CQDs enriches the material with biocompatibility and fluorescence emission characteristics useful for bioimaging [7, 8]. Besides, the rich functional groups on the surface of CQDs could improve the adsorption of pharmaceutical compounds [7].
CQDs have been known as discrete material with size less than 10 nm and various oxygen-containing groups [9]. In addition, the excellent optical properties, good solubility, biocompatibility, and lower toxicity of CQDs could facilitate real-time tracking and biodistribution of CQDs as drug carriers [9, 10]. Functionalization of CQDs is necessary to increase the surface area of oxygen-containing groups, which could induce the maximum loading capacity [10]. The study on combination of HAp and CQDs so far has been performed mainly for bioimaging, pharmaceutical compound sensing, bone regeneration [11], and drug delivery such as synthesized CQDs/α-Fe2O2 for ulvan delivery [12], CQDs/carboxymethyl cellulose/HAp [13] and CQDs/HAp [7] for doxorubicin release, and CQDs/HAp for acetaminophen release [8]. On the other hand, the study on modified CQDs, either functionalized or doped and then combined with HAp for biomedical application, has been limited. The research mainly focuses on their photoluminescence property [14, 15] and bone regeneration [11]. Hence, this study would like to explore the effect of loading of amino-functionalized CQDs on the properties of HAp. The electrostatic interaction and hydrogen bonding between amino-functionalized CQDs and HAp could increase the dispersibility of nanoparticles and enhance their luminescence, surface area, and performance for the slow release of drugs.
2. Methodology
2.1. Materials
Chitosan from shrimp shells 75% deacetylated, sodium hydroxide (NaOH), calcium nitrate tetrahydrate (Ca(NO3)2⋅H2O), diammonium hydrogen phosphate ((NH4)2HPO4), and acetaminophen were purchased from Sigma-Aldrich, USA. Hydrochloric acid (HCl) 37% was purchased from Merck, Germany. Acetic acid glacial was purchased from Junsei, Japan. Phosphate buffer saline (PBS) was purchased from Fisher, USA. All chemicals were used without further purification. Deionized (DI) water (18.2 M𝛺) from PURELAB flex was utilized throughout the experiment.
2.2. Synthesis of amino-functionalized CQDs
The amino-functionalized CQDs were synthesized from chitosan using the hydrothermal method published in previous studies with some modifications [8, 16]. Around 0.5 g of chitosan was added to 50 mL of 1M acetic acid solution and dissolved at room temperature. Then, the mixture was heated hydrothermally at 190 °C for 4 h. The solution was then cooled down to room temperature and centrifuged at 10,000 rpm for 15 min to remove deposits. The supernatant was then dialyzed for 4 h to purify the CQDs solution.
2.3. Synthesis of HAp and amino-functionalized CQDs/HAp
HAp nanoparticles were synthesized using the sol–gel method, published in a previous study [8]. Around 0.025 mol of Ca(NO3)2⋅H2O was dissolved in 50 ml of 0.4 M HCl. Then, 0.015 mol of (NH4)2HPO4 was added dropwise into the solution. Around 500 ml of 0.7 M NaOH solution was then added to form gel solution. To synthesize amino-functionalized CQDs/HAp, the steps follow the synthesis of HAp with an additional step of 10, 20, and 30 mL of amino-functionalized CQDs solution being added in the HCl solution before the formation of gel, to form f-CD10-HAp, f-CD20-HAp, and f-CD30-HAp, respectively. The mixture was stirred at room temperature, filtered using vacuum filtration, and washed with deionized water three times. The precipitates were then dried in the oven at 100 °C overnight. The dried particles were then ground to fine powder using mortar and pestle.
2.4. Characterization
The morphology, size, and elemental analysis of HAp and f-CD-HAp were characterized using Field Emission Scanning Electron Microscopy equipped with Energy Dispersive X-ray Spectroscopy (FESEM-EDX, Zeiss Supra 55 VP). The morphology of f-CD was characterized by using High-Resolution Transmission Electron Microscope (HRTEM) with Field Emission (Tecnai G2 20 S-Twin 200 kV, FEI). The average size of f-CD was determined by using ImageJ software. The functional groups of f-CD, HAp and f-CD-HAp were analysed using Fourier Transform Infrared (FTIR) spectrometer from wavenumber 500 to 4000 cm−1 (Perkin Elmer). The photoluminescence of samples was characterized using spectrophotometer with an excitation source of 420 nm laser (Edinburgh Instrument FLS920), and liquid UV–Vis analysis was performed using Shimadzu UV-1800, while the solid UV–Vis analysis was done using Cary Series UV–Vis Spectrophotometer. The crystallinity of HAp and f-CD-HAp was characterized using X-ray Powder Diffraction (XRD) with Cu Kα irradiation range (diffraction angles (2𝜃)) from 5° to 80° with a step size of 2°/step and exposure time of 1s/step (Bruker D8 Advance). The surface area and pore diameter of HAp and f-CD-HAp were identified using Micrometrics ASAP 2020 Plus. The element compositions of samples were characterized using X-ray Photoelectron Spectrophotometer (XPS) (Thermo Scientific K-Alpha).
2.5. Drug loading and release
During the synthesis of HAp and f-CD-HAp, 1 mg/g of acetaminophen was loaded during the mixing of solution to form gel. The samples were then centrifuged at 5000 rpm for 15 min, washed three times with deionised water, and dried at room temperature. The supernatant and washing fluid were collected, their absorbance results were measured at 340 nm (absorbance of acetaminophen) using Optizen 3220UV UV–Vis spectrophotometer.
The release of acetaminophen was done in 25 mL of PBS at 37 °C. The solution was then shaken at 250 rpm in an orbital shaker. To monitor the release, around 3 mL solution was periodically removed and then replaced with 3 ml of fresh PBS. The cumulative drug release (CDR) was calculated using the equation:
3. Results and discussion
3.1. Morphology, crystallization, and functional groups of synthesized samples
The morphological analysis of f-CD using HRTEM shows spherically shaped particles with average size of 5.93 ± 1.44 nm, which was estimated from 50 particles (Figure 1a). The lattice spacing of f-CD is around 0.209 nm, which is similar to the previous CD synthesized from chitosan and correspond to (001) lattice space of graphitic carbon [16, 17]. Functional groups of f-CD were characterized by using FTIR analysis, which shows strong peak at 3340 cm−1 that represents O–H and N–H stretching vibrations [17, 18]. Small peaks at 2965 and 2882 cm−1 can be assigned to C–H stretching vibrations [18]. The peaks at 1640, 1459, and 1355 cm−1 can be attributed to C=O, C=C, and C–N bonds, respectively [17]. The peaks at 1027 and 951 cm−1 are ascribed to C–H bending vibrations [18].
