

 CC-BY 4.0
CC-BY 4.0
L’histoire du début de l’extension du tableau périodique au-delà de l’uranium est étroitement liée à la physique nucléaire et à la radiochimie des années 1935 à 1945 qui étaient pratiquées dans quelques laboratoires d’Europe et des Etats-Unis. C’est en 1944 que Seaborg a avancé le concept d’actinides. Un article qui englobe ce sujet a été publié aux Comptes Rendus de Physique [1]. Cet article des Comptes Rendus de Chimie traite plus explicitement de la chimie qui a conduit aux découvertes des quatre premiers éléments transuraniens (neptunium, plutonium, américium et curium) et à leur place dans le tableau périodique au sein de la série des actinides, analogue à la série des lanthanides.
Evolution du principe de classification des éléments chimiques
Le tableau périodique des éléments chimique a été construit entre 1869 et 2016. En 1869 Mendeleev propose un système pour classer la soixantaine d’éléments bien connus à cette époque en fonction de leur poids atomique relatif à celui de l’hydrogène. Ce système consiste à ranger les éléments en séries (horizontales) et groupes (verticaux) de façon à faire apparaître les analogies de comportement chimique des éléments classés dans les groupes. A cette époque les oxydes étaient bien connus [2]. Le tableau qui en résulte, même s’il a été précédé d’autres tableaux correspondants à d’autres façons de classer les éléments, a été retenu comme le premier « tableau périodique » en raison de son pouvoir de prédiction. En effet, il comportait des cases vides pour des éléments inconnus qui ont été découverts ultérieurement et dont les propriétés se sont révélées conformes aux prédictions de Mendeleev. En 2016 l’IUPAC a donné des noms aux derniers éléments dont quelques isotopes éphémères avaient été découverts entre 1968 et 2010. Tout ceci est bien connu et a été largement commenté au cours de l’Année Internationale du Tableau Périodique en 2019 [3].
Aujourd’hui le tableau périodique comporte 118 cases réparties en 7 périodes et 18 colonnes, chacune renfermant les isotopes des 118 éléments. Les éléments de chaque colonne sont des homologues. Chaque élément est formé d’atomes. Chaque atome est caractérisé par deux nombres attachés à son noyau : Z, le nombre de protons et A, la somme des nombres de protons et de neutrons, N, (A = Z + N). Pour un élément tous les isotopes ont la même valeur de Z et les valeurs de N varient. La valeur de Z est le numéro atomique de l’élément. Le nombre d’isotopes d’un élément est fixé par les valeurs de N. Le nombre d’électrons d’un atome est égal à Z. A chaque valeur de Z, c’est-à-dire à chaque élément, correspond une structure électronique définie en termes du remplissage des orbitales atomiques de type s, p, d et f. Depuis mai 2019 le poids atomique d’un isotope AZX est le poids de N d’entre eux, N, étant le nombre d’Avogadro, une constante universelle (N = 6,02 × 1023). Le poids atomique d’un élément est le poids pondéré des isotopes qui le constituent.
Les noyaux sont stables pour des valeurs de Z et N fixées. Si N n’a qu’une valeur l’élément est mono-isotopique. Si elles s’en écartent ou si N est très supérieur ou très inférieur à Z ou si Z est supérieur à 83 les noyaux se désintègrent. Les atomes correspondants sont radioactifs et disparaissent alors selon leur période radioactive, T1∕2, en émettant divers rayonnements. Les éléments dont tous les isotopes sont radioactifs sont des radioéléments (Z > 83). Au-delà de Z = 98 les radioéléments n’existent qu’en quantité impondérable et les plus lourds ne sont produits qu’à l’échelle de quelques atomes, qui aussitôt nés se désintègrent.
Toutes ces notions sont enseignées. Elles sont rappelées ici car dans ce qui suit la relation : isotope stable ou radioactif-élément chimique est constamment évoquée.
Avant 1913 un élément chimique était compris comme une entité élémentaire qui se conservait dans les réactions chimiques et son poids atomique était mesuré en déterminant les proportions dans lesquelles il se liait à l’hydrogène et à l’oxygène en prenant comme unité de poids, le poids de l’hydrogène. En 1930 la notion moderne d’élément était acquise et le principe de la construction du tableau périodique pouvait démarrer sur les bases des structures nucléaire et électronique des éléments. Pendant longtemps les chimistes ont seulement expérimenté sur la matière en quantité macroscopique. Avec la synthèse d’isotopes radioactifs (appelés aussi radionucléides) de très faibles périodes, les radiochimistes ont conduit des expériences sur des quantités impondérables de matière dont le comportement pose un problème d’échelle de plusieurs puissances de dix pour en déduire le comportement de l’élément qu’il représente [4].
Construction du tableau périodique
L’attribution des éléments aux cases du tableau périodique s’est faite au cours de quatre transitions (Figure 1) correspondant (1) à la découverte des éléments naturels (Antiquité-1925), (2) des radioéléments naturels en filiation avec 238U, 235U et 232Th (1896–1939), (3) des radioéléments artificiels produits avec des ions légers accélérés (1937–1961) et finalement (4) des radioéléments artificiels produits avec des ions lourds accélérés (1968–2010). Les trois dernières ont été initiées par des découvertes majeures : radioactivité naturelle (1896) pour la transition 2, radioactivité artificielle (1934) et accélération des ions par des cyclotrons à partir de 1930 pour les transitions 3 et 4. La transition 4 est bornée à 2010, car depuis cette date et pour longtemps la quête de l’élément Z = 119 est restée vaine. C’est la découverte de la radioactivité naturelle qui a permis de disposer des premiers ions accélérés à des énergies suffisantes pour découvrir le noyau des atomes et les transformer par réactions nucléaires. La construction du tableau périodique est liée à l’évolution de la conception d’un élément chimique.
