

 CC-BY 4.0
CC-BY 4.0
1. Introduction
Polycyclic compounds based on aromatic hydrocarbons are considered attractive targets for synthesizing medicinal units since they exhibit favorable properties such as stability and ease of synthesis and possess high biological responses, particularly anticancer activity [1]. As a result, many researchers around the world have become interested in the preparation and development of new cytotoxic molecules [2, 3, 4]. In this context, H. Guédouar and co-workers [2] worked on the synthesis of a fairly large number of new phenanthrene skeletons, using a simple procedure, with the aim of preparing promising active compounds for the development of anticancer agents. In fact, they prepared a variety of tricyclic compounds by modifying the central structure of phenanthrene. The compounds were evaluated for their in vitro cytotoxic activity against two tumor cell lines. Interestingly, the analysis of the IC50 values suggests that most compounds exert cytotoxic effects with selectivity. Among them, methyl 8-methyl-9,10-phenanthrenequinone-3-carboxylate D-1 (IC50 = 0.97 μg∕mL) and methyl 8-methyldibenzo[a,c]phenazine-3-carboxylate D-2 (IC50 = 1.09 μg∕mL) are interesting substrates due to their highest potency against the Caco-2 cancer cell (Figure 1).
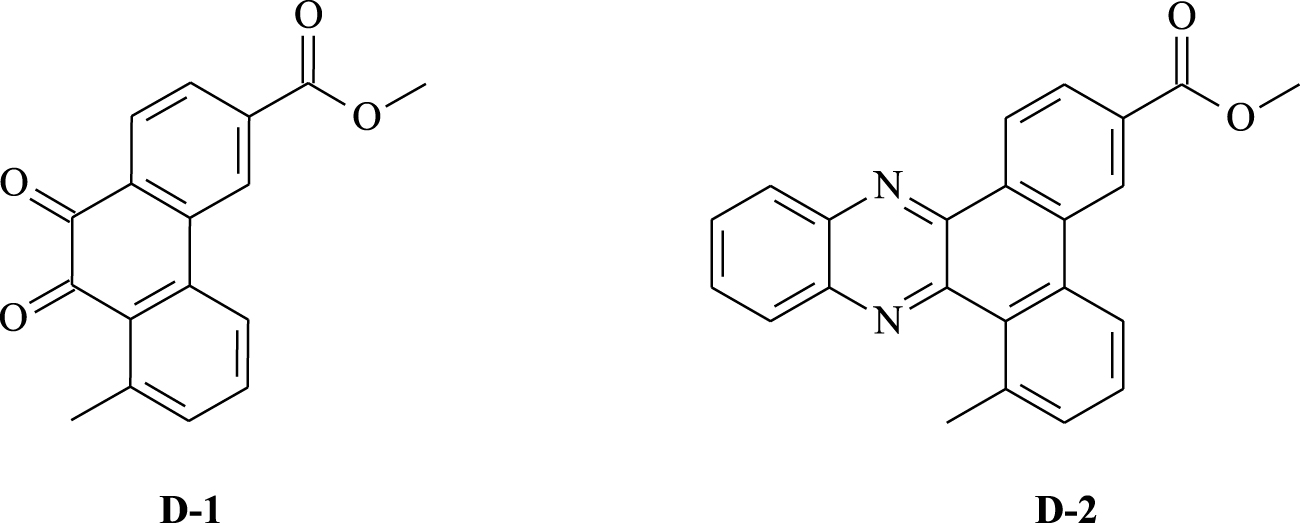
Chemical structures of the studied phenanthrenes D-1 and D-2.
In this study, molecular docking was performed on the two most active O-linked molecules D-1 and D-2, previously prepared by H. Guédouar and co-workers to identify the key structural features required to design new potent candidates of this class. Thus, the results extracted from this study might be useful to design potent antitumor drugs. Before performing the molecular docking, the studied molecules were optimized using the density functional theory (DFT) method. In recent years, the DFT has become the most popular quantum chemical method for computing several molecular properties such as those exhibited by chemical, physical, and biological systems [5, 6, 7]. The reported quantum chemical calculations were performed at the B3LYP/6-31G(d,p) level of theory [8, 9, 10]. The geometry optimization in the gas phases was carried out using the Gaussian 09 suite of programs [11].
2. Molecular docking study
Molecular docking analysis is a reliable method for the evaluation of binding affinity and the prediction of intermolecular interactions of novel compounds containing potential receptors [12, 13]. We performed molecular docking studies for eight vital cancer targets (Table 1). Our research was based on the crystal structures of receptors with bound ligand molecules. This structure was obtained from X-ray crystal data of RCSB Protein Data Bank (PDB) [14, 15, 16].
Protein database used for docking study and their native ligands
| PDB code | Name | Native ligand | Chain |
|---|---|---|---|
| 2HYY | Human Proto-oncogene tyrosine-protein kinase ABL1 | Imatinib | A |
| 3C4C | B-Raf proto-oncogene serine/threonine-protein kinase | PLX4720 | A |
| 3EWH | Vascular endothelial growth factor receptor 2 | Pyridyl-pyrimidine benzimidazole | A |
| 3RCD | Receptor tyrosine-protein kinase erbB-2 | TAK-285 | A |
| 3W2S | Epidermal growth factor receptor | W2R | A |
| 4JT5 | Serine/threonine-protein kinase mTOR | Torkinib (PP242) | A |
| 4U5J | Proto-oncogene tyrosine-protein kinase Src | Ruxolitinib | A |
| 6N2J | GTPase KRas | Tetrahydropyridopyrimidines | A |
In the majority of selected structures, co-crystallized ligand molecules are known drugs with proven action. Thus they were utilized to predict the binding site location [17] as well as to serve as references in our analysis. As previously demonstrated [2], molecules D-1 and D-2, whose 3D-QSAR structures are shown in Figure 2, are the most active. Docking studies of both compounds were carried out to analyze their ability to interact with each target for which the binding site locations are shown in Table 2.
Each enzyme does not necessarily contain a single active site. We chose the active site of interest to the study, which contains the reference inhibitor (which is the drug site); (X, Y and Z) are the three-dimensional coordinates of the active site of interest (in this case, inhibition of the enzyme).
Binding site location for each target
| PDB code | X | Y | Z |
|---|---|---|---|
| 2HYY | 14.251 | 15.282 | 17.632 |
| 3C4C | 0.478 | −2.111 | −19.745 |
| 3EWH | 15.131 | −5.231 | 10.046 |
| 3RCD | 13.047 | 1.810 | 28.168 |
| 3W2S | 5.726 | 0.748 | 12.742 |
| 4JT5 | 51.856 | −0.015 | −49.145 |
| 4U5J | −8.382 | 26.909 | 5.045 |
| 6N2J | 22.52 | 2.591 | −22.481 |
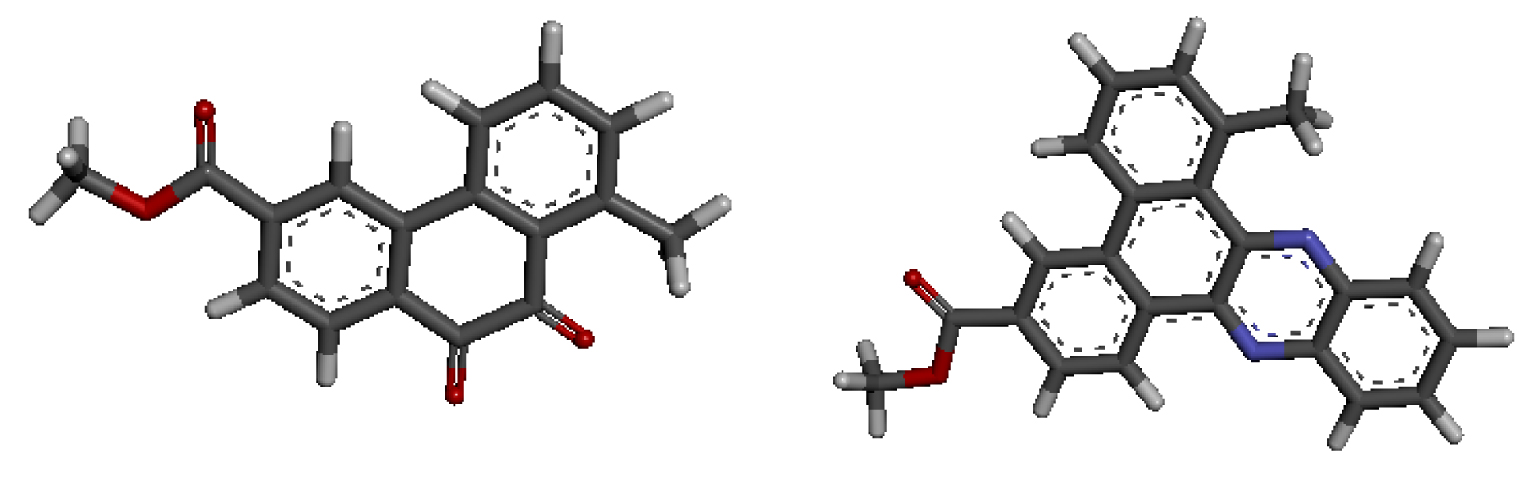
3D-QSAR structures of compounds D-1 (a) and D-2 (b) used for the docking study.
3D structure of the native ligand PLX4720.