(a) and (b) HRTEM images of f-CD, and (c) FTIR spectrum of f-CD.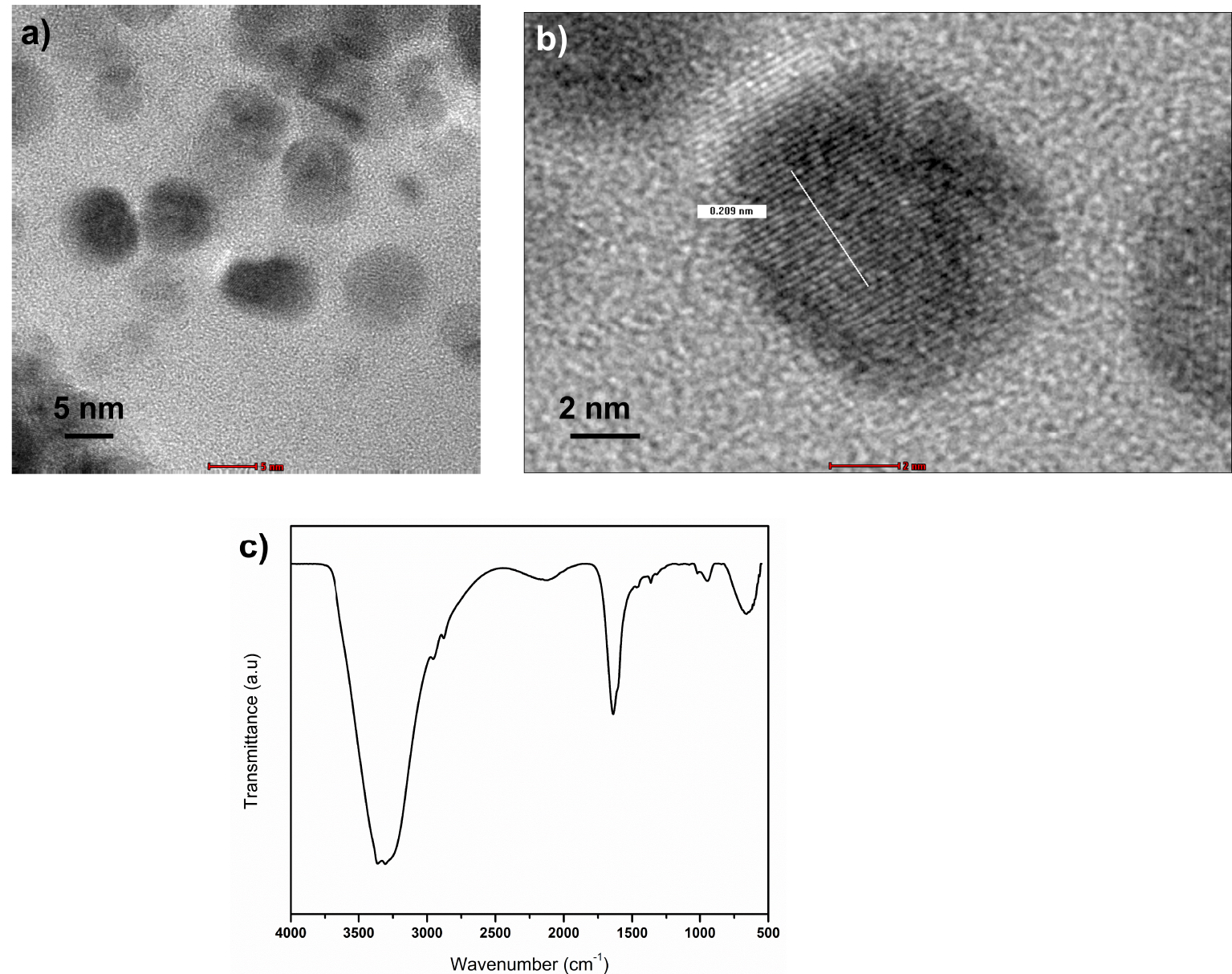
The morphology of synthesized HAp and f-CD-HAp shows clusters of aggregated nanoparticles ranging from 20 to 40 nm (Figure 2). The incorporation of amino-functionalized CQDs into the HAp matrix does not alleviate the morphology [11] but rather exists on the surface of HAp, since carbon and nitrogen peaks can be detected through EDX analysis in HAp (Figure 1c,f,i). According to the EDX spectrum, C (17.26%), O (60.05%), N (12.5%), P (4.91%), and Ca (5.28%) are present for f-CD30-HAp sample (Table 1). The formation of agglomerated nano-HAp can be commonly found when the particles are synthesized by using water as solvent [19]. The Ca/P ratio for the samples is in the range of 1.49 to 1.64, which is near to stoichiometric HAp [19, 20] and close to the results shown in XPS analysis.