Années de début et de fin des transitions de la construction du tableau périodique. L’initiateur de la transition 1, qui est celle de la découverte des éléments naturels, est D. Mendeleev. Les découvertes des rayons X (W. Roëntgen) et de la radioactivité naturelle (H. Becquerel) ont initié la transition 2 correspondant à la découverte des radioéléments naturels. Celle de la radioactivité artificielle (I. et F. Joliot) a initié la transition 3 au cours de laquelle les isotopes radioactifs des radioéléments artificiels ont été produits en utilisant des cyclotrons et des réacteurs nucléaires (objet de cet article). La transition 4 est celle au cours de laquelle les isotopes des éléments 6d et 7p ont été produits avec des accélérateurs d’ions lourds.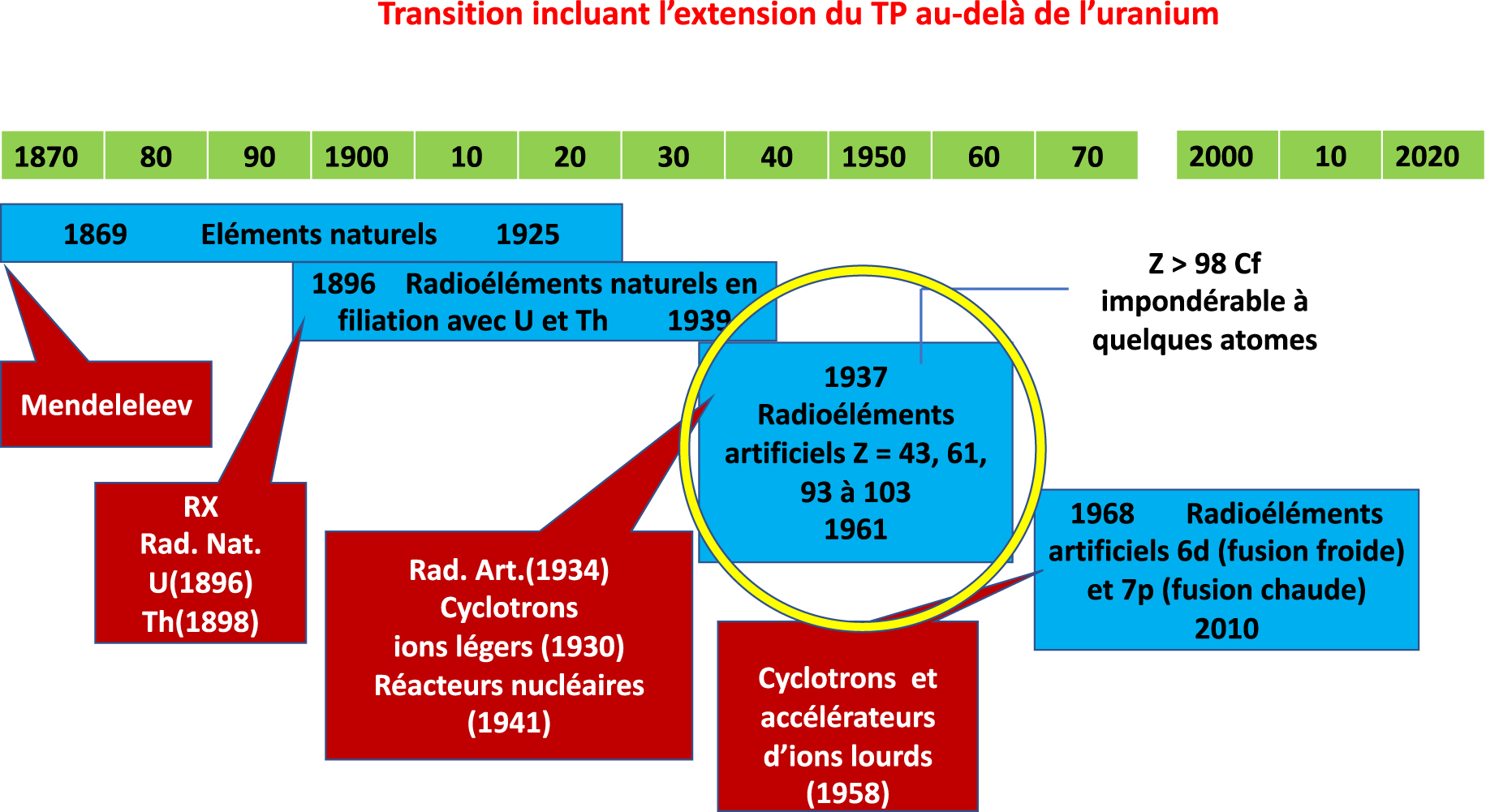
Au-delà de l’uranium
Le premier radioélément transuranien le neptunium a été découvert en 1940 par Edwin McMillan à Berkeley, à la suite d’observations sur les radioactivités produites par irradiation de l’uranium avec des neutrons, irradiations qui avaient débuté en 1934 au lendemain de la découverte de la radioactivité artificielle par Irène et Frédéric Joliot [5]. Ils avaient montré que les rayons alpha du polonium transformaient 27Al mono-isotopique en 30P selon la réaction nucléaire : 27Al(α, n)30P et que 30P se désintégrait par émission beta plus (β+) avec une période de 2,5 minutes en donnant 30Si, stable. Ils avaient aussi irradié d’autres éléments légers avec les particules alpha et montré que l’on pouvait produire des isotopes radioactifs émetteurs beta moins (β−), type de radioactivité bien connue pour les radionucléides naturels. Ils disposaient aussi de sources de neutrons Po/Be tout comme les laboratoires de radiochimie. Dès la découverte des Joliot, Enrico Fermi a, le premier, irradié à Rome de l’uranium avec des neutrons dans le but de synthétiser des isotopes d’éléments transuraniens et il a cru que certaines radioactivités isolées par voie chimique d’une multitude d’autres, étaient la signature de deux isotopes radioactifs d’éléments de Z > 92. Ses résultats ont été critiqués et ses expériences ont été reprises à l’Institut du Radium à Paris et au Kaiser Wilhem Gesellshaft Institute à Berlin. Les expériences d’Irène Joliot-Curie (Paris) et de Otto Hahn (Berlin) ont, en fait, conduit Otto Hahn à la découverte de la fission de l’uranium [6], une découverte majeure dans l’histoire de l’humanité. C’est en étudiant les produits de fission que Joliot a fait une observation essentielle reprise par McMillan.
Avant d’aborder les expériences de McMillan il convient de faire un point sur ce que l’on savait de la physique nucléaire vers 1935 pour synthétiser des noyaux au-delà de Z = 92 et des moyens dont disposaient les expérimentateurs pour identifier les isotopes radioactifs et les attribuer à un élément.