2.1. Docking results
With the aim of confirming the potential cytotoxicity of our phenanthrene derivatives D-1 and D-2, we evaluated the binding mode and explored their intermolecular interactions with appropriate proteins. The docking results are summarized in Table 3. The binding affinity was evaluated by the binding free energy (Kcal∕mol). In fact, all the studied targets can establish binding with the two studied ligands. The tricyclic compound D-1 exhibited binding energies ranging from −9.8 to − 8.3 kcal∕mol. The molecular docking study with molecule D-2 revealed a binding energy ranging from −11.1 to −9.2 kcal∕mol. This slight difference in energy is probably due to the size of the studied molecules. Indeed, molecule D-1 is tricyclic while D-2 is composed of five cycles, one of which is heterocyclic.
Docking score expressed in kcal∕mol
| PDB code | Molecule D-1 | Molecule D-2 |
|---|---|---|
| 2HYY | −8.3 | −9.2 |
| 3C4C | −9.8 | −11.1 |
| 3EWH | −9.4 | −10.1 |
| 3RCD | −9.2 | −10.3 |
| 3W2S | −9.0 | −10.1 |
| 4JT5 | −8.9 | −10.2 |
| 4U5J | −9.4 | −10.4 |
| 6N2J | −9.0 | −9.4 |