FESEM images and EDX analysis of (a–c) HAp, (d–f) f-CD10-Hap, (g–i) f-CD30-HAp.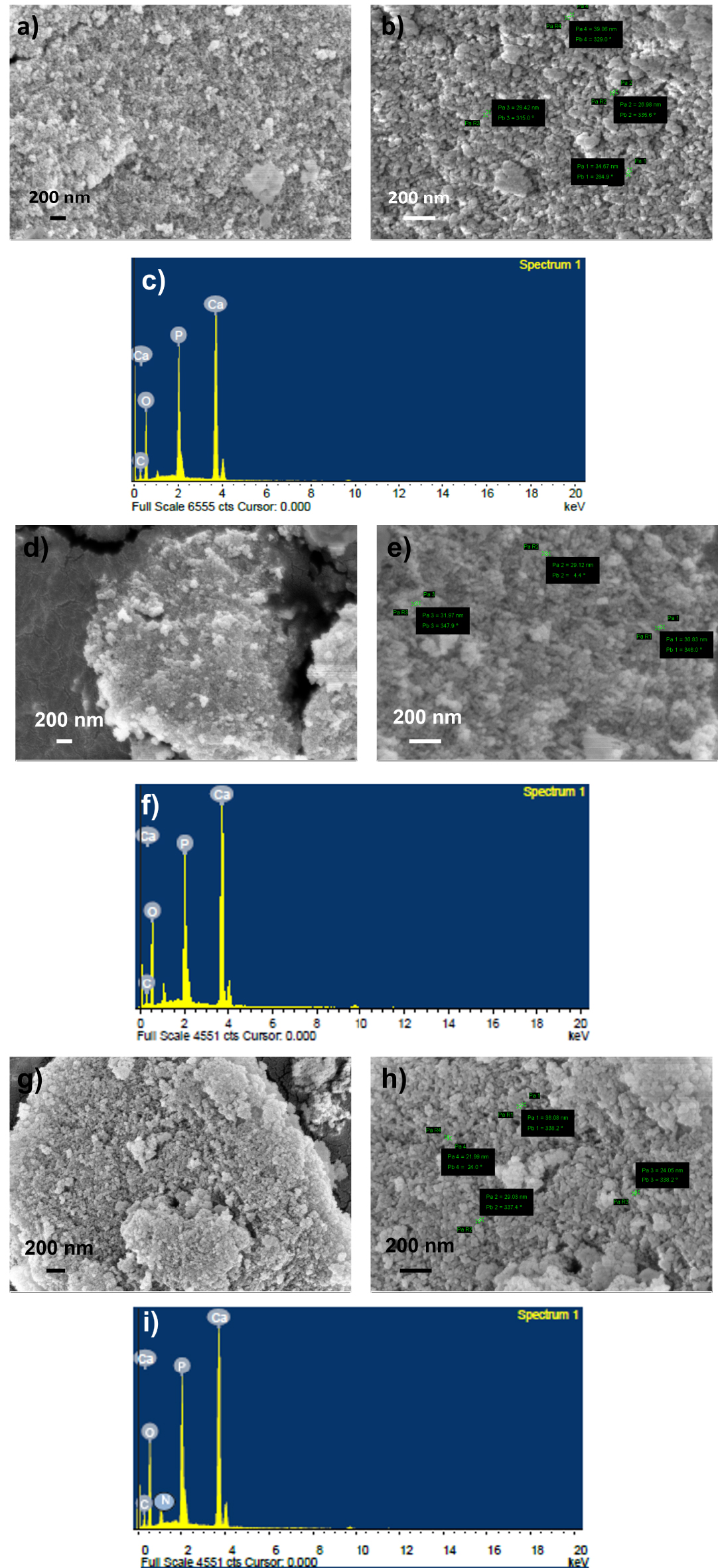
Elemental composition of samples by using EDX
| Sample | Element (%) | ||||
|---|---|---|---|---|---|
| C | O | N | P | Ca | |
| HAp | 7.41 | 49.52 | - | 16.47 | 26.69 |
| f-CD10-HAp | 8.81 | 52.44 | - | 12.43 | 26.33 |
| f-CD30-HAp | 17.26 | 60.05 | 12.5 | 4.91 | 5.28 |
Both infrared and XRD analyses confirm the formation of HAp by showing similar spectra for all samples. The infrared analysis (Figure 3a) of HAp and f-CD-HAp detects a wide peak at 3600–3445 cm−1 corresponding to the hydroxyl bond (O–H) group stretching vibration [21]. The peaks area between 561–605 cm−1 is assigned to internal vibration of group [11]. The asymmetric stretching vibration of the group is represented by the most prominent peak belonging to HAp at around 1036 cm−1 and a small peak at about 877 cm−1 [11, 22]. The asymmetric stretching of the group for HAp is related to peaks 1419 and 1479 cm−1 [23]. The increasing peak intensity in this area might be due to the overlapping with –COO– groups from the interaction of –COOH– in CQDs with HAp [7]. The typical absorption of amino-functionalized CQDs can be shown by the increasing peak intensity at 1640 cm−1 when increasing loading of CQDs (Figure 3b), which can be assigned to C=N stretching vibration [21, 24].
(a) FTIR spectra of samples, and (b) 1750–1550 cm−1 region of infrared spectra of all samples.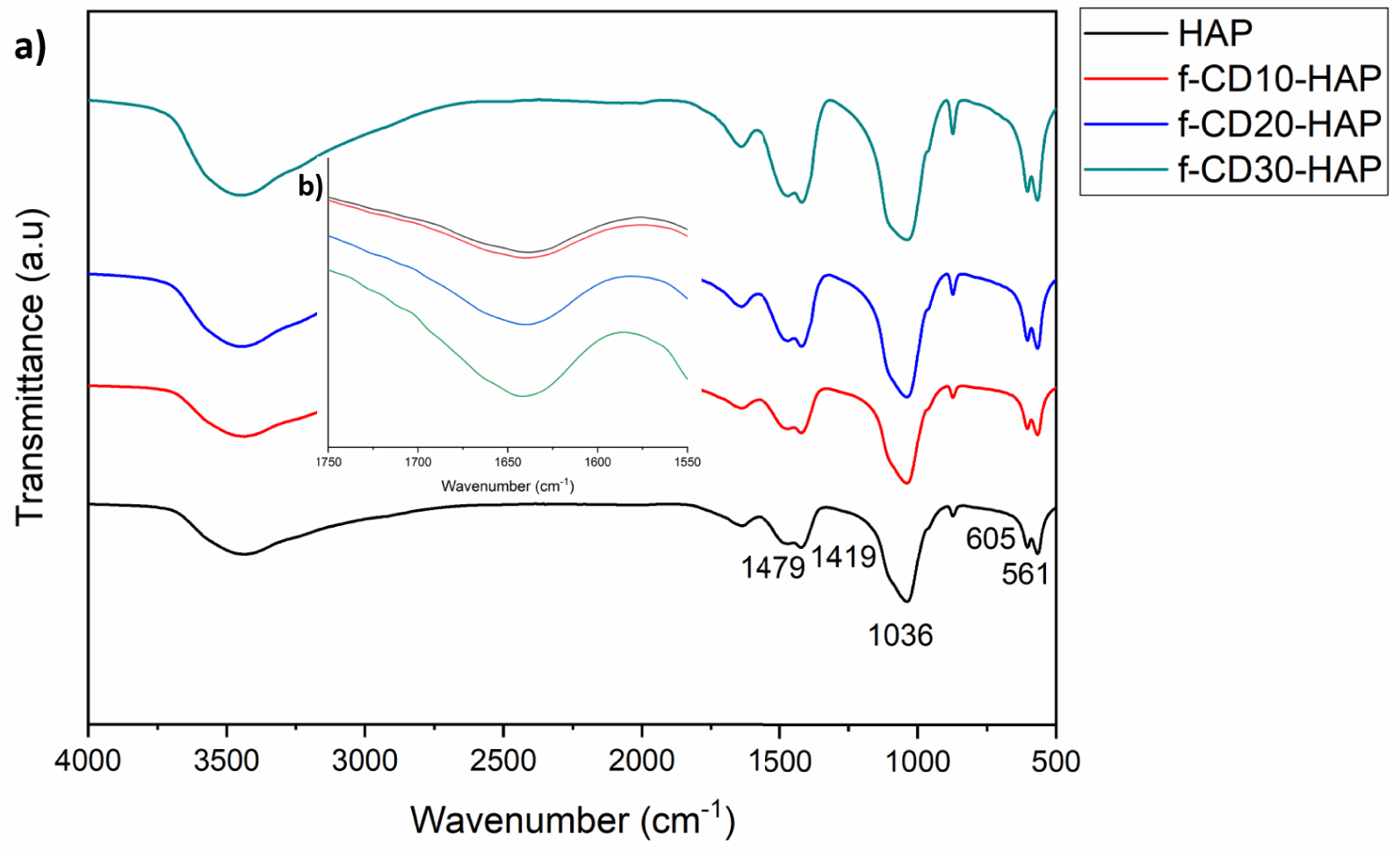