Synthèse nucléaire
Le processus nucléaire : AZX (n, γ) A+1ZX (β−) A+1Z+1X, capture d’un neutron par un isotope lourd avec émission d’un photon gamma, suivi d’une désintégration β− du nouvel isotope formé permet de gagner une unité dans l’échelle des Z, c’est-à-dire de passer d’un élément au suivant dans le tableau périodique. C’est l’ajout du neutron à AZX qui rend, en général, A+1Z+1X radioactif β−. On peut aussi gagner deux unités en Z par irradiation de AZX avec des particules alpha : AZX (He2+, n) A+3Z+2X. C’étaient, dès 1935, les voies les plus faciles à emprunter car on disposait de sources de particules alpha et de neutrons. Les premières à base de sels de 212Po (α, 138 j) ou de 226Ra (α, 1600 a) ou de gaz 222Rn (α, 3,8 j) et de leurs descendants pouvaient avoir une activité alpha de 1 Ci et les autres, mélanges de sels de 226Ra et de béryllium donnaient environ 107 neutrons/s/Ci/4π par le processus : 10Be(α, n) 13C. Les neutrons rapides émis par ce processus pouvaient être ralentis dans la paraffine.
En 1939 les deux cyclotrons de Berkeley aux USA délivraient des ions légers : p (1H+), d (2H+) et α (4He2+) mais aussi des neutrons rapides en bombardant une feuille de Be avec des deutons, ce qui donnait lieu à la réaction 10Be(d, n)11B. On pouvait espérer gagner une ou deux unités en Z par des réactions telles que : AZX (p, n) AZ+1X, AZX (d, n) A+1Z+1X, AZX (d, 2n) AZ+1X ou AZX (He2+,2n) A+2Z+2X. L’espoir de pouvoir synthétiser et détecter dN2 radionucléides en irradiant N1 isotopes (stable ou radioactif) d’une cible, pendant dt, avec un flux Φ de particules est proportionnel à la section efficace de la réaction, σ = dN2∕ΦN1 dt et au rendement, R, de détection de l’activité des radionucléides formés, AN2 = R[0,7∕T1∕2N2], R dépendant de l’instrumentation. Joliot avait donné la preuve de la formation de 30P sur 2,5 × 104 atomes (100 Bq), ce qui correspond sensiblement à la production de quelques atomes par mCi de 210Po dans la source.
A la fin de 1939 on savait que l’irradiation de l’uranium, 238U et 235U (0,7%), avec les neutrons émis par les sources donnaient un effroyable mélange de radioactivités mais les sections efficaces de production des isotopes radioactifs responsables de ces radioactivités étaient très faibles de sorte qu’il fallait irradier longtemps les cibles avec les sources à base de radionucléides naturels. La production de radionucléides artificiels était terriblement augmentée avec des faisceaux de particules accélérées ou avec des neutrons de cyclotrons car les flux étaient sans commune mesure avec ceux des sources. A Berkeley les chimistes avaient accès à deux cyclotrons de 37 et 60 inches construits par Ernest Lawrence et Stanley Livingston. Le flux de neutrons produit par des faisceaux de deutons de 16 MeV et de 10−3 microampères bombardant une feuille de Be étaient équivalent à celui d’une source de neutron de 0,1 Ci en émetteur alpha et des intensités de plusieurs centaines de microampères étaient courantes.
Moyens de détection de la radioactivité
Vers 1939 les moyens de détection et de mesure de l’intensité d’un rayonnement alpha, beta ou gamma était la chambre d’ionisation et le compteur Geiger Muller (GM). Les énergies des rayonnements étaient mesurées par absorption dans des écrans métalliques. La chambre d’ionisation à amplificateur proportionnel, pour mesurer l’énergie des rayonnements alpha était à ses débuts. La chambre de Wilson permettait d’avoir une image photographique des traces d’ionisation des rayonnements dans une atmosphère saturée d’eau. Les rendements des GM étaient variables.
Identification chimique
Depuis Marie Curie la seule méthode d’identification chimique d’un radionucléide en quantité impondérable était la coprécipitation [7] qui consiste, en solution, à entrainer dans un solide un élément trace (radionucléide) par un élément en quantité pondérable (de propriétés chimiques voisines) puis à dissoudre le précipité et à recommencer des coprécipitations successives dans le but d’enrichir les précipités en élément trace (Figure 2). Les résultats essentiels des lois de la coprécititation étaient les suivantes. Si par entraînement d’un élément par un autre on peut enrichir les précipités on a affaire à deux éléments, s’il n’y a pas d’enrichissement on a affaire à deux isotopes. Dans l’application de cette méthode l’idée était que les éléments homologues du tableau périodique ont des propriétés voisines. Elle est facile à mettre en œuvre si l’élément trace est représenté par un isotope radioactif qui est facilement détectable. La coprécipitation avait permis d’identifier tous les descendants des familles radioactives de 238U, 235U et 232Th (deuxième transition du tableau périodique).