Modes and types of bonds between the studied molecules and their potential target
| PDB code | D-1 | D-2 |
|---|---|---|
| 2HYY | 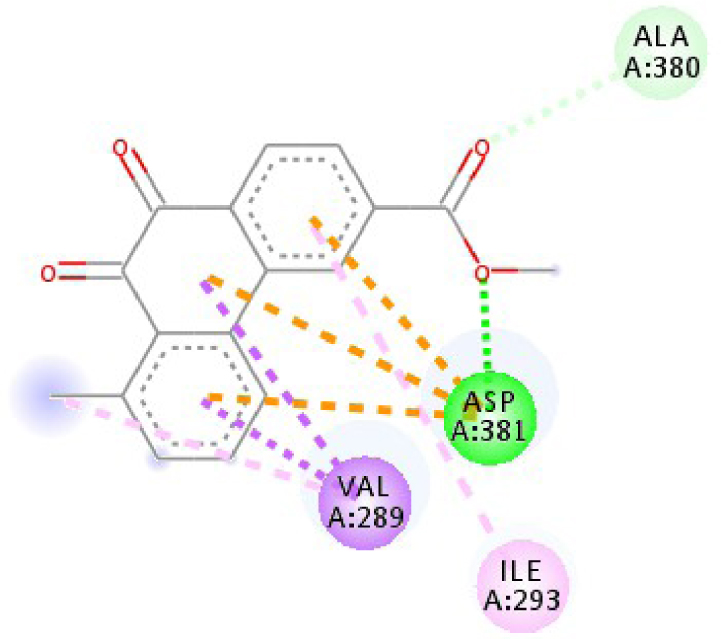 | 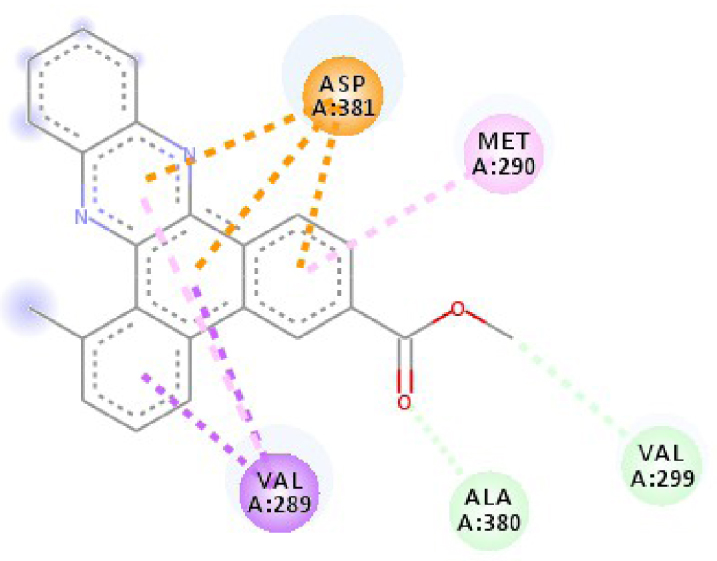 |
| 3C4C | 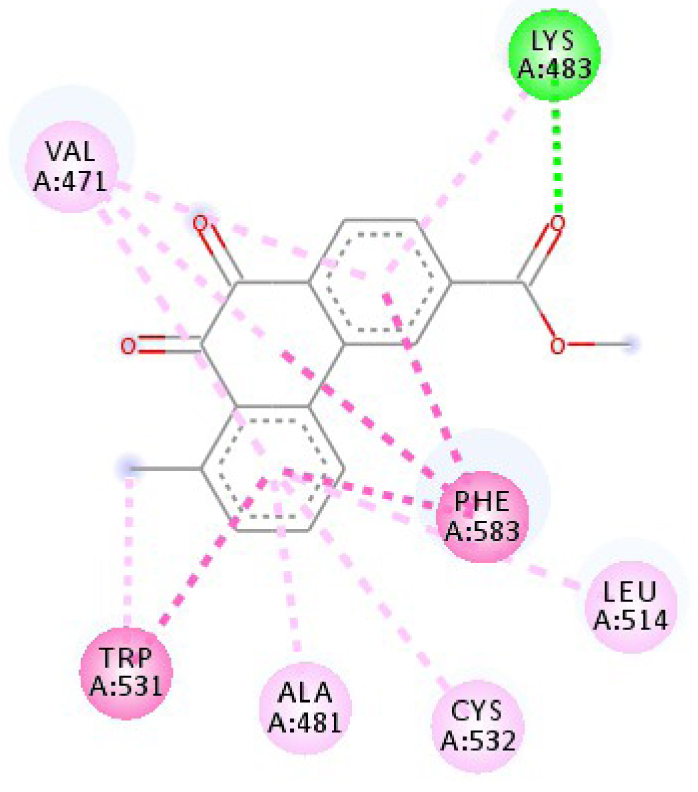 | 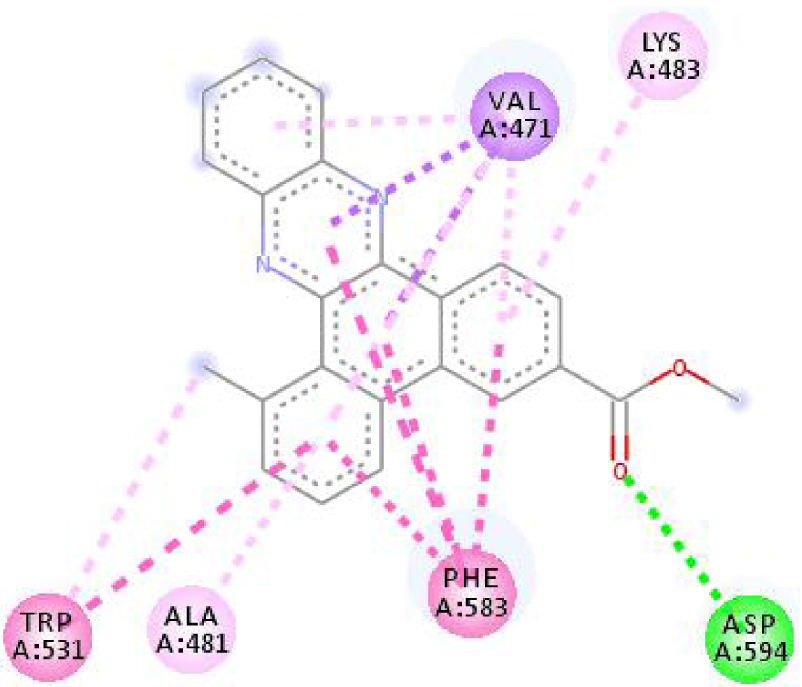 |
| 3EWH | 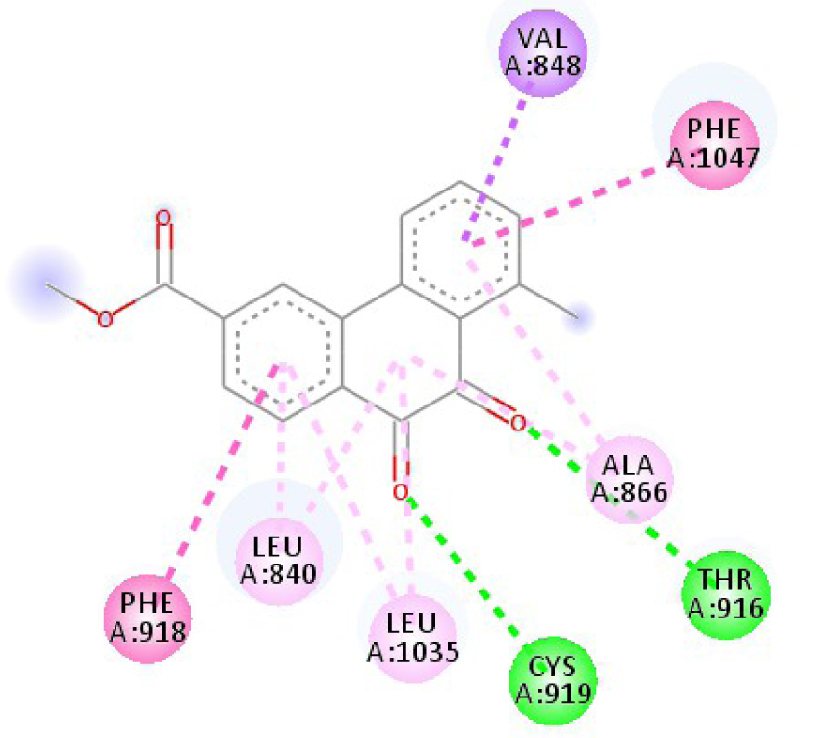 | 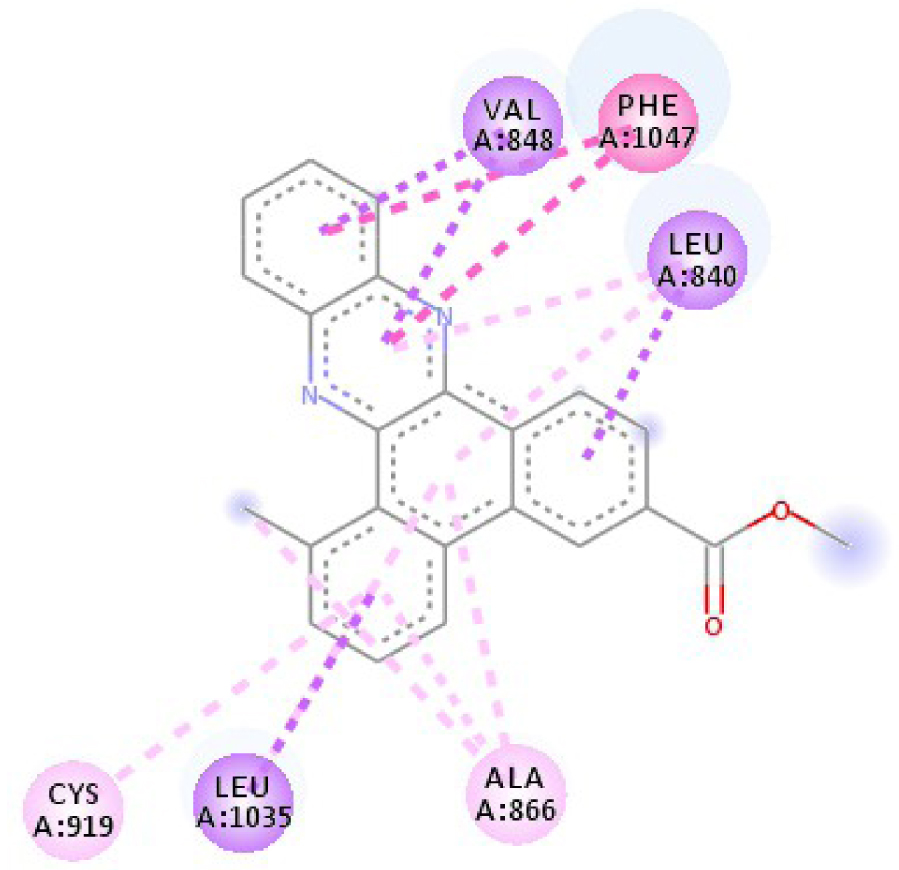 |
| 3RCD | 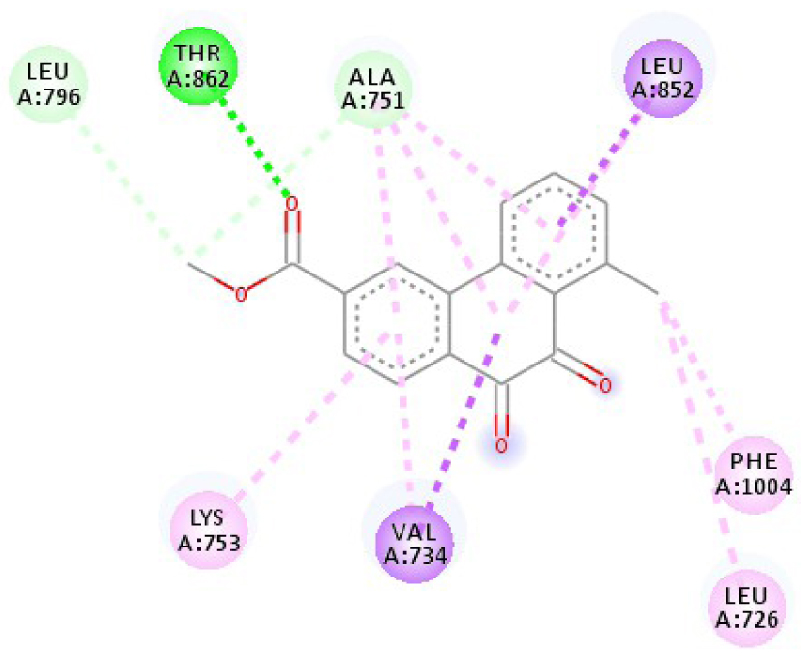 | 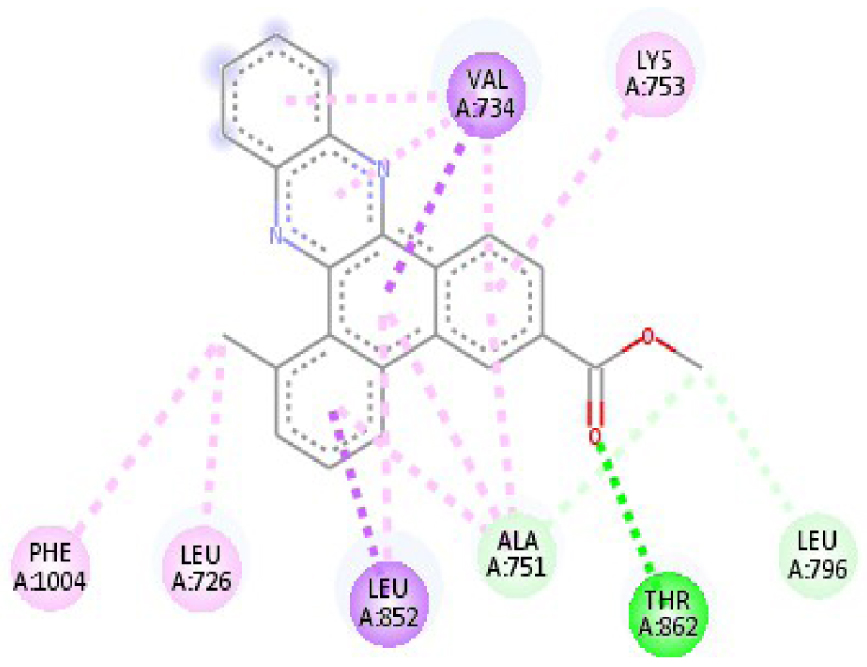 |