XRD spectra of HAp and composites show well-defined diffraction peaks at around 27.8°, 32.09°, 39.8°, 46.8°, 49.4°, 53.07°, and 63.9° which correspond to Miller indices of (002), (211), (310), (222), (213), (004), and (304), respectively (Figure 4). The peaks match JCPDS card no. 9-432 of pure crystalline hydroxyapatite. Previous studies using the wet precipitation method to synthesize HAp show similar agreement with the exhibited spectra [13, 25, 26], and the incorporation of f-CD did not change the crystal structure of HAp. The spectra exhibit widening peaks which can be due to low crystallinity of HAp from the wet precipitation method.
XRD spectra of samples.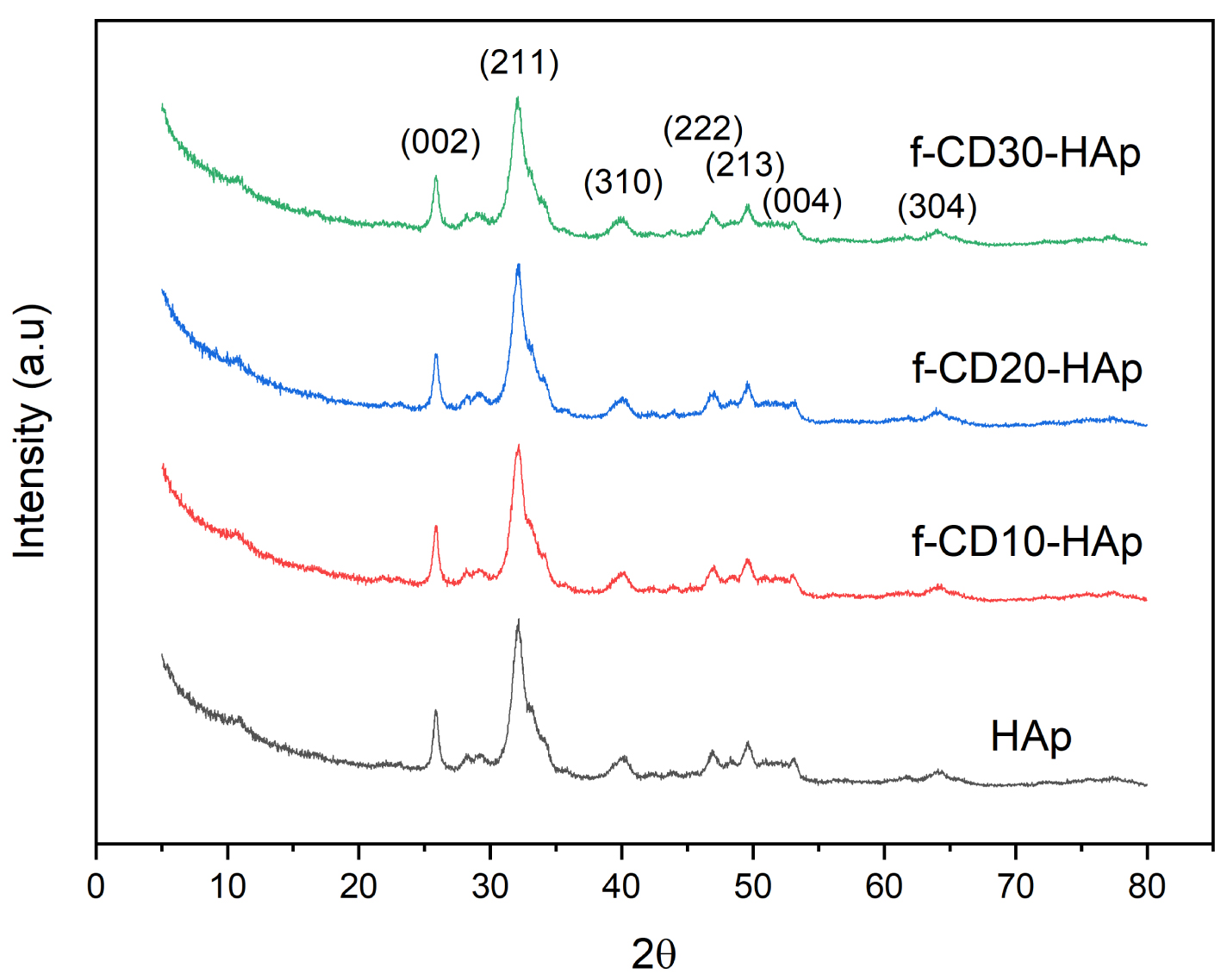
3.2. Optical properties
The loading of amino-functionalized CQDs could improve the photoluminescence intensity of HAp, as can be seen in Figure 5(a). The maximum emission peak for all samples can be found within the range of 438 to 451 nm, which can be associated with /tetrahedral anion or structural defect from oxygen vacancies or interstitial atoms [27, 28]. Shoulder peak at around 600 nm can be attributed to defects or impurities such as O2− ions in the matrix of HAp, which can act as luminescence center [27, 29]. For the f-CD-HAp composite, the widening emission peak at 550 nm can be due to amino-functionalized CQDs loading in the HAp matrix [13]. The UV–Vis spectrum of amino-functionalized CQDs (Figure 5c) shows the appearance of absorbance at around 260 and 480 nm. The absorbance peak at 260 nm can be assigned to the electronic π–π∗ transition of sp2 domain in carbon core [21, 30] while the peak at 480 nm can be attributed to n–π∗ transition of C=O or C–N/C=N bond [21]. HAp mainly does not have a UV–Vis absorption band [28], hence by increasing loading of CQDs, the absorbance intensity is also increasing (Figure 5b). The absorption band at around 480 nm can be observed in composite spectra, especially for f-CD20-HAp and f-CD30-HAp (Figure 5b).
