



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Considerable developments are underway to develop alternative sustainable and competitive pathways for methionine and hydroxy analogue production. Indeed, methionine is the most essential amino acid in human and animal organisms. It plays an important role in cellular survival and protein biosynthesis. Added to food supplements, it improves the quantity and quality of products (meat, milk) from herbivorous animals [1, 2, 3, 4, 5]. Methionine can be obtained synthetically by cyclisation of homoserine followed by an opening of the lactone using mercaptan salts [6]. Methionine may be given as a racemic compound as well as one of its analogue, the 2-hydroxy-4-(methylthio)butyric acid (HMB). Indeed the latter and 2-keto-4-(methylthio)butyric acid (KMB) (Figure 1) are biotransformed in the animal organisms to form methionine in vivo [7, 8, 9, 10]. The synthesis of these derivatives is therefore an alternative to the preparation of methionine at industrial level.
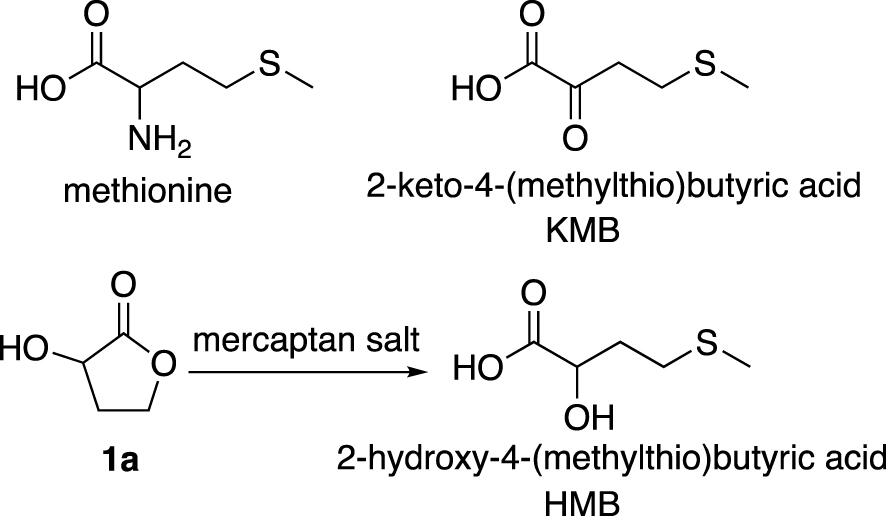
Structures of methionine, HMB and KMB derivatives.
KMB is synthetized by a 1,4 Michael addition on an α,β-unsaturated ketone [1, 2, 3, 4, 5, 11] while the opening of 2-hydroxybutyrolactone (1a) using mercaptan salts is the classic pathway to HMB [12], which are energy consuming and generates a significant amount of solid waste (notably sodium salts). To overcome this problem, the replacement of mercaptan salts by soft nucleophiles in the presence of Lewis acids is promising. In this context, several methods of (α-substituted) lactones ring opening have been carried out, in particular with the use of a aluminium halides as Lewis acids [13, 14], but they still have a limitation in the application scope in terms of the nature of the substitution [13, 14, 15, 16, 17]. The ring-opening reaction of 2-hydroxy-substituted lactones such as 1a remains relatively underdeveloped despite its great usefulness to lead to HMB with minimum waste. To our knowledge, scarce reports of 1a opening reactions have appeared where the reaction occurs at the carbon atom C(sp3)–O with various strong or soft nucleophiles like nitrogen [15], selenium [18, 19, 20], sulphur [1, 16, 21], or iodide atoms [22, 23].
In this work, we developed a ring-opening methodology of 2-hydroxybutyrolactone (1a) catalysed by a Lewis acid, with soft sulphur nucleophile to obtain 2-hydroxy-4-(ethylthio)butyric acid (2a), an HMB analogue (Scheme 1).
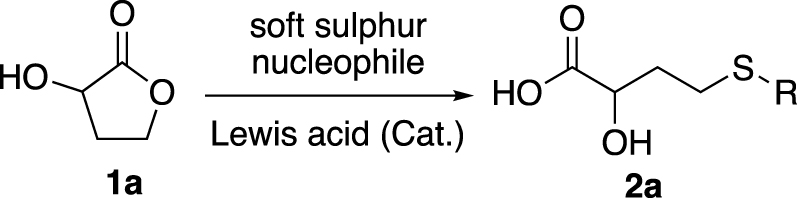
Ring opening of 1a catalysed by Lewis acid.
Here we describe a general route to the desired product, in which a direct coupling between 1a and (ethylthio)trimethylsilane was effectively catalysed by InI3 in the presence of K2CO3 under drastic conditions (InI3 is stored and weighted in glove-box, Schlenck techniques are used, see Chapter 4: experimental section). Our method was inspired by the methodology initially developed by Baba and co-workers [21] (Scheme 2). They first studied the formation of thioether starting from ester. Among several Lewis acid catalyst, the best result was achieved with InI3 (77%), which showed a better ability to promote the substitution than InBr3 or InCl3. Then, they applied their best catalytic conditions to the ring opening reaction of γ-butyrolactone (1b), which gave the corresponding thioethercarboxylic acid in 73% yield, using InI3 as a catalyst and (phenylthiol)trimethylsilane as a nucleophile. Better yields can be obtained with larger lactones which highlights the relative difficulty to open butyrolactones by the cleavage of the C(sp3)–O bond.