Illustrations de l’identification des radionucléides produits en quantités impondérables. (A) coprécipitation. L’ajout de réactif (H2S ou HCl) à une solution 10−11 M de 210Po ou de 228Ra ne provoque pas de précipité. Après ajout d’un sel de Bi ou de Ba 10−3 M, l’ajout de réactif (H2S ou HCl) précipite Si3Bi2 et Po ou BaCl2 et Ra. Les quantités x et y de micro-composant et de macro-composant dans les précipités sont liées au facteur de séparation F. Ces exemples correspondent aux découvertes de Po et Ra par M. Curie. B) Chromatographie. Elution goutte à goutte des radionucléides M3+, Z = 63 à 71 et Z = 96 à 102 à partir d’une résine échangeuse de cations Dowex-50 par l’alpha-hydroxy-isobutyrate d’ammonium.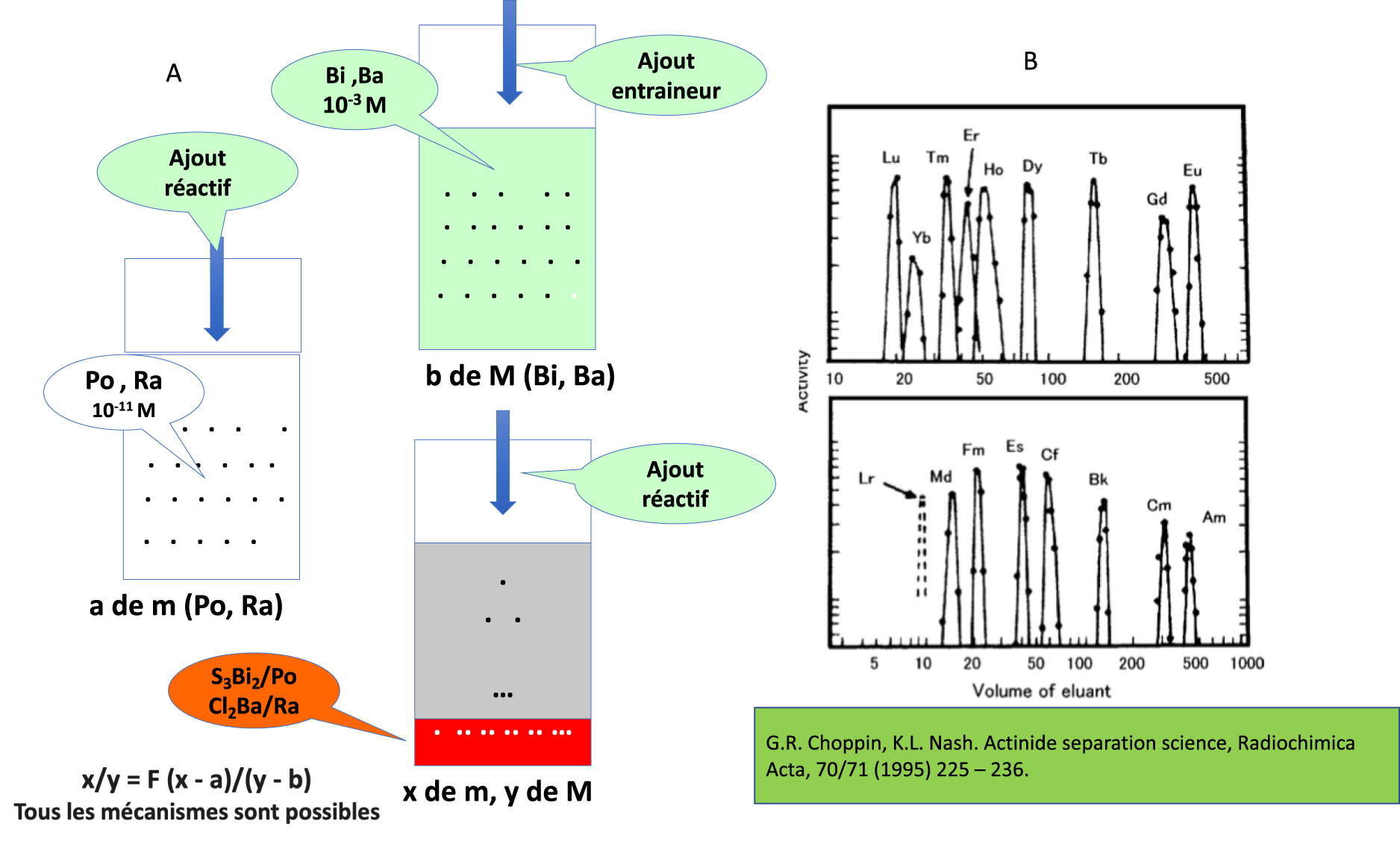
Situation sur l’irradiation de l’uranium par les neutrons en 1939
Les expériences d’irradiation de l’uranium par des neutrons lents ou rapides conduites avant 1939 avaient conduit à de nombreuses observations par exemple que les neutrons lents étaient plus efficaces que les rapides et que la fission était 15 fois plus probable que la capture d’un neutron. Deux ont été particulièrement importantes dans le contexte de cet article. Les faibles activités des radionucléides formés par irradiation avec des sources, mesurées au GM, n’étaient que 4 à 5 fois supérieures à celles des descendants de 238U : 234Th (β, 24,1 j) et 234mPa (β, 1,2 min) émetteurs beta. Irène Joliot-Curie a montré en 1938 [8] que si on séparait Th et Pa de l’uranium l’identification des radionucléides formés était facilitée. Aussi après 1938 a-t-on irradié de l’uranium radiochimiquement pur, c’est à dire exempt de ses descendants, mais pour une vingtaine de jours seulement. Pour cela Th et Pa étaient entrainés par Fe(OH)3 d’une solution carbonatée d’uranium et l’uranium était précipité sous forme de U2O7(NH4)2. Hahn a suivi ce protocole pour montrer la fission de U. Il a identifié le produit de fission 140Ba (β, 12, 8 j) sur 3,6 106 atomes coprécipités avec BaCl2. Joliot a montré[9, 10] en 1939 que la fission de 238,235U par les neutrons donnait 2 à 3 neutrons rapides libres et que les produits de fission s’échappaient d’une fine couche de U2O7(NH4)2 irradiée aux neutrons, en emportant et se partageant sous forme d’énergie cinétique les 200 MeV d’énergie de fission.
Découverte de neptunium
Avant 1939 toutes les expériences d’irradiation de l’uranium étaient faites en Europe et suivies par les chercheurs de Berkeley. En confirmant les expériences de Joliot, McMillan remarque au printemps 1940 qu’une radioactivité 𝛽 de 2,3 j, facilement mesurable au GM, reste dans la fine couche de U2O7(NH4)2 irradié aux neutrons [11]. Manifestement elle n’est pas due à un produit de fission, mais elle est compatible avec une émission β d’un radionucléide AZX dans laquelle l’énergie de recul du fils AZ+1X est quasi nulle (100 eV), par exemple celle de 23992U émetteur beta, formé par capture radiative d’un neutron par 23892U, et donnant 23993X, lui-même émetteur beta. Selon cette hypothèse 23993X devait être un isotope d’un homologue du rhénium. McMillan et Philip Abelson [12] irradient U2O7(NH4)2 au cyclotron de 60 inches de Berkeley, pendant une heure avec des neutrons produits par un faisceau de deutons de 100 microampères. Dans ces conditions environ quelques 107 atomes de nouveaux isotopes avaient été formés.
Sur la base de mesures de radioactivité ils montrent que l’activité de 2, 3 j de X provient en fait de la décroissance d’une activité 𝛽 de 23 min, d’énergie plus élevée que la première. Elle est attribuée à 239U une activité déjà mesurée par Fermi dès 1935. Le processus de formation de 23993X selon la séquence : 23892U (n,γ) 23992U (β, 23 min, Emax = 1 MeV) donnant 23993X (β, 2,3 j, Emax = 0,5 MeV ) est confirmé. Jusqu’à ces expériences la période de 239U n’était pas claire, Fermi et Hahn avait proposé des valeurs différentes.