| 3W2S | 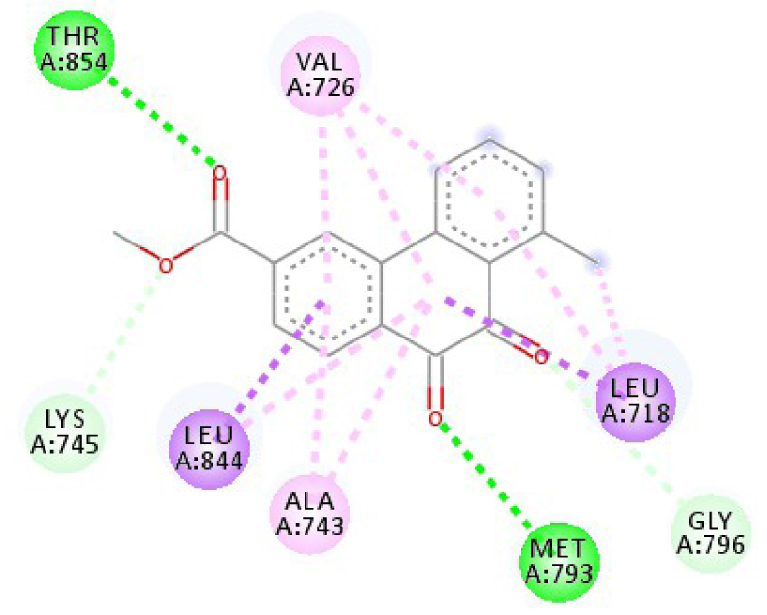 | 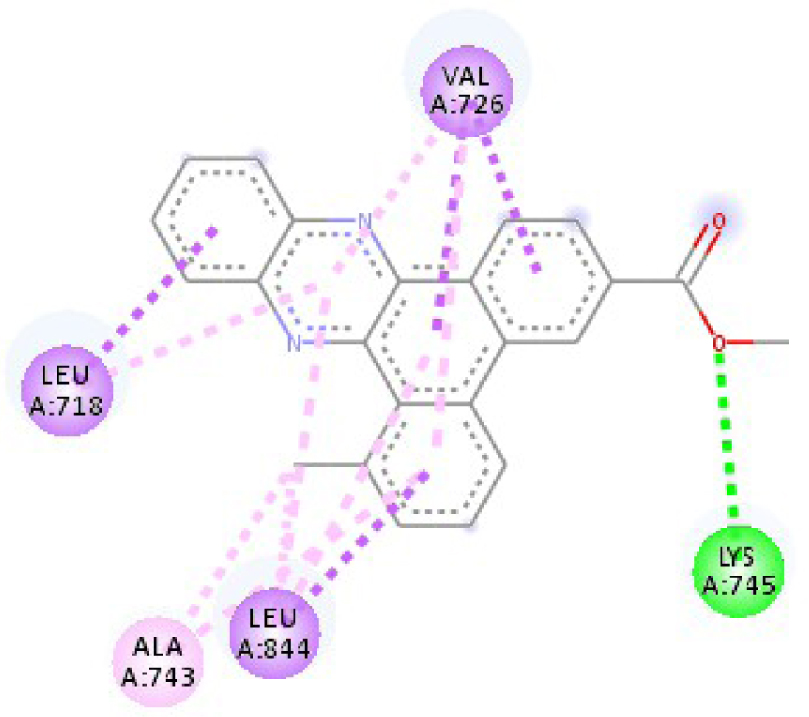 |
| 4JT5 | 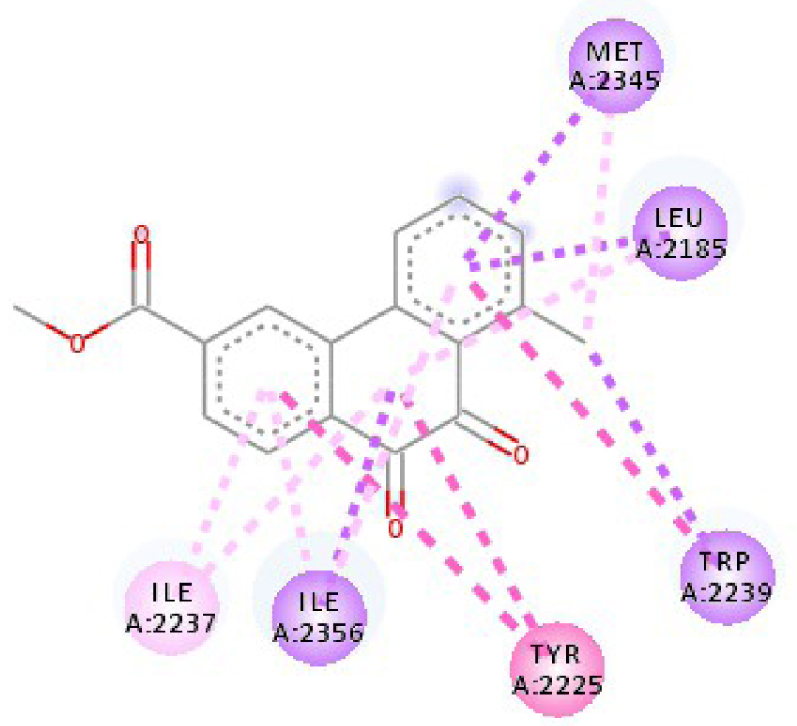 | 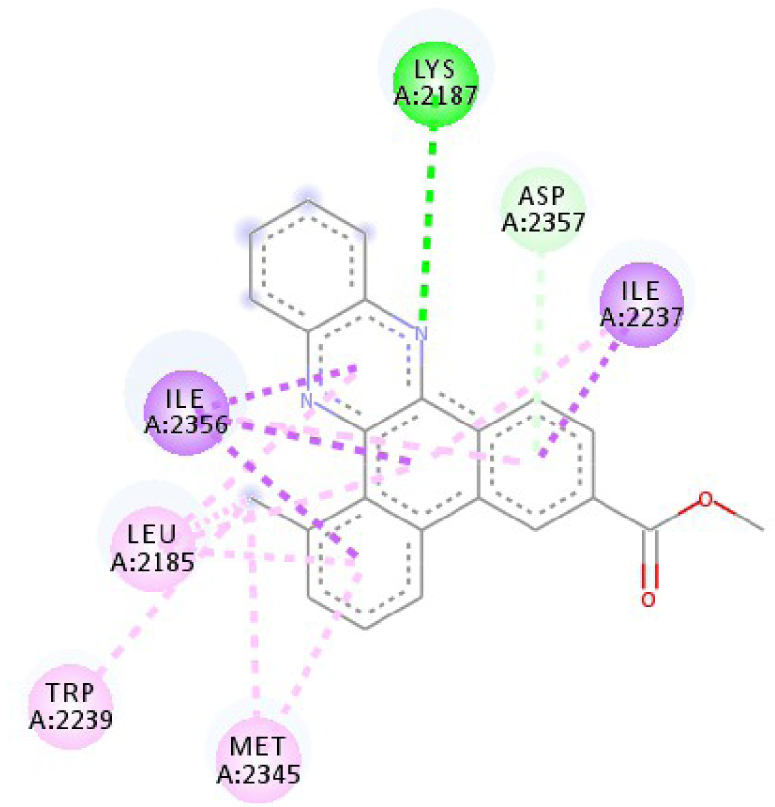 |
| 4U5J | 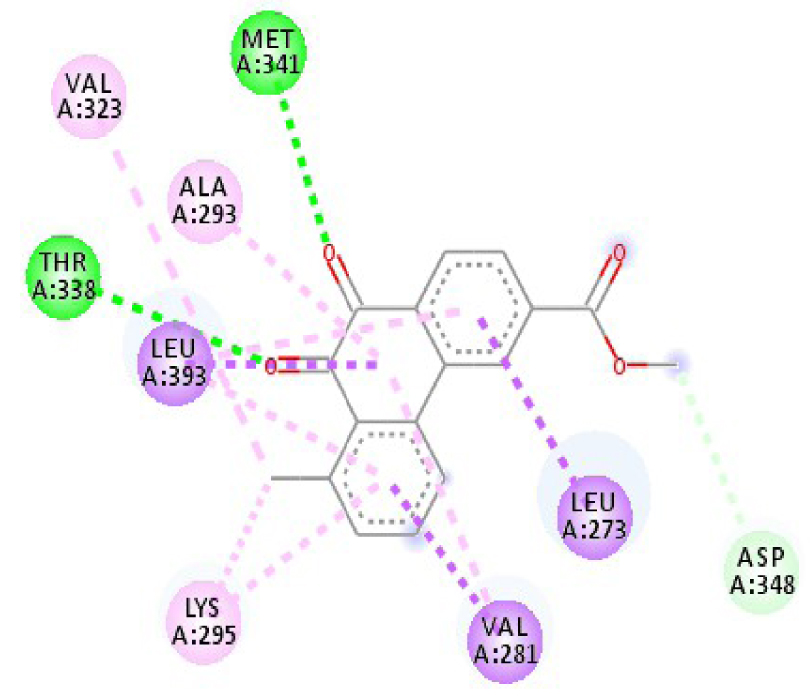 | 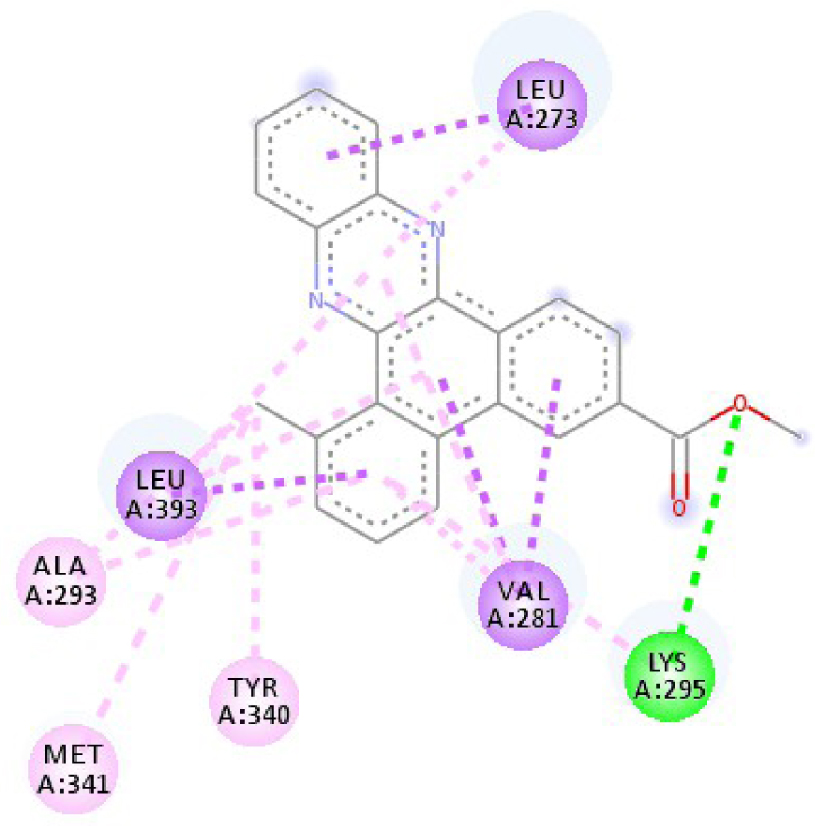 |
| 6N2J | 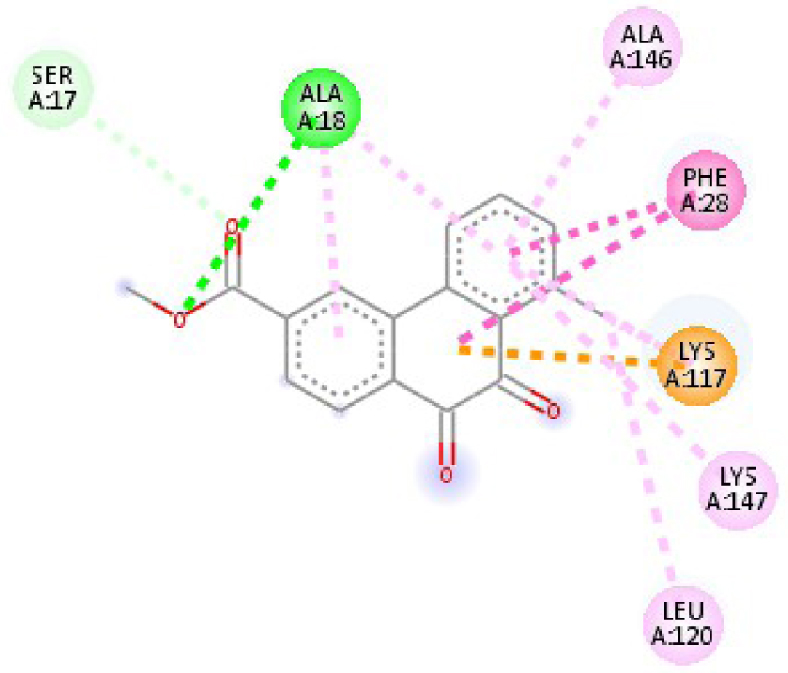 | 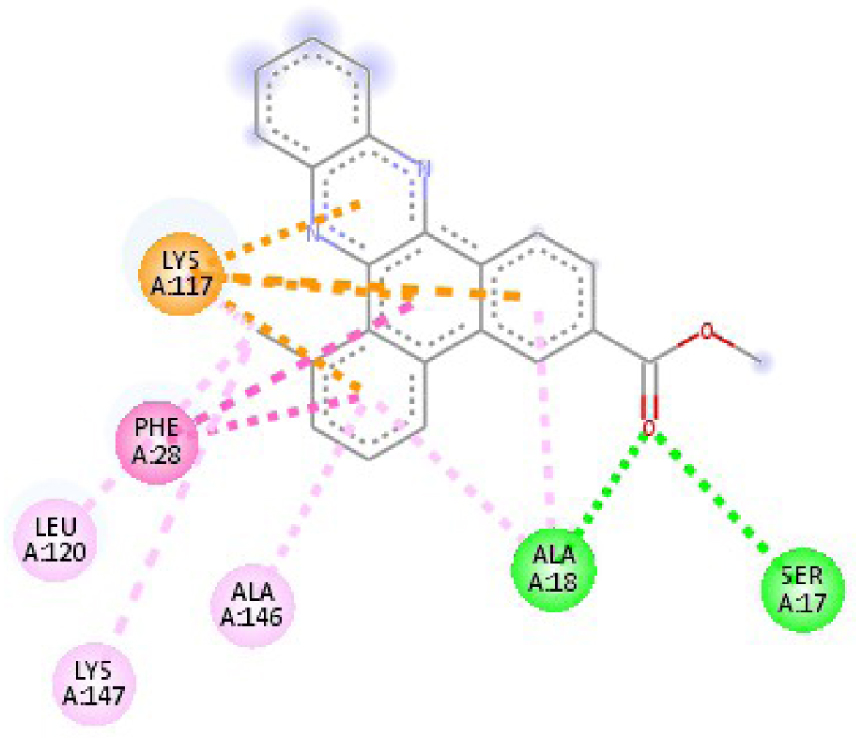 |
 | ||
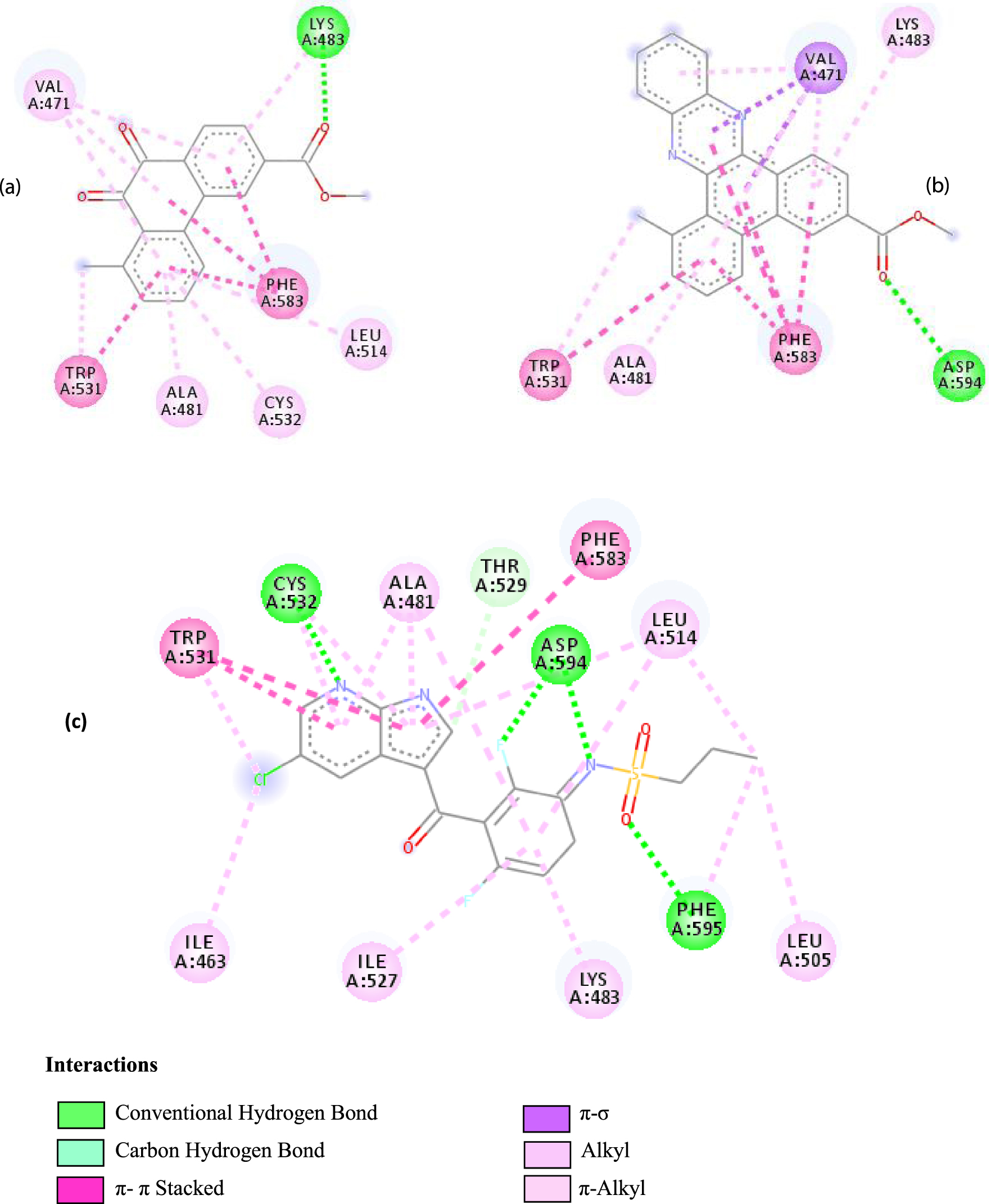
Interactions between the protein B-Raf proto-oncogene serine/threonine-protein kinase and molecule D-1 (a), molecule D-2 (b), and the native ligand PLX4720 (c).