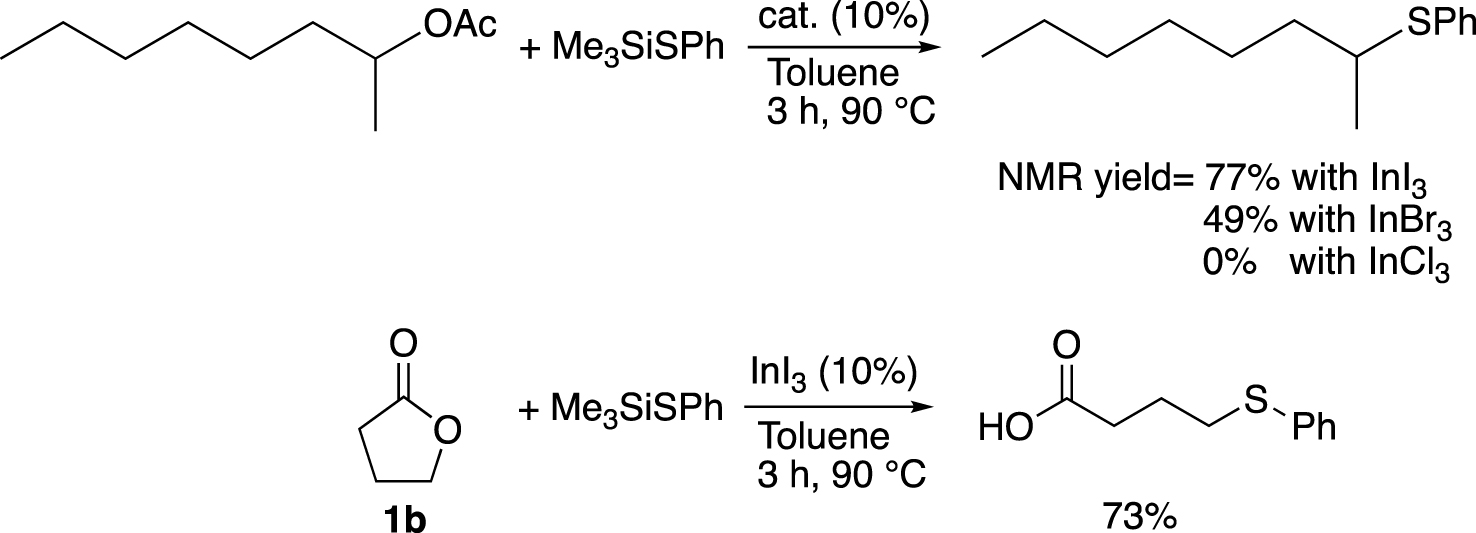
Formation of thioether from ester and γ-butyrolactone (1b) catalysed by InI3 developed by Baba and coworkers [21].
2. Results and discussion
We first began our investigations by reproducing the ring-opening reaction of 1b, but by replacing (phenylthiol)trimethylsilane with ethanethiol or (ethylthio)trimethylsilane (Table 1, entries 1 and 2).1 Ethanethiol was stirred in toluene with 1b and a catalytic amount of InI3 at 90 °C to obtain 2b in only 15% yield after 20 h of stirring. However, the use of (ethylthio)trimethylsilane as a nucleophile under the same conditions led to a full conversion after only 3 h of stirring (Entry 2). When 1a was used, no ring opening was observed with EtSH even after increasing the amounts of both nucleophile and catalyst, as well as the reaction time (Entry 3). So, we leaned towards the use of EtSSiMe3 because it gave an excellent result with 1b (Entry 2) and its boiling point is higher than the reaction temperature. To our delight, the ring opening of 1a carried out with this soft nucleophile led to the desired product 2a in 30% yield along with 3a in 7% yield (ring opening and thioesterification product, entry 4). At this point, we concluded that our system could be different from Baba’s one due to the presence of the hydroxy group at the α-position of the lactone. Indeed, the addition of a stoichiometric amount of K2CO3 to the reaction mixture allowed to increase the yield of 2a, although the formation of by-product 4a, resulting from the nucleophilic attack on the C-1 position (ester), was also observed (Entry 5).