(a) PL, (b) UV–Vis spectra of samples, and (c) UV–Vis spectrum of amino-functionalized CQDs.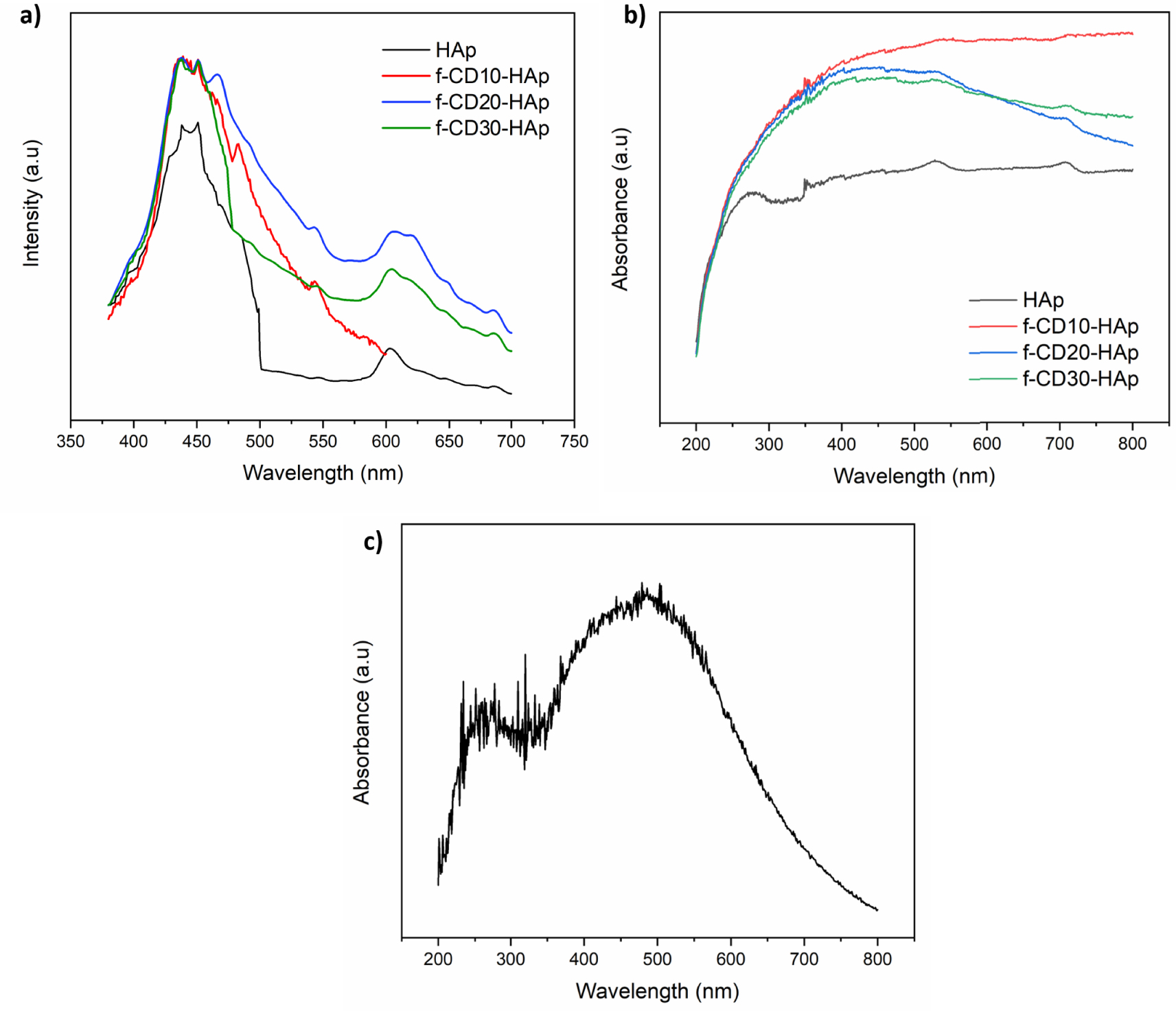
3.3. XPS analysis
XPS analysis determined the elemental compositions and chemical bonding states presented in the synthesized HAp and f-CD-HAp samples. Figure 6 shows the XPS survey spectra of the synthesized HAp and f-CD-HAp samples. Elemental compositions of the samples are presented in Table 2. From the survey spectra, peaks of Ca 2p, P 2p, O 1s, C 1s, and N 1s were observed at binding energy of 350, 135, 530, 290, and 405 eV, respectively. From the elemental compositions determined from XPS analysis, the Ca/P ratio of HAp and f-CD30-HAp were found to be 1.48 and 1.70, respectively. XPS high-resolution spectra of the samples illustrated in the Ca 2p spectra (Figure 7a) are deconvoluted into two major peaks located at 347.70 eV and 350–356 eV, which correspond to Ca 2p3∕2 and Ca 2p1∕2 of Ca2+ ions that belong to HAp and calcium carbonate, CaCO3 formed from the HAp and CQDs, respectively [31, 32]. P 2p spectra for HAp and f-CD30-HAp samples are depicted in Figure 7(b). The peaks are determined at binding energies of 134.16 eV and 136–140 eV, which can be ascribed to the P 2p3∕2 and P 2p1∕2 signals, representing the and P–O bonding in the HAp [31, 33]. On the other hand, from the O 1s spectra (Figure 7c), peaks are found at binding energies of 531.93 eV, 533–535 eV, and 536–540 eV, which correspond to the O–Ca bonding, O–P bonding, and adsorbed molecular oxygen of HAp, respectively [34, 35]. Figure 7d shows the C 1s spectra of the synthesized HAp and f-CD30-HAp samples. The C 1s spectra are deconvoluted into peaks at binding energies of 287.79 eV, 288.52 eV, 291–293.50 eV, corresponding to C=O bonding, O–C=O bonding, and π–π∗ transition of the CQDs [36, 37]. Peaks at binding energies of 294–296 eV are ascribed to CO3 bonding of calcium carbonate formed from the HAp and CQDs [37]. N 1s spectra are depicted in Figure 7e. It is observed that XPS analysis of pure HAp produces a very weak signal of nitrogen element. Upon adding amino-functionalized carbon dots into HAp, peaks are detected at a binding energy of 402.67 eV and 405.08 eV, corresponding to C–N bonding and N–O bonding, respectively [38, 39]. Thus, the bonding and elemental compositions determined from XPS analysis have proven the successful incorporation of amino-functionalized CQDs into HAp matrix.