Pour vérifier ces conclusions et identifier les propriétés chimiques de 23993X = X (pour simplifier) ils commencent les expériences de coprécipitation sur la solution de dissolution de la cible dans HCl en prenant Re comme entraîneur. Pour cela du ReO4NH4 est ajouté à la solution. Les expériences sont négatives, X n’est entrainé ni par ReS2 (traitement par H2S) ni par Re métal (réduction par Zn) ni même par volatilisation avec ReO3 (oxydation par chauffage avec H2SO4). X serait-il particulier pour ne pas être classé comme homologue de Re ? et les éléments transuraniens en général seraient- ils aussi particuliers, avec des propriétés ? plus proches de celles de U que de celles des éléments classés dans les colonnes 8 (Z = 94), 9 (Z = 95) et 10 (Z = 96) du tableau périodique ?
McMillan et Abelson comparent alors les propriétés sommaires de X et de U lors d’un cycle redox en particulier pour explorer les degrés d’oxydation ou les valences de X. Cela est possible puisque la solution de dissolution de la cible contient 238U en quantité pondérable, 239U et X en quantités impondérables. Ils procèdent à des coprécipitations sachant que l’acide acétique précipite U(VI) sous forme d’acétate double de sodium, que les fluorures de Ce (III,IV) ou Th(IV) entrainent respectivement les éléments trivalent M(III) et M(IV), que BrO3K (couple ) est oxydant en milieu acide et que SO2 (couple ) est réducteur dans ce même milieu. Les conditions dans lesquelles les expériences historiques ont été conduite ne sont pas toujours bien précisées. Par ailleurs il s’est agi de chimie qualitative. Les résultats qui suivent correspondent à des conditions idéalisées pour la clarté de l’exposé.
L’ajout d’acide acétique à la solution de dissolution de la cible précipite l’acétate double de 238U(VI) et de sodium qui entraîne 239U, X reste en solution. L’ajout de BrO3K ne change rien au comportement des isotopes d’uranium vis-à-vis de l’acide acétique mais l’acétate de 238,239U(VI) entraîne X : cela indique que X est oxydé de X(?) à X(VI). Le traitement de la solution par SO2 ne change rien non plus au comportement de l’uranium mais X reste en solution : cela n’indique rien sur la réduction possible de X.
Pour la coprécipitation sous forme de fluorures le milieu réactionnel est préparé par ajouts de sels solubles de cérium et/ou thorium à la solution de dissolution. L’ajout de HF ne conduit à aucun entraînement de X par CeF3 ou ThF4 : X n’est ni trivalent, ni tétravalent mais comme il peut être oxydé en X(VI) cela montre que dans HCl il est sous la forme de X(V). Après oxydation par le bromate il en est de même : X est oxydé en X(VI). Après réduction par barbotage de SO2 dans le milieu, X est entrainé par ThF4 : X est réduit en X(IV).
Ces simples expériences ont conduit à établir l’existence de 23993X(V) isotope d’un nouvel élément dont le degré d’oxydation stable est 5 et qui peut être oxydé et réduit. X est rapidement nommé neptunium par McMillan et Abelson.
Ils montrent qu’on peut séparer Np et U d’un cible d’uranium irradiée aux neutrons par un cycle redox : BrO3K + SO2 qui amène Np au degré d’oxydation 4, Np(IV), lequel peut alors être coprécipité avec ThF4. Np est pur et séparé des produits de fission.
Découverte de plutonium
A la suite de la découverte de Np, McMillan et Abelson émettent l’hypothèse d’une « Série des Uranides » et Seaborg [13] entre dans la course aux éléments de Z > 93. Ils suspectent que 239Np (β, 2,3 j) peut donner, par décroissance, un isotope de Z = 94, émetteur α. Effectivement une très faible activité α de très longue période est détectée avec un compteur proportionnel. Mais pour en avoir suffisamment afin d’en mesurer l’activité il faut augmenter la production de 239Np et cela est sans espoir compte tenu de la faible section efficace de la réaction de capture radiative de 238U et des flux de neutrons disponibles avec les cyclotrons de Berkeley. Pour franchir deux unités en Z à partir de 238U il faut chercher une autre voie, par exemple l’irradiation de l’uranium, avec des particules chargées : une réaction (d,2n) suivie d’une émission β semble possible pour passer à Z = 92 à Z = 94.
En août 1940 Glen Seaborg, McMillan, Joseph Kennedy, Arthur Walh lancent l’irradiation de 5,5 g de U3O8 (purifié) avec un faisceau mixte de neutrons et de deutons au cyclotron de Berkeley : une heure et 200 microampères. L’idée est de produire au moins 239Np (connu) comme référence, formé par les neutrons, et d’autres isotopes de Z = 94 formés par les deutons et émissions beta.
La cible de U est traitée par le cycle redox connu BrO3K/SO2 pour isoler Np dans ThF4. Après ce traitement Np est relativement pur.
En décembre 1940 ils montrent sur la base de mesures de radioactivité que ThF4 coprécipite bien 239Np (β, 2,3 j, Emax = 1 MeV) mais aussi un autre radionucléide donnant une activité beta X (β, 2,1 j, Emax = 1 MeV), qui décroit en donnant une activité α, laquelle augmente avec le temps : Y(α). Les deux radioactivités ont sensiblement même période et même énergie. Ils comparent alors les comportements redox des radionucléides associés à X (β, 2,1 j) et Y(α) avec le comportement connu de 239Np (β, 2,3 j) qui sert de référence.
Le précipité ThF4 est dissous en milieu HCl concentré. Après oxydation de la solution par le bromate Np(β, 2,3 j) et X (β, 2,1 j) ne sont pas entrainés par ThF4 mais Y(α) l’est : cela indique que X (β, 2,1 j) est probablement un isotope de Np et qu’il est bien oxydé en Np(VI) comme Np(β, 2,3 j) tandis que Y(α) ne l’est pas. En revanche si la solution est traitée par le persulfate de sodium en présence d’argent, Y(α) n’est pas entrainé par ThF4 (et les isotopes de Np non plus) : Y(α) est oxydé au degré d’oxydation 6 mais plus difficilement que Np. En milieu réducteur, imposé par SO2, tous les radionucléides sont coprécités par ThF4 : Y(α) est au degré d’oxydation 4 comme les isotopes de Np. De ces expériences l’équipe de Seaborg déduit que le degré d’oxydation stable de Y(α) est 4. En 1940 le réactif S2O8Na2/Ag (couple ) était le plus puissant oxydant connu.