InI3-catalysed ring opening of 1a and 1b
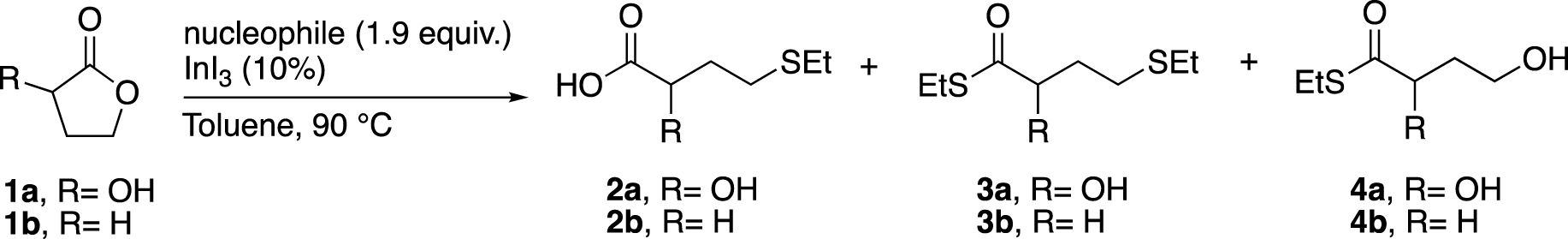
| Entry | Substrate | Nucleophile | t (h) | Isolated yields (%) | ||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | ||||
| 1 | 1b | EtSH | 20 | 2b: 15a | 0 | 0 |
| 2 | 1b | EtSSiMe3 | 3 | 2b: 99 | 0 | 0 |
| 3 | 1a | EtSHb | 48 | 0 | 0 | 0 |
| 4 | 1a | EtSSiMe3 | 22 | 2a: 30 | 3a: 7 | 0 |
| 5 | 1a | EtSSiMe3c | 22 | 2a: 42 | 0 | 4a: 15 |
aNMR yields. bReaction performed with 3.8 equiv. of EtSH and 20% of InI3. cReaction in the presence of 1.1 equiv. of K2CO3.
EtSH has a boiling point about 35 °C, which is below the reaction temperature. Its volatility can lead to a loss of reagent while heating at 90 °C. To this end, reactions with EtSH were performed in an autoclave system (closed Schlenk tube of 5 mL) containing only 2/5 of dead volume instead of a conventional reflux system. No real improvement was observed with this nucleophile. But when reactions were carried out with EtSSiMe3 (boiling point about 134 °C, above the reaction temperature) using the same conditions, the yields have been improved (Table 2).
InI3-catalysed ring opening of 1a in an autoclave system

| Entry | Nucleophile | InI3 (%) | K2CO3 (equiv.) | Isolated yields (%) | ||
|---|---|---|---|---|---|---|
| 2a | 3a | 4a | ||||
| 1 | EtSSiMe3 | - | - | 0 | 0 | 0 |
| 2 | EtSSiMe3 | - | 1.1 | 0 | 0 | 0 |
| 3 | EtSH | - | 1.1 | Traces | 0 | 0 |
| 4 | EtSSiMe3 | 10 | - | 35 | 4 | Traces |
| 5 | EtSSiMe3 | 10 | 1.1a | 66 | 0 | 9 |
| 6 | EtSH | 10 | - | 0 | 0 | 0 |
| 7 | EtSH | 10 | 1.1a | 18 | 0 | 0 |
| 8 | EtSH | 10 | 1.1b | 26 | 0 | 0 |
| 9 | EtSSiMe3 | 10 | 1.1b | 85 | 0 | 11 |
| 10 | EtSSiMe3 | 10 | 1.1c | 16 | 0 | 9 |
aWithout preliminary drying of 1a and K2CO3. bWith preliminary drying of 1a and K2CO3. cWith addition of 0.25 mL of water in the reaction mixture.
First, a control experiment in the absence of the catalyst did not lead to the formation of products 2–4a (Entries 1–3). On the other hand, with 10% of InI3 and in the absence of K2CO3, there was no significant efficiency compared to a conventional reflux system (Entry 4, compared to entry 4 in Table 1). When the base was present (Entry 5), the yield of 2a was increased significantly (66%). As expected, EtSH instead of EtSSiMe3 was less efficient in producing 2a (18% yield) in the presence of K2CO3 (Entry 7) while it did not generate the desired product without a base (Entry 6). In addition, the presence of a residual amount of water in 1a or K2CO3 is suspected to decrease the reactivity of the nucleophile reagent, and consequently, of leading to lower yields. Drying 1a and K2CO3 before their use confirmed this hypothesis (Entries 8 and 9). Finally with optimised conditions, EtSSiMe3 gave the desired product. Indeed, 2a was successfully isolated with 85% yield along with by-product 4a in 11% yield, which corresponds to an overall conversion of 96%. The introduction of additional water into the reaction mixture logically led to a dramatic decrease in yield (Entry 10).
With optimal reaction conditions in hand, based on the Baba’s study [21], further screening of catalysts in autoclave systems was realized (Table 3).2 As expected, InI3 provided the best result for the ring opening of 1a (Entry 1). But In(OTf)3 (less hard LA than InI3) also showed good activity (Entry 2). However, other Lewis acids based on metal triflate have been tested. The soft and larger Bi(OTf)3 was found to be unsatisfactory (Entry 3), as well as the unreactive oxophile Al(OTf)3 (Entry 4). Also, AlBr3 allowed the formation of only a small quantity of the thioester 4a (10%). Therefore, the reactivities are clearly dependent on the nature of the Lewis acid as observed by Baba.