XPS survey spectra of HAp and f-CD30-HAp samples.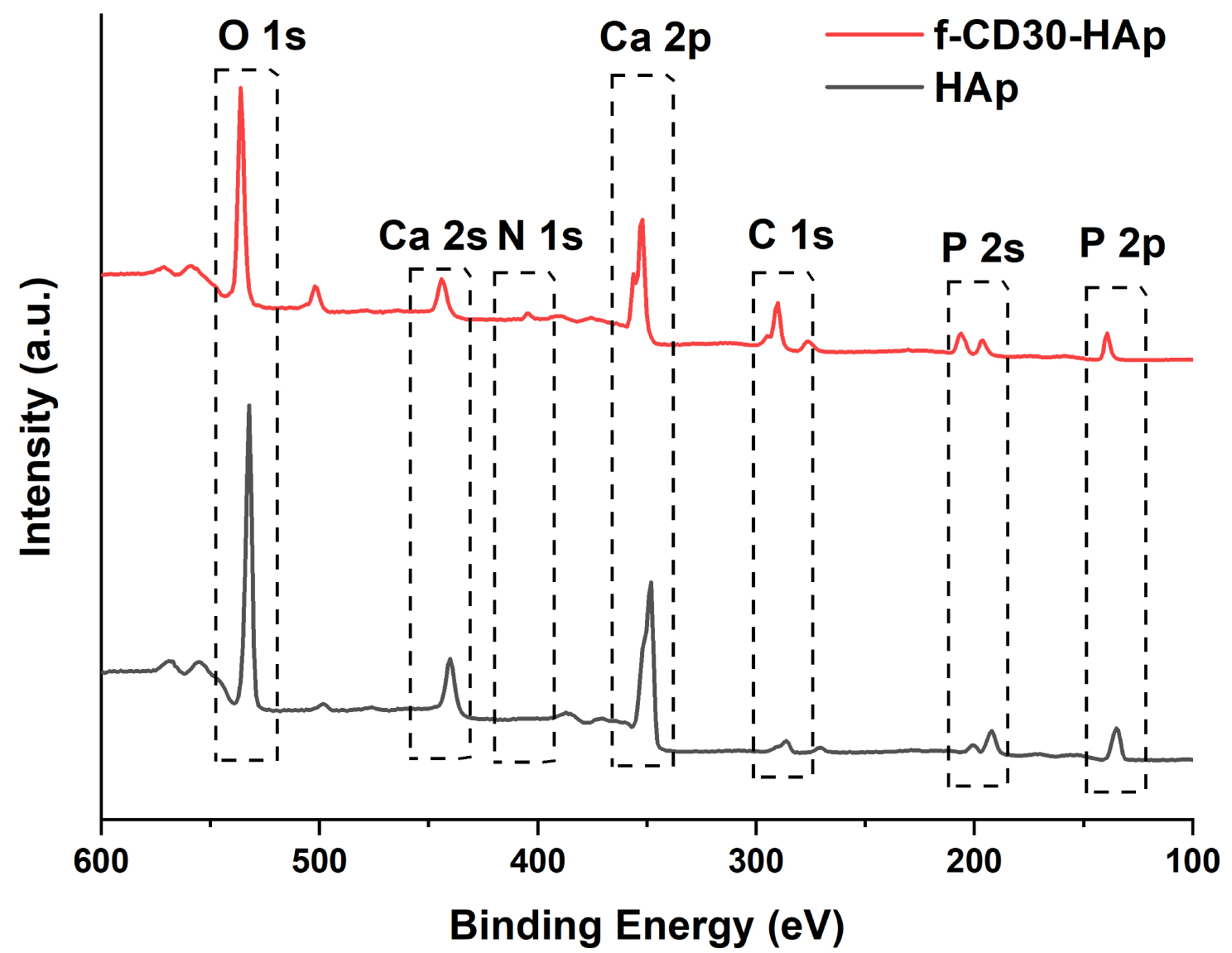
(a) Ca 2p high-resolution spectra for HAp and f-CD30-HAp samples, (b) P 2p high-resolution spectra for HAp and f-CD30-HAp samples, (c) O 1s high-resolution spectra for HAp and f-CD30-HAp samples, (d) C 1s high-resolution spectra for HAp and f-CD30-HAp samples, (e) N 1s high-resolution spectra for HAp and f-CD30-HAp samples.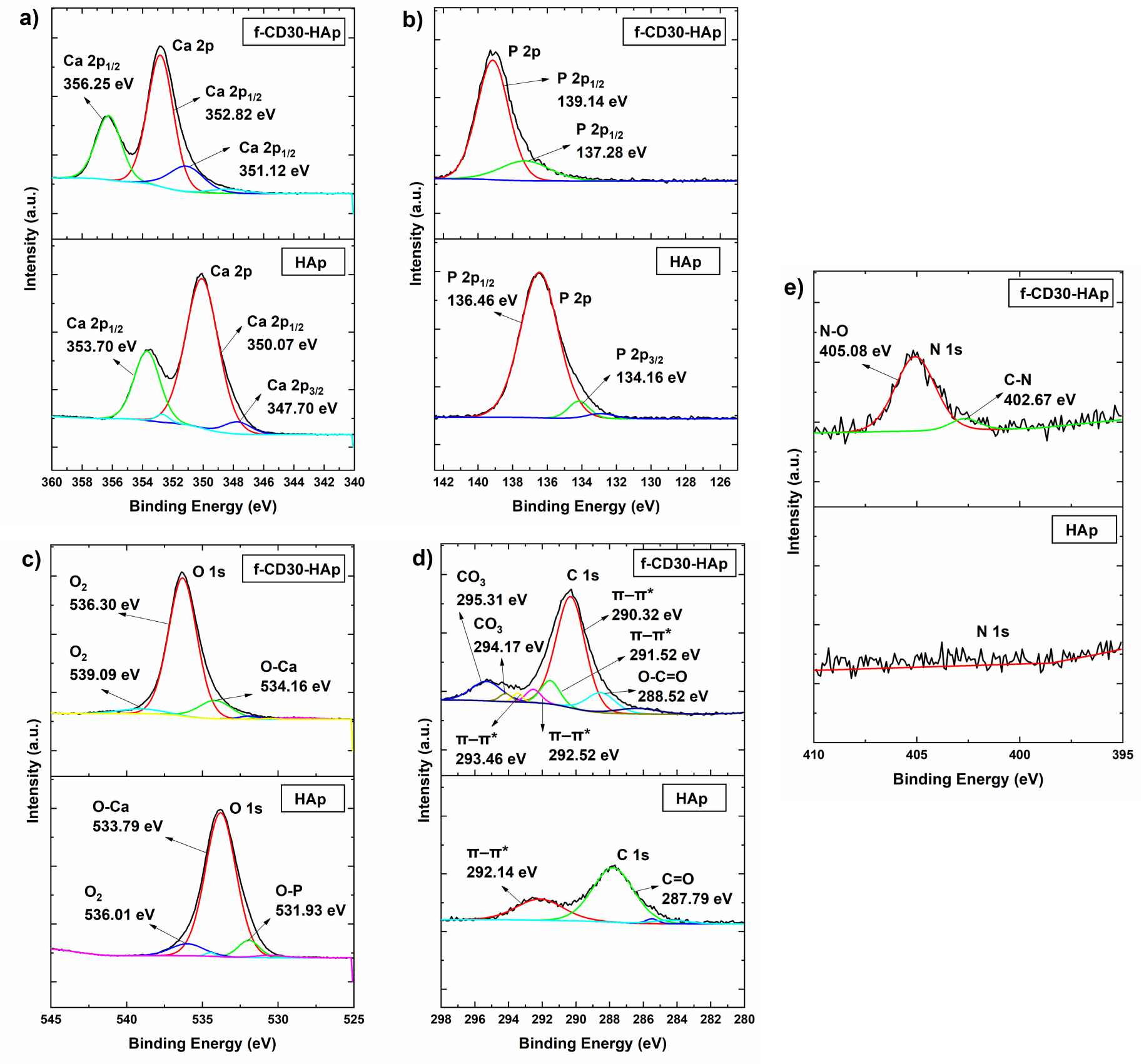
Elemental compositions of HAp and f-CD30-HAp samples
| Sample | Ca (at%) | P (at%) | O (at%) | C (at%) | N (at%) |
|---|---|---|---|---|---|
| HAp | 22.02 | 14.84 | 54.91 | 8.18 | 0.05 |
| f-CD30-HAp | 16.66 | 9.82 | 44.02 | 27.74 | 1.76 |
3.4. Surface area analysis
The samples exhibit a narrow hysteresis loop from adsorption and desorption of N2, which indicates the mesoporous type IV isotherm (Figure 8). The inclusion of additives into the matrix of HAp has been shown to increase its surface area. The previous study of combining CQDs in HAp could increase its surface area from 41.6 to 78.7 m2/g [8], and the inclusion of another type of additive such as Fe3O4 could increase the surface area of HAp from 125 to 175 m2/g [40]. In this study, the samples have large surface area at 84.16, 91.29, 105.16, and 129.94 m2/g for HAp, f-CD10-HAp, f-CD20-HAp, and f-CD30-HAp, respectively, while the pore size of samples indicates mesoporous structure with the value at 22.6, 23.16, 18.5, and 13.3 nm for