Details of the different interactions
| Target | D-1 | D-2 | Target | D-1 | D-2 |
|---|---|---|---|---|---|
| 2HYY | ASP 381 NH–O ligand | ALA 380 C–O ligand | 3W2S | MET 793 NH–O ligand | LYS 745 NH–O ligand |
| ALA 330 C–O ligand | VAL 299 O–C ligand | THR 854 OH–O ligand | LEU 718 C–π ligand | ||
| ASP 381 O–π ligand | ASP 381 O–π ligand | LYS 745 C–O ligand | VAL 726 C–π ligand | ||
| ASP 381 O–π ligand | ASP 381 O–π ligand | GLY 796 C–O ligand | VAL 726 C–π ligand | ||
| ASP 381 O–π ligand | ASP 381 O–π ligand | LEU 718 C–π ligand | LEU 844 C–π ligand | ||
| VAL 289 C–π ligand | VAL 289 C–π ligand | LEU 844 C–π ligand | ALA 743 π–C ligand | ||
| VAL 289 π–C ligand | VAL 289 C–π ligand | LEU 718 π–C ligand | LEU 844 π–C ligand | ||
| ILE 293 π–π ligand | MET 290 π–π ligand | VAL 726 π–π ligand | LEU 844 π–π ligand | ||
| VAL 289 π–π ligand | ALA 743 π–π ligand | LEU 718 π–π ligand | |||
| VAL 726 π–π ligand | VAL 726 π–π ligand | ||||
| ALA 743 π–π ligand | LEU 844 π–π ligand | ||||
| LEU 844 π–π ligand | VAL 726 π–π ligand | ||||
| VAL 726 π–π ligand | ALA 743 π–π ligand | ||||
| 3C4C | LYS 483 NH–O ligand | ASP 594 NH–O ligand | 4JT5 | LEU 2185 C–π ligand | LYS 2187 NH–N ligand |
| TRP 531 π–π ligand | VAL 471 C–π ligand | MET 2345 C–π ligand | ASP 2357 N–π ligand | ||
| PHE 583 π–π ligand | VAL 471 C–π ligand | ILE 2356 C–π ligand | ILE 2237 C–π ligand | ||
| PHE 583 π–π ligand | TRP 531 π–π ligand | TRP 2239 π–π ligand | ILE 2356 C–π ligand | ||
| PHE 583 π–π ligand | PHE 583 π–π ligand | TYR 2225 π–π ligand | ILE 2356 C–π ligand | ||
| TRP 531 π–C ligand | PHE 583 π–π ligand | TYR 2225 π–π ligand | ILE 2356 C–π ligand | ||
| VAL 471 π–π ligand | PHE 583 π–π ligand | MET 2345 π–C ligand | ILE 2356 C–π ligand | ||
| LYS 483 π–π ligand | PHE 583 π–π ligand | ILE 2237 π–π ligand | LEU 2195 π–C ligand | ||
| VAL 471 π–π ligand | TRP 531 π–C ligand | ILE 2356 π–π ligand | MET 2345 π–C ligand | ||
| VAL 471 π–π ligand | VAL 471 π–π ligand | LEU 2285 π–π ligand | TRP 2239 π–C ligand | ||
| ALA 481 π–π ligand | VAL 471 π–π ligand | ILE 2237 π–π ligand | ILE 2356 π–π ligand | ||
| LEU 514 π–π ligand | VAL 471 π–π ligand | ILE 2356 π–π ligand | LEU 2185 π–π ligand | ||
| CYS 532 π–π ligand | LYS 483 π–π ligand | ILE 2237 π–π ligand | |||
| ALA 481 π–π ligand | LEU 2185 π–π ligand | ||||
| CYS 532 π–π ligand | LEU 2185 π–π ligand | ||||
| MET 2345 π–π ligand | |||||
| 3EWH | THR 916 OH–O ligand | LEU 840 C–π ligand | 4U5J | THR 338 OH–O ligand | LYS 295 NH–O ligand |
| CYS 919 NH–O ligand | VAL 848 C–π ligand | MET 341 NH–O ligand | LEU 273 C–π ligand | ||
| VAL 848 C–π ligand | VAL 848 C–π ligand | ASP 348 OH–C ligand | VAL 281 C–π ligand | ||
| PHE 918 π–π ligand | LEU 1035 C–π ligand | LEU 273 C–π ligand | VAL 281 C–π ligand | ||
| PHE 1074 π–π ligand | PHE 1047 π–π ligand | VAL 281 C–π ligand | LEU 393 C–π ligand | ||
| LEU 840 π–π ligand | PHE 1047 π–π ligand | LEU 393 C–π ligand | ALA 293 π–C ligand | ||
| LEU 1035 π–π ligand | ALA 866 π–C ligand | LYS 295 π–C ligand | MET 341 π–C ligand | ||
| LEU 1035 π–π ligand | LEU 840 π–π ligand | VAL 323 π–C ligand | LEU 393 π–C ligand | ||
| LEU 840 π–π ligand | ALA 866 π–π ligand | LEU 393 π–π ligand | TYR 340 C–π ligand | ||
| ALA 866 π–π ligand | LEU 1035 π–π ligand | VAL 281 π–π ligand | LEU 393 π–π ligand | ||
| ALA 866 π–π ligand | LEU 840 π–π ligand | ALA 293 π–π ligand | LEU 273 π–π ligand | ||
| ALA 866 π–π ligand | LYS 295 π–π ligand | VAL 281 π–π ligand | |||
| CYS 919 π–π ligand | LEU 393 π–π ligand | LEU 393 π–π ligand | |||
| VAL 281 π–π ligand | |||||
| VAL 293 π–π ligand | |||||
| LYS 295 π–π ligand | |||||
| 3RCD | THR 862 OH–O ligand | THR 862 OH–O ligand | 6N2J | ALA 18 NH–O ligand | SER17 N–O ligand |
| ALA 751 OH–C ligand | ALA 571 O–C ligand | SER 17 C–O ligand | ALA 18 N–O ligand | ||
| LEU 796 O–C ligand | LEU 796 O–C ligand | LYS 117 N–π ligand | LYS 117 N–π ligand | ||
| VAL 734 C–π ligand | VAL 734 C–π ligand | PHE 28 π–π ligand | LYS 117 N–π ligand | ||
| LEU 852 C–π ligand | LEU 852 C–π ligand | PHE 28 π–π ligand | LYS 117 N–π ligand | ||
| LEU 726 π–C ligand | LEU 726 π–C ligand | LYS 117 π–C ligand | LYS 117 N–π ligand | ||
| PHE 1004 π–C ligand | PHE 1004 π–C ligand | LEU 120 π–C ligand | PHE 28 π–π ligand | ||
| VAL 734 π–π ligand | VAL 734 π–π ligand | ALA 18 π–π ligand | PHE 28 π–π ligand | ||
| ALA 751 π–π ligand | ALA 751 π–π ligand | LYS 117 π–π ligand | LYS 117 π–C ligand | ||
| LYS 753 π–π ligand | LYS 753 π–π ligand | ALA 18 π–π ligand | LEU 120 π–C ligand | ||
| ALA 751 π–π ligand | ALA 751 π–π ligand | LYS 117 π–π ligand | LYS 147 π–C ligand | ||
| LEU 852 π–π ligand | LEU 852 π–π ligand | ALA 146 π–π ligand | ALA 18 π–π ligand | ||
| ALA 751 π–π ligand | VAL 734 π–π ligand | LYS 147 π–π ligand | LYS 117 π–π ligand | ||
| ALA 751 π–π ligand | ALA 18 π–π ligand | ||||
| VAL 734 π–π ligand | LYS 117 π–π ligand | ||||
| ALA 146 π–π ligand | |||||
In addition, the data shown in Table 3 indicate that both molecules show a very high affinity to and stability with all the studied targets. In particular, the highest affinity is noted for the protein B-Raf proto-oncogene serine/threonine-protein kinase (PDB code 3C4C) [18, 19]. In fact, the affinity between molecule D-1 and 3C4C is found to be about −9.8 Kcal∕mol. The affinity between 3C4C and molecule D-2 is about −11.1 Kcal∕mol. We can conclude from these results that the B-Raf protein is the most likely target for this molecule. Thus, it is worth noting that this protein is involved in sending signals inside cells that are involved in directing cell growth. It regulates cell proliferation and growth, cell survival, cell mobility, protein biosynthesis, and transcription, which suggests that it is the most targeted protein by our tricyclic molecule.
Moreover, it is believed that the remarkable activity of molecules D-1 and D-2 is related to their stability, which is explained by the numbers and types of bonds established with the studied potential targets. These descriptors are mentioned in Table 4, and the details of the interactions are presented in Table 5. In this regard, the amino acids VAL 471, PHE 583, LYS 483, ALA 481, and TRP 531 are found to be important for the antiproliferative activity of our molecules since they form bonds in both cases. As expected for 3C4C, it appears that the hydrogen bond formed with the amino acid ASP 594 increases affinity to the target, which in turn increases the activity of both molecules.
The positioning of each molecule in the active site and the binding pocket are shown in Table 6.