Catalysts screening for the ring opening of 1a

| Entry | Catalyst | Isolated yields (%) | ||
|---|---|---|---|---|
| 2a | 3a | 4a | ||
| 1 | InI3 | 85 | 0 | 11 |
| 2 | In(OTf)3 | 58 | 8 | 0 |
| 3 | Bi(OTf)3 | 4 | 7 | 0 |
| 4 | Al(OTf)3 | 0 | 0 | 0 |
| 5 | AlBr3 | 0 | 0 | 10 |
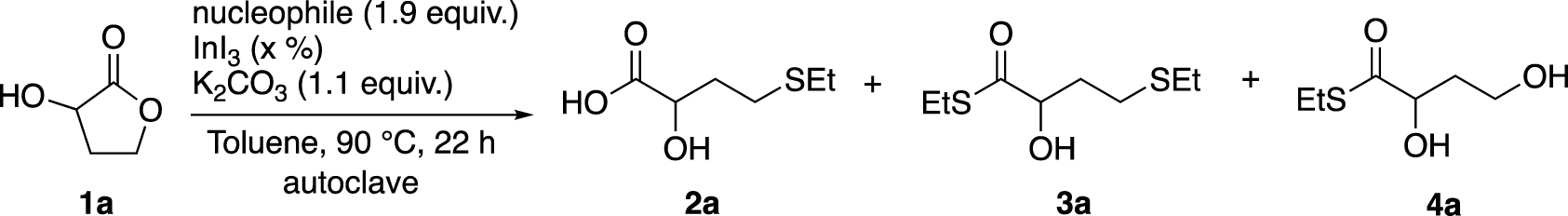
We studied in more detail the loading of InI3 on the outcome of the reaction. As shown in Table 4, the ring opening of 1a was examined with different catalytic amounts of this Lewis acid.
Ring opening of 1a with various loadings of InI3
| Entry | Nucleophile | InI3 (%) | Isolated yields (%) | ||
|---|---|---|---|---|---|
| 2a | 3a | 4a | |||
| 1 | EtSH | 10 | 26 | 0 | 0 |
| 2 | EtSH | 20 | 23 | 0 | 0 |
| 3 | EtSSiMe3 | 5 | 59 | 6 | 0 |
| 4 | EtSSiMe3 | 10 | 85 | 0 | 11 |
| 5 | EtSSiMe3 (1.1 equiv) | 10 | 24a | 15a | 0 |
| 6 | EtSSiMe3 (0.3 equiv) | 10 | Tracesa | Tracesa | 0 |
| 7 | EtSSiMe3 | 20 | 91 | Traces | 3 |
| 8 | EtSSiMe3 | 33 | 61 | 6 | 0 |
| 9 | EtSSiMe3b | 33 | 47 | 2 | 0 |
| 10 | EtSSiMe3 (3 equiv) | 33 | 90 | 0 | 0 |
aNMR observations. bThe putative catalytic intermediate InI(3−n)(SEt)n was heated to 90 °C (2 h) then cooled to r.t. before the addition of 1a and K2CO3. Formation of InI(3−n)(SEt)n: InI3 + n EtSSiMe3→InI(3−n)(SEt)n + n ISiMe3.
The InI3 quantity has no influence on the yield when EtSH was used (Table 4, entries 1 and 2). Nevertheless, with the EtSSiMe3 and 5% of InI3 (Entry 3), the desired product 2a was isolated with 59% yield, while the yield reached 91% with 20% of Lewis acid (Entry 7). Surprisingly, a remarkable decrease in yield was observed with 33% of the catalyst loading (Entry 8), which could indicate either an insufficient activation of plausible intermediates InI(3−n)(SEt)n formed in situ, or an insufficient amount of nucleophile. The second hypothesis was confirmed by a rise in the yield with 3 equivalents of EtSSiMe3 (Entries 9 and 10). This suggests that likely at least (3 ∗ n (catalyst) + 1) equivalents of the nucleophile are required to achieve the maximum conversion. This hypothesis was also confirmed by the experiment with 0.3 equiv. of EtSSiMe3 and 10% of InI3 (Entry 6), where only traces (instead of expected 20–30%) of expected products were observed. This lack of conversion suggests a plausible interaction between the catalyst and the nucleophile. The ratio nucleophile/catalyst appears to be important and the putative InI(3−n)(SEt)n species could be formed before acting as a catalyst. It should be noted that Baba and co-workers used an InI3/ester/PhSSiMe3 = 1/10/20 ratio in their experiments, so more than (3*n (catalyst) +1) equivalents of nucleophile.