HAp, f-CD10-HAp, f-CD20-HAp, and f-CD30-HAp, respectively. Hence, the results show increasing surface area value by incorporating amino-functionalized CQDs. Low dispersion of CQDs in the matrix of HAp may induce inter-particular porosity between amino-functionalized CQDs and HAp or between the same compounds, which may increase the surface area [40]. Composites with high surface area could facilitate better drug adsorption, and decoration of its surface with functional groups can induce hydrogen bonding and electrostatic interaction between composite and drug [41, 42].
N2 adsorption-desorption isotherms of samples.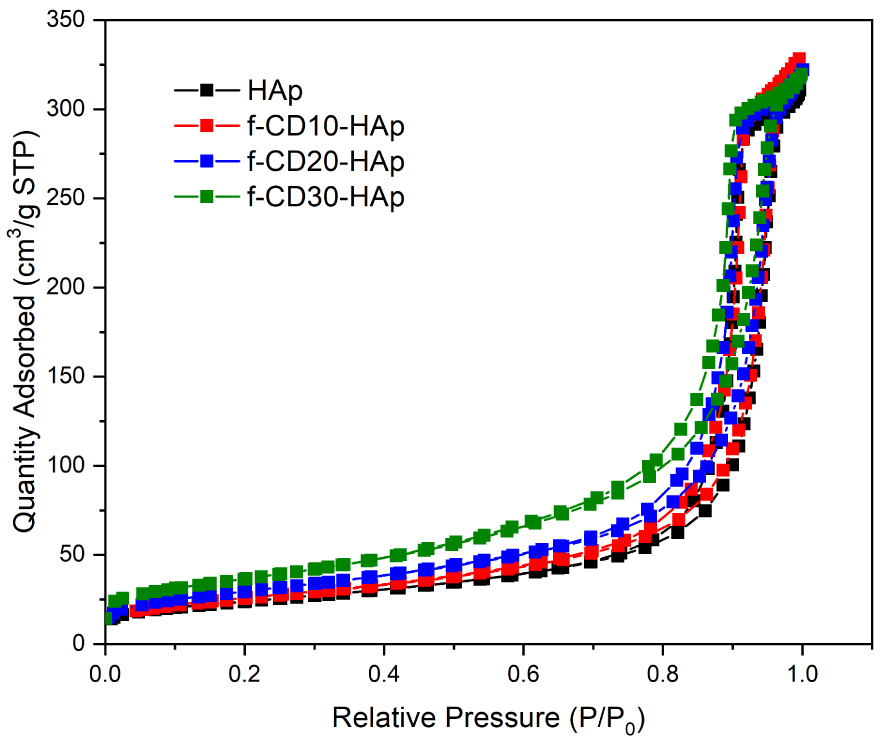
3.5. Drug release behavior
The loading of acetaminophen was done through encapsulation during the synthesis process of composite. The release of acetaminophen from the composite (Figure 9a) shows the fully released drug after 24 h. After 4 h, the drug release is around 89, 84, 73, and 60% for HAp, f-CD10-HAp, f-CD20-HAp, and f-CD30-HAp, respectively, while after 8 h, HAp has fully released acetaminophen, and other samples of f-CD10-HAp, f-CD20-HAp, and f-CD30-HAp have released around 96, 92, and 80% acetaminophen, respectively. The current study shows slower release profile in comparison with previous results in releasing doxorubicin from CQD-HAp nanorod, in which full release of drug was achieved after around 10 h [7], and release of acetaminophen using CQD/HAp was fully achieved after 8 h [8].