En février 1941 les conclusions de l’équipe de Seaborg sont les suivantes. Au plan nucléaire X(β, 2,1 j) est le radionucléide 238Np formé par le processus 238U(d,2n) 238Np, Y(α) est le fils de 238Np : 23894Y(α), c’est un isotope du deuxième élément transuranien. 238Np est un nouvel isotope de Np. La formation de 239Np provient de la voie neutronique et de la réaction nucléiare 238U(d, n) 239Np. Au plan chimique 23894Y(𝛼) est tétravalent alors que Np est pentavalent et il est beaucoup plus difficile à oxyder que Np. L’élément correspondant à 23894Y(α, 80 a) sera nommé Pu en 1942 et la publication dans la littérature ouverte sur ce sujet date de 1946 [14]. En effet dès 1941 les recherches sur l’élément 94 sont plus ou moins secrètes et à partir du début de 1942 elles sont couvertes par le secret militaire dans le cadre du « Manhattan Project ». Dès janvier 1941 Seaborg informe Washington par notes des avancées sur l’élément 94. En 1946 quand seront dévoilées les recherches on en saura d’ailleurs plus sur les propriétés de Pu que ne l’indique la publication.
Très rapidement Seaborg et al montrent que l’on peut séparer Pu et Np de U et des produits de fission par un cycle redox en deux étapes : (1) dissolution de l’uranium irradié en milieu acide, traitement par S2O8Na2 puis par SO2 suivi par la coprécipitation de Np(IV) et Pu(IV) avec ThF4, (2) reprise de ThF4 en milieu acide puis oxydation de Np(IV) en Np(V) par BrO3K et coprécipitation de Pu(IV) avec ThF4, Np(V) restant en solution.
La découverte des propriétés chimiques de 238Pu proches mais différentes de celle de Np et de U a conforté l’idée de l’existence d’une série d’uranides, regroupant U et les éléments transuraniens mais toutes les expériences allant dans ce sens avaient porté sur des quantités impondérables. Pour obtenir des quantités pondérables de Pu Seaborg emprunte la voie neutronique qui conduit à 239Pu par l’intermédiaire de 239Np. En mars 1941 1,2 kg de UO2(NO3)2, enfermé dans un bloc de paraffine, est irradié 2 jours au cyclotron de Berkeley avec des neutrons lents pour obtenir 239Np. Le nitrate est dissous dans HNO3, l’uranium est extrait par solvant au diéthyléther et la solution aqueuse est oxydée par BrO3K. 239Pu qui reste au degré d’oxydation 4 est coprécipité avec LaF3. Il faut l’enrichir en Pu. Après 6 cycles : coprécipation-dissolution-coprécipation successifs, le précipité est du 239PuF4 quasi pur, il pèse 0,5 μg. Seaborg montre que 239Pu fissionne 50 fois mieux que 235U. C’est une découverte qui laisse entrevoir la possibilité de réaliser un engin nucléaire par une voie différente de l’obtention de 235U quasi pur par enrichissement isotopique de 238,235UF6. 235U avait été découvert en 1935 et rendu visible en 1940. En septembre 1942 les recherches sur les propriétés chimique de Pu continuent dans le « Plutonium Project » à Chicago, partie intégrante du Manhattan Project.
Elles commencent par l’irradiation de 10 kg de U3O8 aux cyclotrons de Berkeley et de St Louis. Les lots d’oxyde d’uranium sont traités à Chicago. On étudie des protocoles industrialisables de séparation de Pu. Le premier composé visible de Pu obtenu en 1942 est un échantillon de 2,8 μg de PuO2. Il est obtenu à partir de 10 microlitres d’une solution concentrée de 239Pu. La quantité totale de 239Pu produite au cyclotron n’a été que de 2 mg.
La production industrielle de 239Pu, pour faire une bombe, qui demande quelques kilogrammes de cet isotope, passe par le traitement du combustible d’un réacteur nucléaire plutonigène. Fin 1944, à Hanford, 3 réacteurs plutonigène et une usine de traitement donnent 50 à 100 g/j de « Pu militaire », 239Pu pur à 97,3%. La première bombe atomique au Pu est testée à Alamogordo en juillet 1945. Aujourd’hui 1 tonne de combustible usé des réacteurs électrogènes contient environ 10 kg de « Pu civil » de composition isotopique variable, 238 à 243Pu, selon le taux de combustion [15].
Découvertes de curium et américium
En 1944 McMillan et Seaborg pensent que les éléments Z = 95 et Z = 96 auront les mêmes propriétés que Np et Pu. Ils irradient une cible de 239Pu au cyclotron de Berkeley par des deutons dans l’espoir de produire un isotope de Z = 96 et par des neutrons dans l’espoir d’avoir un isotope de Z = 95. Cela conduit bien à des radioactivités nouvelles mais elles sont insensibles aux cycles redox. Seaborg émet alors l’hypothèse de l’existence d’une série d’éléments similaires aux lanthanides : les actinides. Alors les éléments Z = 95 et Z = 96 sont homologues de Eu et Gd et sont trivalents [16]. Cette intuition géniale va se vérifier.
En 1944 Seaborg, Ralph James, Léon Morgan et Albert Ghiorso irradient 239Pu avec des He2+ pour obtenir des isotopes de l’élément Z = 96 car les neutrons fissionnent 239Pu et compliquent les mesures. Ils identifient le radionucléide 24296X (α, 163 j) par coprécipitation avec LaF3. Il est formé par la réaction : 239Pu(He2+,n) 242Cm . X est nommé curium. Cm est bien trivalent. Ils montrent par ailleurs que 242Cm donne, par émission α, 238Pu qui était déjà connu. Dans ces expériences il n’est pas possible d’isoler un isotope de Z = 95.
Entre temps une intense source de neutrons était devenue disponible à Chicago : le cœur du réacteur nucléaire historique de Fermi de 1942 qui avait été reconstruit.
En 1945 Ghiorso, James, Morgan, Seaborg [17] irradient 239Pu dans ce réacteur. En réacteur les processus nucléaires s’enchaînent et tout devient plus compliqué pour trier les radionucléides. Ils identifient 24195X (α, 443 a) par coprécipitation avec LaF3. Il est formé par la suite de processus : 239Pu (n, γ) 240Pu(n, γ) 241Pu(β, 14 a) 241Am . X est nommé américium. Am est bien trivalent. Ils montrent que 242Cm qui venait d’être découvert est aussi produit en réacteur selon : 241Am(n, γ) 242Am(β, 14 a) 242Cm.