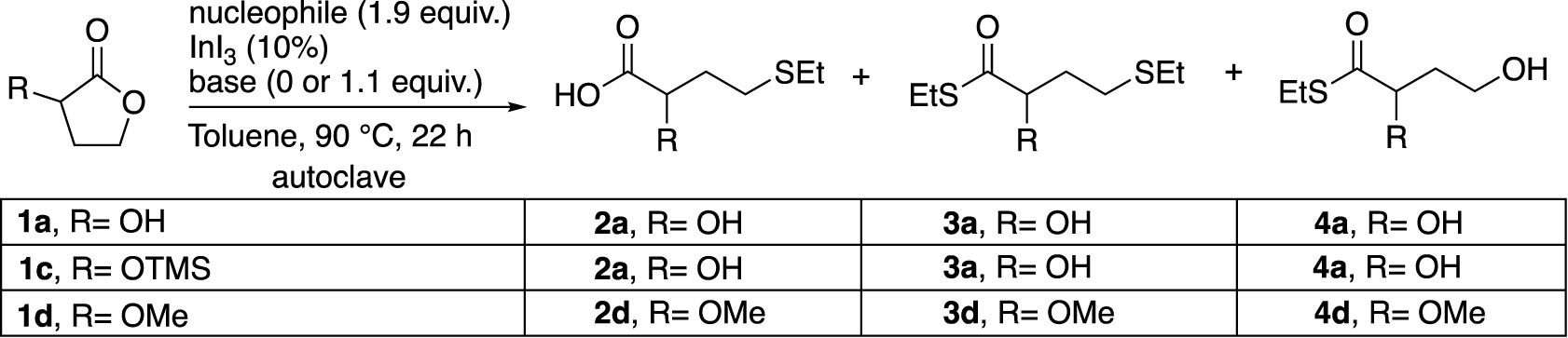
Finally, the outcome of the ring opening of several α-substituted-γ-butyrolactones was examined under various reaction conditions (Table 5).
Reactivity study of different α-substituted-γ-butyrolactones in InI3-catalysed ring-opening reactions
| Entry | Substrate | Nucleophile | Base (equiv.) | Isolated yields (%) | ||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | ||||
| 1 | 1a | EtSH | K2CO3 (1.1) | 2a: 26 | 0 | 0 |
| 2 | 1c | EtSH | K2CO3 (1.1) | 2a: 21 | 3a: 4 | 4a: Traces |
| 3 | 1d | EtSH | K2CO3 (1.1) | 2d: 39 | 0 | 0 |
| 4 | 1a | EtSSiMe3 | - | 2a: 35 | 3a: 4 | 4a: Traces |
| 5a | 1a | EtSSiMe3 | - | 2a: 34 | 3a: 4 | 4a: Traces |
| 6 | 1c | EtSSiMe3 | - | 2a: 55 | 3a: 5 | 4a: 17 |
| 7 | 1d | EtSSiMe3 | - | 2d: 2 | 3d: 17 | 4d: 39 |
| 8 | 1a | EtSSiMe3 | K2CO3 (1.1) | 2a: 85 | 0 | 4a: 11 |
| 9 | 1c | EtSSiMe3 | K2CO3 (1.1) | 2a: 47 | 0 | 4a: Traces |
| 10 | 1d | EtSSiMe3 | K2CO3 (1.1) | 2d: 11b | 0 | 4d: 53b |
| 11 | 1a | EtSSiMe3 | DBU (1.1) | 2a: 40 | 0 | 4a: Traces |
aReaction performed with the addition of molecular sieves. bNMR yields.
1c [24] and 1d [25, 26] were prepared from 1a according to the protocols of the literature and isolated with 96% and 86% yields respectively. Of the two compounds, the ether 1d was found to be the most reactive with EtSH in the presence of K2CO3 (Table 5, entries 1–3). On the other hand, when the nucleophile is EtSSiMe3 and in the absence of a base, 1c was found to be the most reactive substrate with a yield of 55% of 2a (Entries 4–7). Moreover, the addition of K2CO3 promoted the reactivity of 1a to 2a, while 1d mainly provided the thioester 4d (Entries 8–10). The presence of the -OTMS group on the substrate 1c (Entries 2, 6 and 9) did not seem to have a beneficial influence in the selective formation of the compound 2a in these conditions. It is important to note that this type of K2CO3 base is not crippling for industrial purposes due to the possibility of regeneration [27]. Based on the above results, we decided to keep a base, but we replaced K2CO3 with the more soluble DBU (Entry 11), which resulted in a disappointing loss of yield.
3. Conclusions
In conclusion, we have developed an efficient and practical synthesis of one methionine analogue, the HMB, by the InI3-catalysed ring-opening reaction between 2-hydroxybutyrolactone (1a) and a thiosilane in the presence of a base (K2CO3) which can be regenerated. This could be advantageous for further industrial developments. The reaction occurred selectively at the carbon atom C(sp3)–O. The method allows easy access to the desired product 2a in a high isolated yield (85%) with 10% InI3 catalyst, when the reaction is carried out in an autoclave and in the absence of water. To our knowledge, the current one-pot reaction system is the only one, among the reported procedures, that produces HMB analogues in excellent yields from 1a using catalytic electrophilic activation.