(a) Drug release, and (b) first-order kinetics profiles of samples.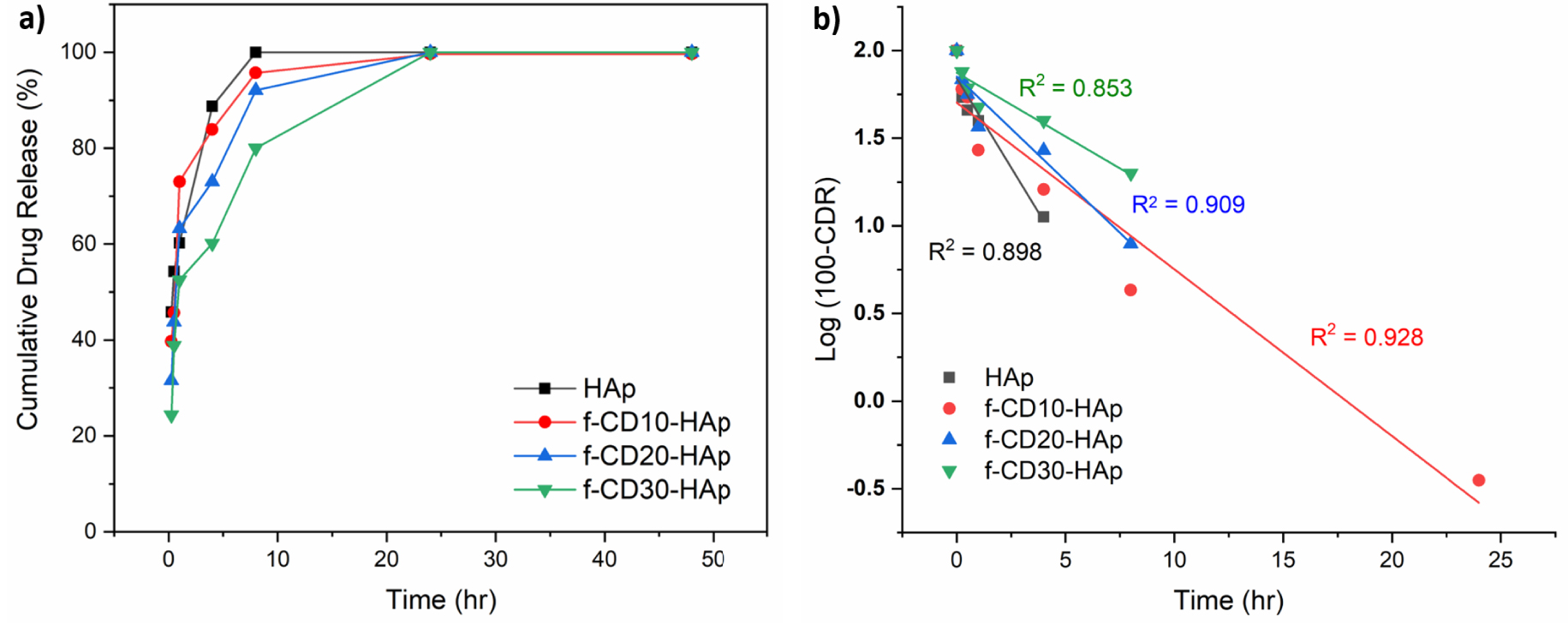
The improvement of controlled release of acetaminophen can be observed by increasing the loading of amino-functionalized CQDs in the HAp matrix. The availability of higher amount of functional groups from amino-functionalized CQDs provides higher electrostatic and π–π interactions, which help slow the release of acetaminophen [13, 43]. Additionally, the high surface area of the composite helps to load and retain the drug, which sustains the release of the drug. The results show agreement with previous study on the synthesis of N-CQD/dox/HAp hydrogel, which shows slow release of doxorubicin from the composite hydrogel [44]. The ionic interaction and hydrogen bonding can happen between N-CQD and HAp, also with N-CQD and doxorubicin, which affects the prolonged release of the drug [44]. The release of acetaminophen fits the first-order drug release profile (Figure 9b), which is time-dependent at constant rate of around 0.204, 0.095, 0.119, and 0.073 h−1 for HAp, f-CD10-HAp, f-CD20-HAp, and f-CD30-HAp, respectively.
4. Conclusion
The loading of amino-functionalized CQDs in HAp enhanced the photoluminescence intensity and absorption of composite, which can benefit imaging in drug delivery applications. The dispersion of amino-functionalized CQDs in HAp induced the porosity in the HAp matrix, which can appear due to inter-particular interaction between CQDs and HAp, increasing the surface area of HAp to 129.94 m2/g. The availability of functional groups in CQDs complements HAp in maintaining the electrostatic interactions and addition of π–π interaction, which slows the release of acetaminophen.
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgement
The work was financially supported by Universiti Teknologi PETRONAS–Universitas Muhammadiyah Surakarta Collaborative research grant 015ME0-256.