Positioning and the binding pocket of each molecule in the active site
| PDB code | D-1 | D-2 |
|---|---|---|
| 2HYY | 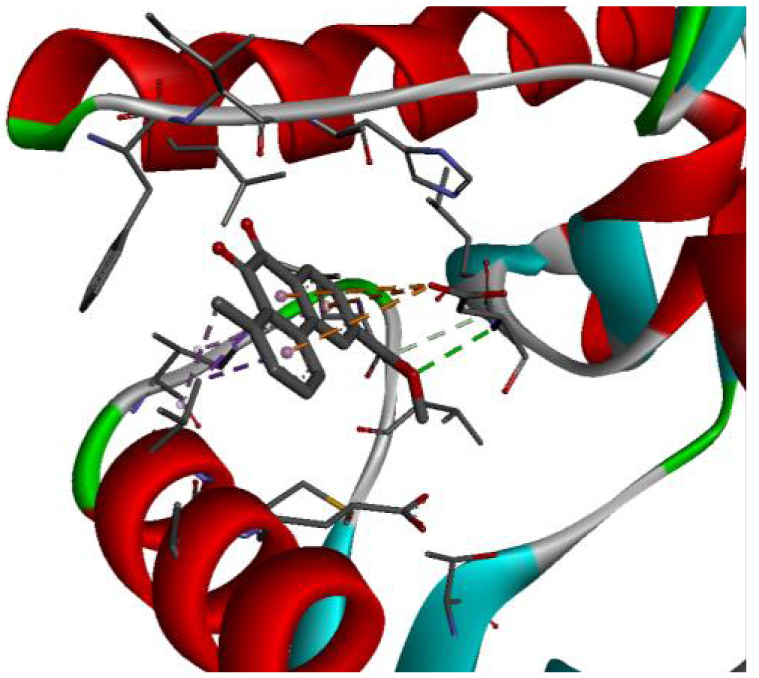 | 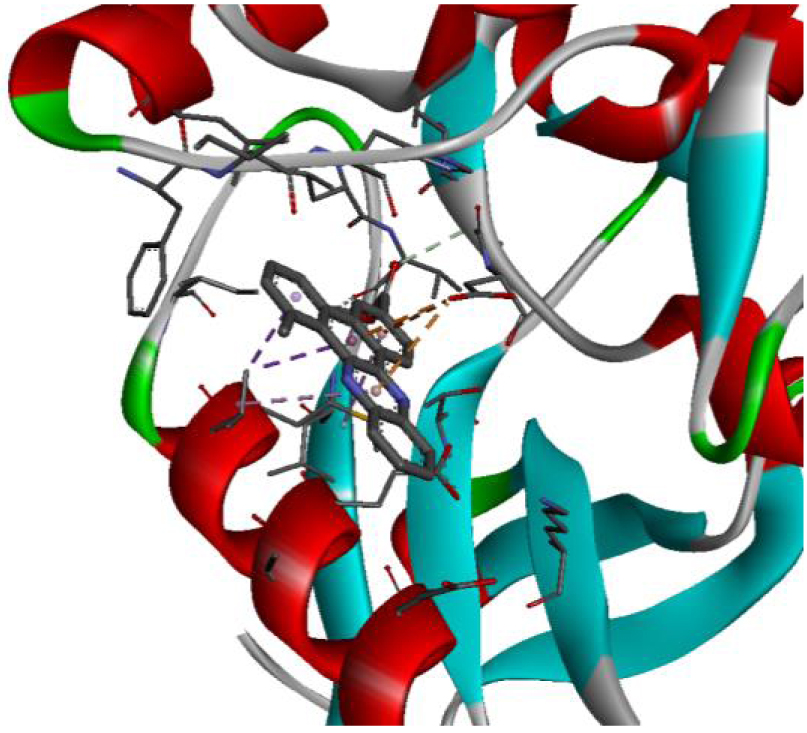 |
| 3C4C | 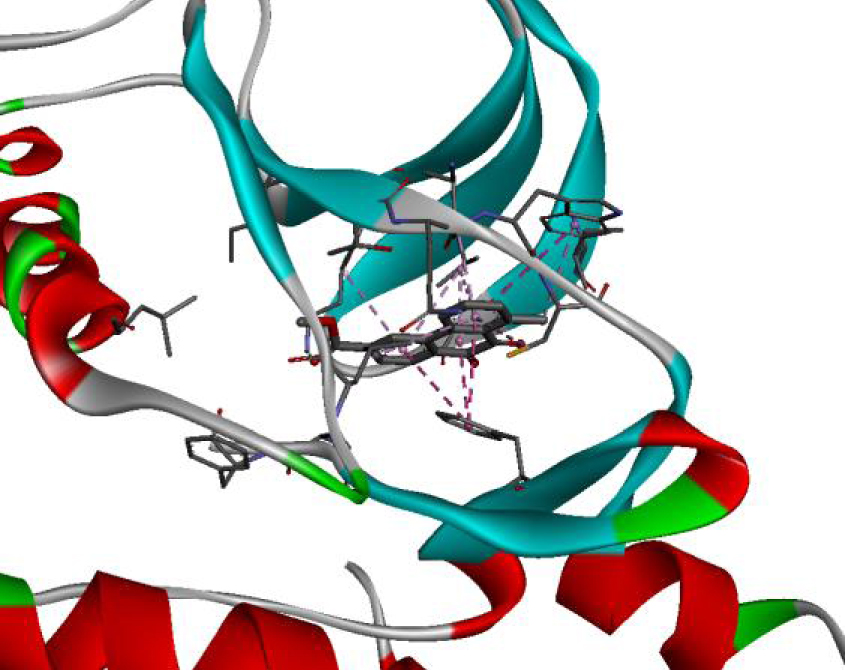 | 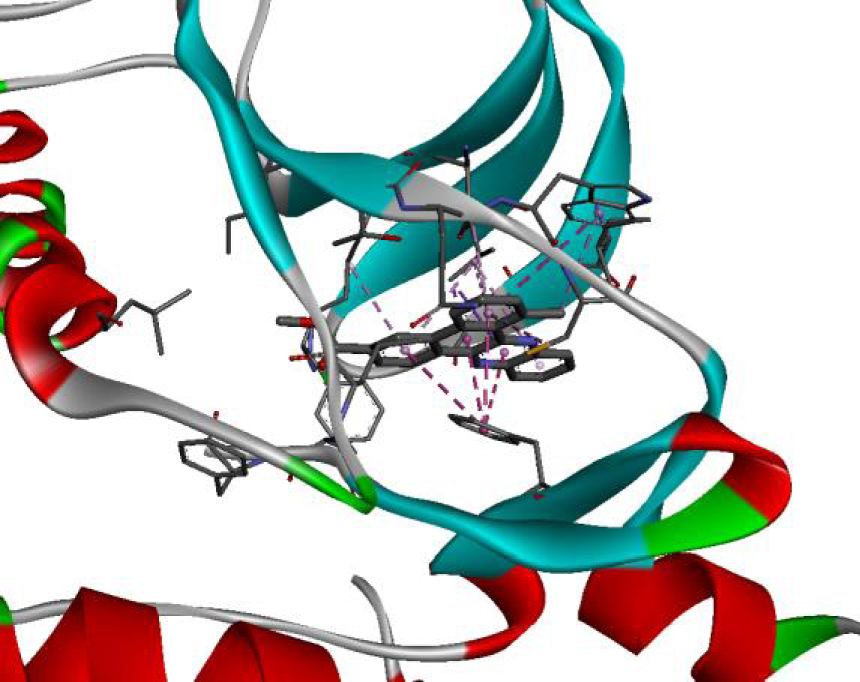 |