4. Experimental section
4.1. General methods
All reagents were obtained from commercial sources and used as received unless otherwise noted. K2CO3 was dried at 85 °C under vacuum in a Büchi glass oven B-585. THF and toluene were purified and dried over Braun solvent purification system (MB-SPS-800). InI3 and the other Lewis acids were stored and weighted in glove-box in oven-dried glassware. All reactions were performed in oven-dried glassware under an atmosphere of argon using Schlenk techniques unless otherwise noted. Analytical Thin Layer Chromatography (TLC) was carried out on Merck silica gel60 F254. Products were stained with potassium permanganate dyeing reagent solution followed by gentle heating. Flash chromatography was performed on Combiflash® Companion or with Merck silica gel 60 (230–400 mesh). 1H and 13C NMR spectra were recorded in CDCl3, at ambient temperature on Bruker Avance III 300 or 400 spectrometers operating at 300 and 400 MHz respectively for 1H. 13C nuclei were observed with 1H decoupling. Solvent residual signals were used as internal standard [28]. Chemical shifts (δ) and coupling constants (J) are given in ppm and Hz respectively. The peaks patterns are indicated as the following format multiplicity (s: singlet; d: doublet; t: triplet; q: quartet; qt: quintuplet; m: multiplet; dd: doublet of doublet). The prefix br. indicates a broadened signal. IR spectra were obtained using a Bruker Alpha Platinum ATR. HRMS were recorded on SYNAPT G2 HDMS (Waters) or on QStar Elite (Applied Biosystems SGIEX) equipped with an Atmospheric Pressure Ionization (API) source or on MaXiS 4G (Bruker) equipped with an Electron Spray Ionization (ESI) source. Mass spectra were obtained using a Time Of Flight (TOF) analyser. Elementary analyses were recorded on a Thermo Finnigan EA 1112 apparatus.
4.2. Starting materials
The 2-hydroxybutyrolactone 1a is commercially available and was given by our industrial partner Adisseo. It is dry over 3 Å MS for 72 h.
4.2.1. 3-(Trimethylsilyl)oxydihydrofuran-2(3H)-one (1c)
This compound was prepared according to a procedure reported elsewhere [24]. To a stirred solution of 1a (0.074 mL, 1 mmol, 1.0 equiv.), in CH3NO2 (1 mL) under argon was added freshly distilled HMDS (0.21 mL, 1 mmol, 1.0 equiv.). The reaction mixture was stirred for 30 min at room temperature. After concentration, the crude product was purified by column chromatography using a gradient eluent (from 100% petroleum ether to 90:10 = petroleum ether:ethylacetate). 1c was recovered as colorless oil (167 mg, 96%). These data are consistent with those described in the literature [29].
1H NMR (400 MHz, CDCl3) δ 4.43–4.36 (m, 2H, CH2OC(O)), 4.23–4.16 (m,1H, CHOSi), 2.51–2.43 (m, 1H, CH2CHOSi), 2.28–2.18 (m, 1H, CH2CHOSi), 0.20 (s, 9H, Si(CH3)3).
4.2.2. 3-Methoxydihydrofuran-2(3H)-one (1d)
NaH (60% in oil, 302 mg, 7.54 mmol, 1.5 equiv.) was introduced in a double-neck round bottom flask. The oil is removed by several washings with pentane. Then, dry THF (10 mL) under argon atmosphere is added. At 0 °C, 1a (0.37 mL, 5.02 mmol, 1.0 equiv.) is added dropwise to the stirred solution. After 1 h, while warming to room temperature, freshly distilled MeI (0.47 mL, 7.54 mmol, 1.5 equiv.) is added. Subsequently, the reaction mixture was stirred for 22 h and then, 20 mL of saturated NH4Cl aqueous solution is added. The layers were separated, and the aqueous layer was extracted with 3 × 25 mL of Et2O. The combined organic layer was dried over Na2SO4, filtered and concentrated. The crude product was purified by column chromatography using a gradient eluent (from 100% petroleum ether to 60:40 = petroleum ether:ethylacetate). 1d (501 mg, 86%) was obtained as yellow oil.
Rf = 0.55 (AcOEt/petroleum ether 8:2). 1H NMR (400 MHz, CDCl3) δ 4.39–4.34 (m, 1H, CH2OC(O)), 4.23–4.17 (m,1H, CH2OC(O)), 4.00 (t, J = 7.8 Hz, 1H, CHOCH3), 3.51 (s, 3H, OCH3), 2.53–2.46 (m, 1H, CH2CHOCH3), 2.23–2.14 (m, 1H, CH2CHOCH3). 13C NMR (100 MHz, CDCl3): δ 174.8, 75.0, 65.4, 58.2, 29.4. These data are consistent with those described in the literature [25, 26].
4.3. Experimental procedures and characterizations data
4.3.1. 4-(Ethylthio)butyric acid (2b)
To a stirred suspension of InI3 (100 mg, 0.2 mmol, 0.1 equiv.) in anhydrous toluene (2 mL) was added EtSSiMe3 [0.680 mL (90% purity), 3.8 mmol, 1.9 equiv.]. The reaction mixture was stirred for a few minutes under N2 atmosphere at room temperature before the addition of γ-butyrolactone 1b (0.15 mL, 2 mmol, 1.0 equiv.). The mixture was then heated to 90 °C and stirred for 3 h. After cooling to room temperature, the resulting mixture was poured into 20 mL of HClaq (1M) and the layers were separated. The aqueous layer was extracted with 3 × 20 mL of ethylacetate, and the combined organic layers were washed with brine, dried over Na2SO4 and filtered. After concentration in vacuo, carboxylic acid 2b was obtained as a yellow liquid (295 mg, 99%) without further purification. Rf = 0.63 (AcOEt/petroleum ether: 8:2). 1H NMR (400 MHz, CDCl3) δ 10.66 (CO2H), 2.59–2.47 (m, 6H, CH2SCH2 and CH2CO2H), 1.91 (qt, J = 7.19, 7.25 Hz, 2H, CH2CH2CH2), 1.24 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 179.7, 32.9, 30.8, 25.8, 24.4, 14.8. MS (ESI+): m∕z = 155 [M+Li]+; 171 [M+Na]+. These data are consistent with those described in the literature [14].