En 1945 Seaborg publie un tableau périodique incluant le début de la série des actinides [16].
Découverte des éléments transcurien
Entrer dans les détails demanderait un long développement. L’idée en 1945 était qu’on pouvait synthétiser progressivement des isotopes des éléments de Z > 96 par des réactions nucléaires appropriées tout en sachant que leur production allait diminuer drastiquement avec l’augmentation de Z. Il fallait avoir : le bon isotope à irradier, des faisceaux intenses de particules, des détecteurs appropriés très sensibles, des procédés de séparations et d’identifications plus rapides que la coprécipitation et des techniques nucléaires rapides pour une chimie à l’échelle de l’atome. Pendant les 10 ans entre 1949 et 1961 ces conditions seront peu à peu réunies et on découvrira les éléments Bk jusqu’à Lr (Tableau 1). Il convient de noter ici une avancée majeure dans l’identification des éléments Am à Lr qui confirme également l’hypothèse des actinides. Il s’agit de l’ordre d’élution de ces éléments au degré d’oxydation 3, fixés sur une colonne de résine échangeuse d’ions, par un complexant très sélectif, qui commençait à être utilisé à l’époque, l’alpha-hydroxy-isobutyrate-d’ammonium. L’élution se fait en ordre inverse de Z comme pour les lanthanides de Eu à Lu. Elle est plus rapide que la coprécipitation.
Conditions des découvertes des éléments transcurien [13]
| Symbole et renvois à l’article de la découverte | Date de découverte | Processus de découverte | Conditions de découverte | Article de la découverte |
|---|---|---|---|---|
| Bk [22] | 1949 | 241Am(He,2n) 243Bk(α, ) 241Am(He,n) 244Bk(CE, 4.5 h) EHe = 35 MeV, 10 μA/cm2, Berkeley | 7 mg de 241Am, plusieurs milliers d’atomes de Bk | [22] |
| Cf [23] | 1950 | 242Cm(He, n) 245Cf(α, 44 min) EHe = 35 MeV, 10 μA/cm2, Berkeley | μg de 242Cm, 5000 atoms de Cf | [23] |
| Es Fm [24] | 1955 | 238U(15n, 7e-) 253Es(α, 20 j) 238U(18n, 8e-) 255Fm(α, 22 h) Argonne National Laboratory, Chicago, Los Alamos Scientific Laboratory, Albuquerque | kg de 238U, centaines d’atomes de Es et Fm. Mike explosion, U absorbe plusieure fois 1024 neutrons en quelques (quelques moles de neutrons). Produits aussi par irradiation aux neutrons de grammes de Pu dans le réacteur à haut flux MTR à Idaho. | [24] |
| Md [25] | 1955 | 253Es(He, n) 256Md(EC, 1.3 j) 256Md donne 256Fm(SF, 2.6 h) EHe = 41 MeV, 100 μA/cm2, Berkeley | 109 atomes of 253Es(α, 20 h) produits à Idaho. 17 atomes de 256Md produits “one atom at a time”. Réaction parasites (α,n) sur 244Cm donnant 246Cf | [25] |
| No [26] | 1958 | 246Cm(12C,4n) 254No and 244Cm(12C,4n) 252No. 254No donne 250Fm(α, ) 252No donne 248Fm(α, 36 s) HILAC Berkeley | Le groupe de Berkeley identifie sans ambiguité 254No sur la base de l’existence de 250Fm (11 atomes détectés à la position correcte d’elution) et les caractéristiques de décroissance de 252No. D’autres groupes utilisant 244Cm et 13C - Stockholm - ou 239Pu et 16O - Dubna - n’ont pas identifié les produits de réaction. En 1964, le groupe Dubna a identifié 256No(α, ) comme parent de 252Fm. L’existence de No a été confirmée. | [26] |
| Lr [27] | 1961 | 249to252Cf(10,11B, xn) 257Lr(α,4.3 s) HILAC, Berkeley | 3 μg de Cf, quelques atomes de 257Lr détectés parmi d’autres radionucléides lourds. Existence de 256Lr(α, 45 s) confirmée en 1965 à Dubna suite à la réaction 243Am(18O, 5n) | [27] |
Le mendélévium
Cet élément est le dernier dont on peut produire des isotopes avec des ions légers car au-delà il n’y a plus d’émission beta qui puisse faire gagner une unité en Z. C’est la capture électronique qui prévaut quand N ≫ Z et elle conduit à un radionucléide de Z-1. C’est aussi le premier élément pour lequel les atomes ont été produits « One atom at a time » et pour lequel on a fait de la chimie statistique (répétition d’expériences). En 1955 la production de l’isotope 256Md (CE, 1,5 h) à partir de 106 atomes de 253Es (α, 20 j) était de 2 atomes toutes les 3 heures d’irradiation. Il était détecté par la fission spontanée de son fils 256Fm (2, 3 h). En 1975 on disposait de microgrammes de 253Es et on produisait 106 atomes/h de quoi faire de la chimie sur des quantités impondérables. La chimie à ces échelles de concentration pose de nombreux problèmes instrumentaux, de détection et des problèmes fondamentaux de chimie. Les expériences de partage sur le Md(III) et Md(II) ont permis de lancer les équipements pour identifier et faire de la chimie sur les éléments de Z > 103.