4.3.2. General procedure for the ring opening of compounds 1
In an autoclave system (5 mL closed Schlenk tube) with a minimised dead volume, the nucleophile was added to a stirred suspension of Lewis acid in anhydrous toluene (2.4 mL). The reaction mixture was stirred for a few minutes under N2 atmosphere at room temperature before the addition of dried 1 (2.44 mmol, 1.0 equiv.) followed by the eventual addition of base. The mixture was then heated to 90 °C and stirred for 22 h. After cooling to room temperature, the resulting mixture was filtered, and then was poured into 20 mL of HClaq (1M) and the layers were separated. After extraction of the aqueous phase with 3 × 20 mL of ethylacetate, the combined organic layer was washed with brine, and dried over Na2SO4, filtered and concentrated. The crude product was purified by column chromatography using a gradient eluent (from 100% petroleum ether to 100% ethyl acetate).
4.3.3. 4-(Ethylthio)-2-hydroxybutyric acid (2a)
This compound was prepared following the general procedure starting from EtSSiMe3 (0.83 mL, 4.64 mmol, 1.9 equiv. (90% purity)), InI3 (242 mg, 0.48 mmol, 0.2 equiv.), 1a (0.18 mL, 2.44 mmol, 1.0 equiv.) and anhydrous K2CO3 (371 mg, 2.68 mmol, 1.1 equiv.). The crude product was purified by column chromatography using a gradient eluent (from 100% petroleum ether to 100% ethyl acetate).
2a (365 mg, 91%) was obtained as yellowish oil. Rf = 0.52 (AcOEt/ petroleum ether: 8:2). 1H NMR (400 MHz, CDCl3) δ 5.52 (br s, 2H, OH and CO2H), 4.43 (dd, J = 8.0, 3.8 Hz,1H, CHOH), 2.70 (t, J = 6.7 Hz, 2H, SCH2CH2), 2.56 (q, J = 7.4 Hz, 2H, CH3CH2S), 2.18–2.09 (m, 1H, CH2CHOH), 2.01–1.93 (m, 1H, CH2CHOH), 1.26 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 179.1, 69.4, 33.6, 27.3, 25.9, 14.8. IR (film): 3400, 2966, 2926, 1718, 1261, 1213, 1166, 1089 cm−1. HRMS (ESI): m∕z [M+H]+ Calcd for C6H12O3S 165.0580; Found 165.0579.
4.3.4. S-Ethyl 4-(ethylthio)-2-hydroxybutanethioate (3a)
This compound is a by-product formed during the synthesis of 2a and was isolated during the chromatography. 3a (traces, <1%) was obtained as colorless oil.
Rf = 0.37 (AcOEt/ petroleum ether 1:9). 1H NMR (300 MHz, CDCl3) δ 4.41 (dd, J = 8.3, 3.6 Hz, 1H, CHOH), 2.92 (q, J = 7.4 Hz, 2H, CH3CH2S(O)), 2.84 (br s, 1H, OH), 2.70–2.65 (m, 2H, SCH2CH2), 2.56 (q, J = 7.4 Hz, 2H, CH3CH2SCH2), 2.17–2.06 (m, 1H, CH2CHOH), 1.98–1.87 (m, 1H, CH2CHOH), 1.29–1.23 (m, 6H, 2CH3). 13C NMR (100 MHz, CDCl3): δ 203.8, 76.9, 34.5, 27.2, 25.9, 23.1, 14.8, 14.7. IR (film): 3420, 2966, 2929, 1678, 1263, 1088 cm−1. HRMS (ESI): m∕z [M+Na]+ Calcd for C8H16O2S2 231.0484; Found 231.0484.
4.3.5. S-Ethyl 2,4-dihydroxybutanethioate (4a)
This compound is a by-product formed during the synthesis of 2a and was isolated during the chromatography. 4a (10 mg, 3%) was obtained as colorless oil.
Rf = 0.55 (AcOEt/ petroleum ether 8:2). 1H NMR (500 MHz, CDCl3) δ 4.41 (dd, J = 9.3, 3.0 Hz,1H, CHOH), 3.83 (t, J = 4.1 Hz, 2H, CH2OH), 2.84 (q, J = 7.5 Hz, 2H, CH3CH2S(O)), 2.10–2.04 (m, 1H, CH2CHOH), 1.85–1.79 (m, 1H, CH2CHOH), 1.22 (t, J = 7.3 Hz, 3H, CH3). 13C NMR (125 MHz, CDCl3): δ 206.3, 75.9, 59.4, 35.8, 22.8, 14.6. IR (film): 3328, 2973, 1417, 1086, 1044, 879 cm−1. HRMS (ESI+): m∕z [M+Na]+ Calcd for C6H12O3SNa+ 187.0399; found 187.0398.
4.3.6. 4-(ethylthio)-2-methoxybutanoic acid (2d)
This compound was prepared following the general procedure starting from 1d (290 mg, 2.5 mmol, 1.0 equiv.), EtSH (0.355 mL, 4.75 mmol, 1.9 equiv.) and 0.1 equiv. of InI3 (124 mg, 0.25 mmol). The crude product was purified by column chromatography using DCM, then DCM with 5% MeOH as the eluent. 2d (172 mg, 39%) was obtained as yellowish oil.
Rf = 0.56 (DCM/MeOH 9:1). 1H NMR (400 MHz, CDCl3) δ 3.98 (dd, J = 7.0, 5.0 Hz, 1H, CHOCH3), 3.45 (s, 3H, OCH3), 2.68–2.60 (m, 2H, SCH2CH2), 2.53 (q, J = 7.5 Hz, 2H, CH3CH2S), 2.08–2.00 (m, 2H, CH2CHOCH3), 1.24 (t, J = 7.4 Hz, 3H, CH3CH2S). 13C NMR (100 MHz, CDCl3): δ 177.7, 78.7, 58.6, 32.4, 27.1, 25.8, 14.8. IR (film): 2928, 1718, 1450, 1193, 1117 cm−1. HRMS (ESI+): m∕z [M+H]+ Calcd for C7H14O3S 179.0736; Found 179.0737.
4.3.7. S-Ethyl 4-(ethylthio)-2-methoxybutanethioate (3d)
This compound is a by-product formed during the synthesis of 2d. This compound was prepared following the general procedure starting from 1d (290 mg, 2.5 mmol, 1.0 equiv.), 0.1 equiv. of InI3 (124 mg, 0.25 mmol) and no base was added. The crude product was purified by column chromatography using a gradient eluent (from 100% petroleum ether to 100% ethylacetate). 3d (93 mg, 17%) was obtained as yellow oil.
Rf = 0.75 (AcOEt/ petroleum ether 1:9). 1H NMR (400 MHz, CDCl3) δ 3.89 (dd, J = 7.8, 4.8 Hz, 1H, CHOCH3), 3.46 (s, 3H, OCH3), 2.86 (q, J = 7.4 Hz, 2H, CH3CH2S(O)), 2.69–2.58 (m, 2H, SCH2CH2), 2.52 (q, J = 7.3 Hz, 2H, CH3CH2SCH2), 2.01–1.89 (m, 2H, CH2CHOCH3), 1.27–1.22 (m, 6H, 2CH3CH2S). 13C NMR (100 MHz, CDCl3): δ 203.5, 85.9, 59.4, 33.7, 27.1, 25.7, 22.4, 14.8, 14.7. IR (film): 2929, 1678, 1452, 1264, 1116, 1100 cm−1. HRMS (ESI+): m∕z [M+Na]+ Calcd for C9H18O2S2 245.0640; Found 245.0643.
4.3.8. S-ethyl 4-hydroxy-2-methoxybutanethioate (4d)
This compound is another by-product formed during the synthesis of 2d and was isolated during the chromatography. 4d (174 mg, 39%) was obtained as brown oil.
Rf = 0.65 (AcOEt/petroleum ether 8:2). 1H NMR (400 MHz, CDCl3) δ 3.93–3.85 (m, 1H, CHOCH3), 3.73–3.65 (m, 2H, CH2OH), 3.42 (s, 3H, OCH3), 2.81 (q, J = 7.4 Hz, 2H, CH2CH3), 1.91–1.76 (m, 2H, CH2CH2OH), 1.20 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (101 MHz, CDCl3): δ 174.7, 75.0, 65.3, 58.1, 29.4, 19.5, 19.0. IR (film): 3408, 2931, 2880, 1417, 1376, 1177, 1105 cm−1. HRMS (ESI+): m∕z [M+Na]+ Calcd for C7H14O3SNa+ 201.0556; found 201.0562.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
We thank Dr. Christophe Chendo and Dr. Valérie Monnier for mass spectrometry analyses (Spectropole, Fédération des Sciences Chimiques de Marseille), Arnaud Treuvey (Centrale Marseille) for IR spectroscopy and Yoann Cotelle and Maxime Huchede for reading the manuscript.
1 In the classical synthesis of methionine analogues, methanethiol is used. But for practical reasons (odor and handling), we used EtSH and more convenient TMSSEt in this study.
2 In comparison to the study of Yamakasi and Shibuya [22, 23], the Brönsted acid HI (1 equiv.) was used in EtSH as solvent (C = 0.5 M), at 35 °C without base. Mixture of iodide substituted compounds were detected by mass spectroscopy, but no desired SEt substituted products were observed.