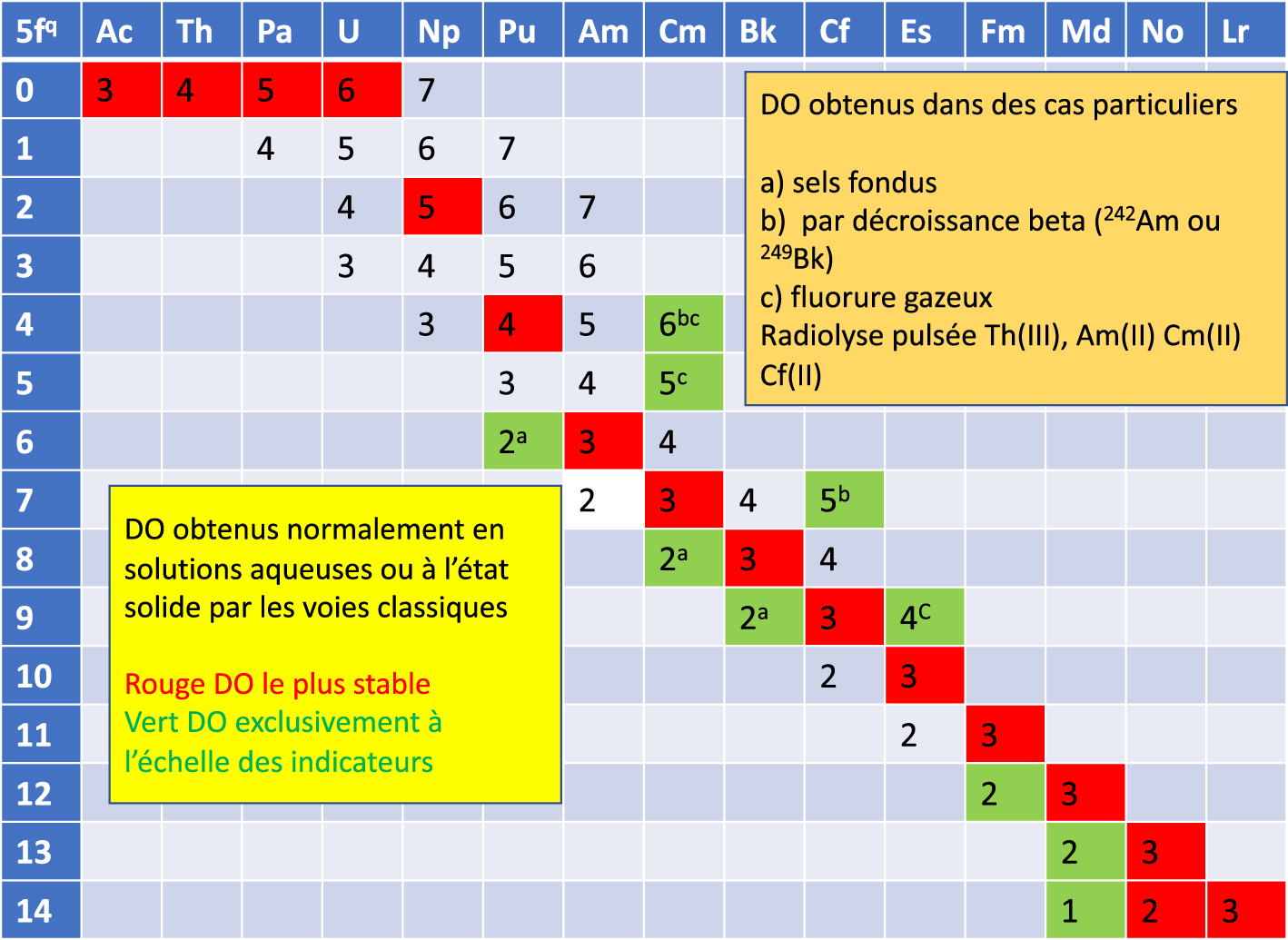
Degré d’oxydation (DO) des actinides ou éléments 5f dans diverses conditions.
Les actinides
De nombreuses données confirment aujourd’hui le bien fondé de la série des actinides analogue à la série des lanthanides et elle est admise par la communauté scientifique. Les propriétés physicochimiques de tous les actinides sont bien connues [18]. Toutefois il n’y a pas de stricte correspondance entre lanthanides et actinides. Par exemple Th, Pa et U sont vraiment des homologues de Hf, Ta et W, les structures électroniques des actinides sont fondées sur le remplissage des sous-couches relativistes 5f5/2 et 5f7/2 dont l’énergie et l’extension différent des sous-couches 5f. Les énergies de ces sous couches sont très proches de celle des sous couches 6d alors que les sous couches 4f et 5d sont séparées en énergie. Les premiers électrons 5f apparaissent dans l’atome de Pa, alors que c’est dans celui de Ce pour les lanthanides. Il s’ensuit une multiplicité des degrés d’oxydation (Figure 3).
Le Pu est en l’élément le plus connu au plan médiatique [19]. Pour les physicochimistes c’est un élément fascinant. Il présente sept formes allotropiques dans l’état métallique et il forme des alliages avec de nombreux autres métaux. La phase delta, stabilisée au Ga, Pu-Ga, est stable de la température ambiante à 550 °C, sa densité augmente avec T et sa compressibilité est remarquable. Ces propriétés permettent de déclencher une réaction en chaîne explosive dans les bombes avec 239Pu par compression. Le plutonium donne une large variété de composés solides et de nombreuses espèces ioniques en solution avec la possibilité de créer jusqu’à douze liaisons. En milieu acide, quatre degrés d’oxydation (3, 4, 5 et 6) peuvent coexister en équilibre. 238Pu (α, 80 a) est utilisé comme source d’énergie dans les sondes spatiales.
Conclusion
La découverte des éléments transuraniens rangés dans la série des actinides a été une histoire assez courte (1943 à 1960), mais compliquée dans laquelle la radiochimie a joué un rôle majeur ainsi que des moyens expérimentaux lourds d’irradiation comme les cyclotrons et les réacteurs nucléaires. La période de temps autour 1939–1945 a été exceptionnellement fertile en découvertes, mais révélées après la guerre. Elle a été précédée par la période 1934–1939 non moins riche en découvertes comme la fission de l’uranium. Plusieurs grands scientifiques impliqués dans la « science nucléaire » de ces époques ont reçu un prix Nobel : I. et F. Joliot (1935), E. Fermi (1938), E.O. Lawrence (1939), O. Hahn (1944), E. McMillan et G.T. Seaborg (1951).
Au plan historique la période de la deuxième guerre mondiale a été la période charnière pendant laquelle la science nucléaire est passée de l’Europe aux USA, avec le début de la deuxième guerre mondiale. Au plan technologique c’est la période du passage des sources radioactives aux accélérateurs de particules. Au plan scientifique (Physique/Chimie) c’est celle où on passe des éléments naturels aux éléments artificiels et où on ouvre de nombreux champs de recherches et d’applications. Enfin au plan sociétal pendant cette période on produit un élément pivot de la mise en œuvre de l’énergie nucléaire, le plutonium.
La découverte des actinides a ouvert la voie à la synthèse des éléments trans-lawrencium, Z = 104 à 118 [20, 21]. La quatrième transition de construction du Tableau Périodique débute en 1968 et correspondant aux découvertes des éléments 6d et 7p, ce sont des éléments éphémères.