| 3EWH | 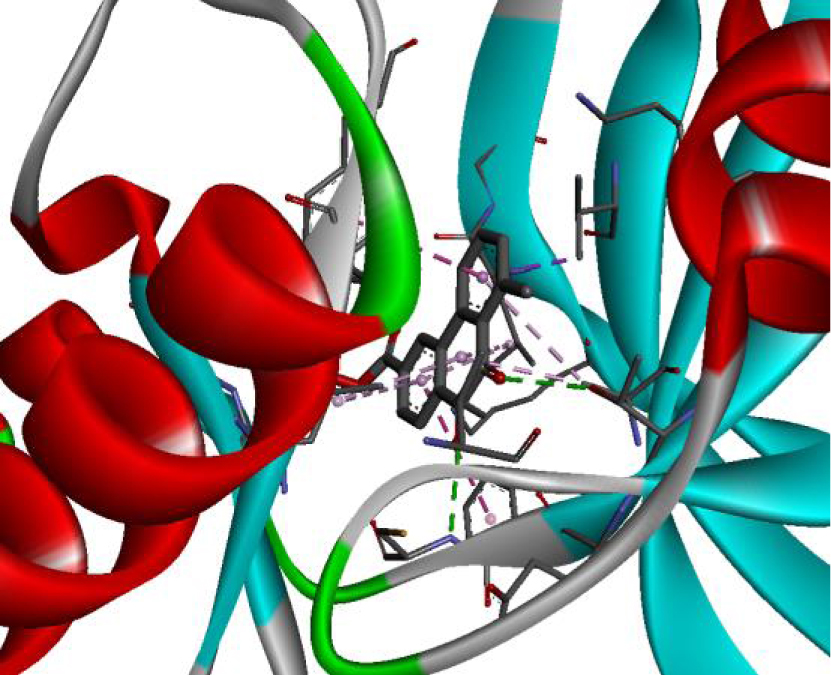 | 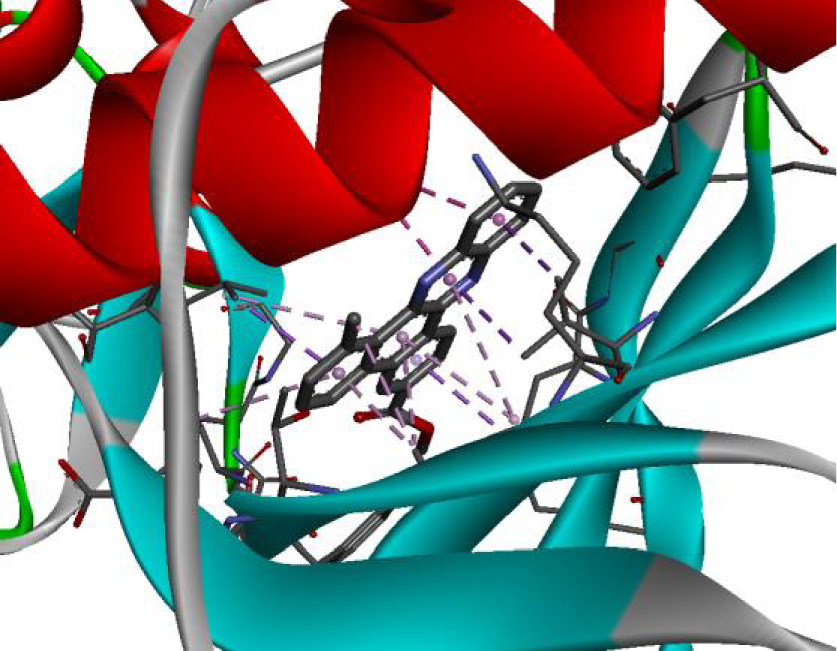 |
| 3RCD | 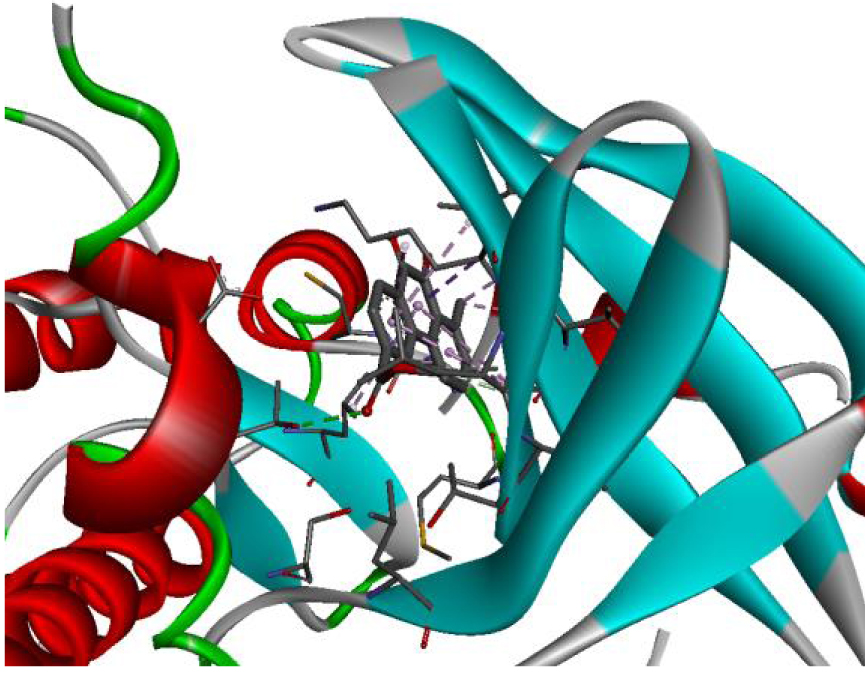 | 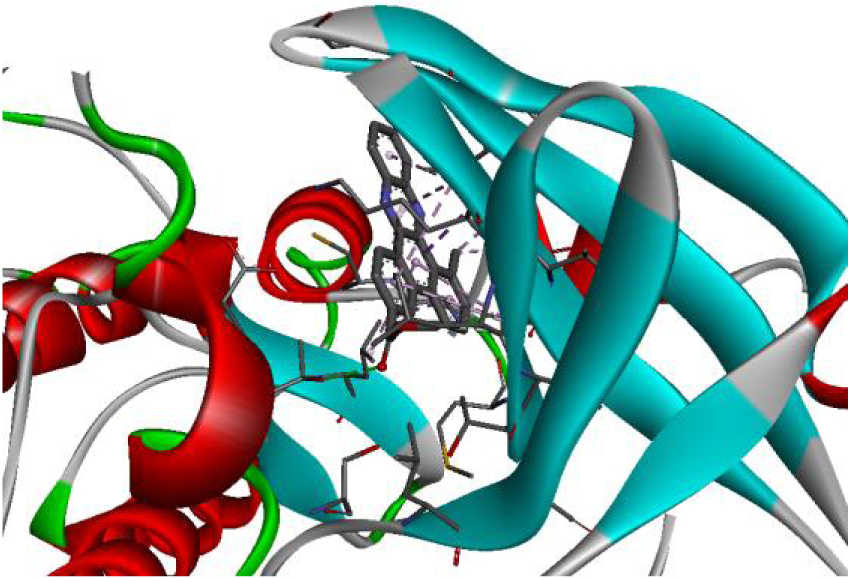 |
| 3W2S | 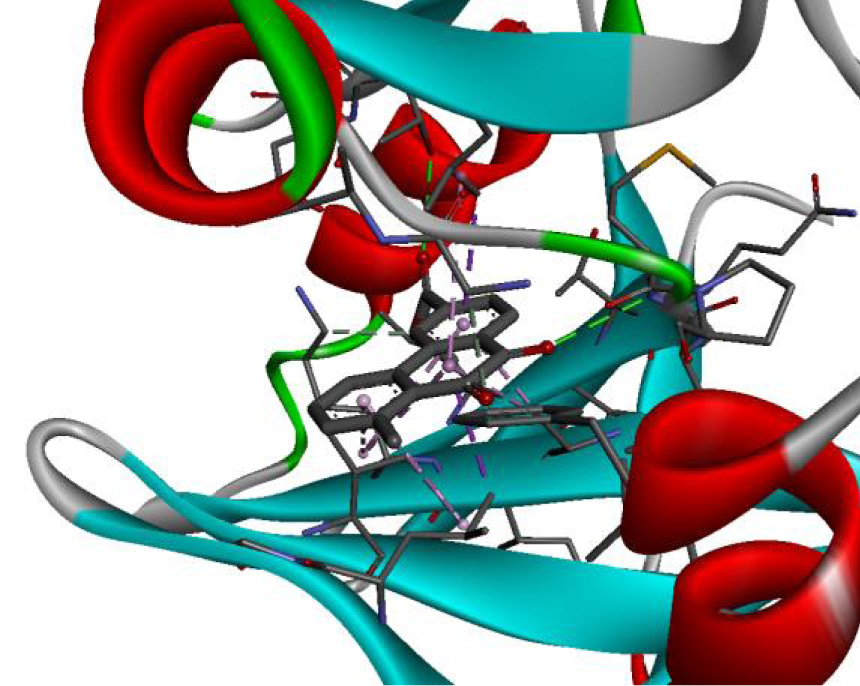 | 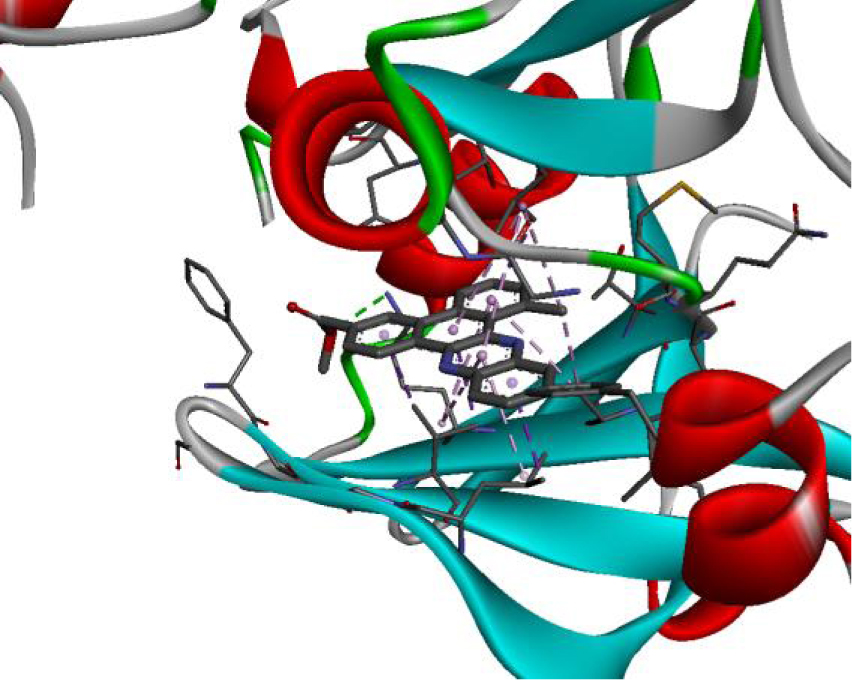 |
| 4JT5 | 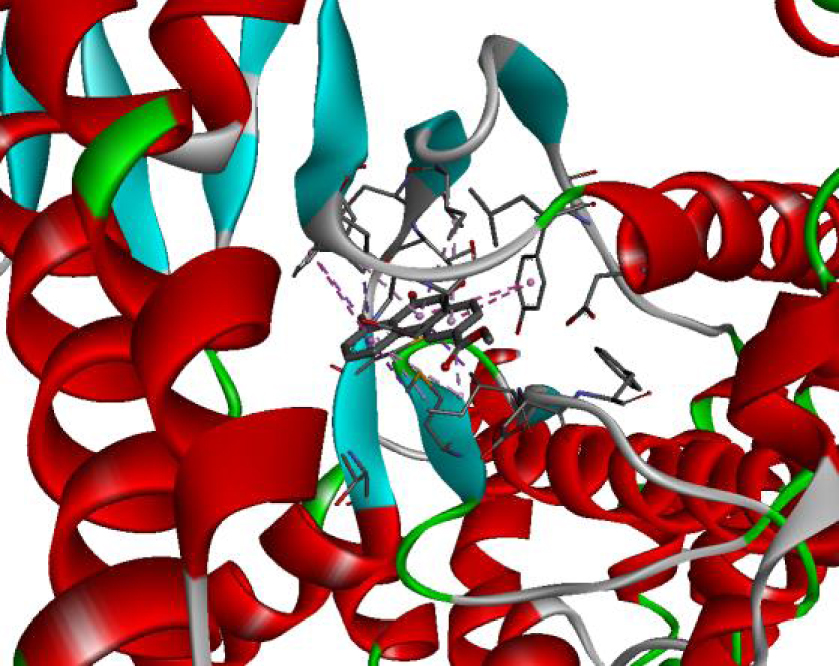 | 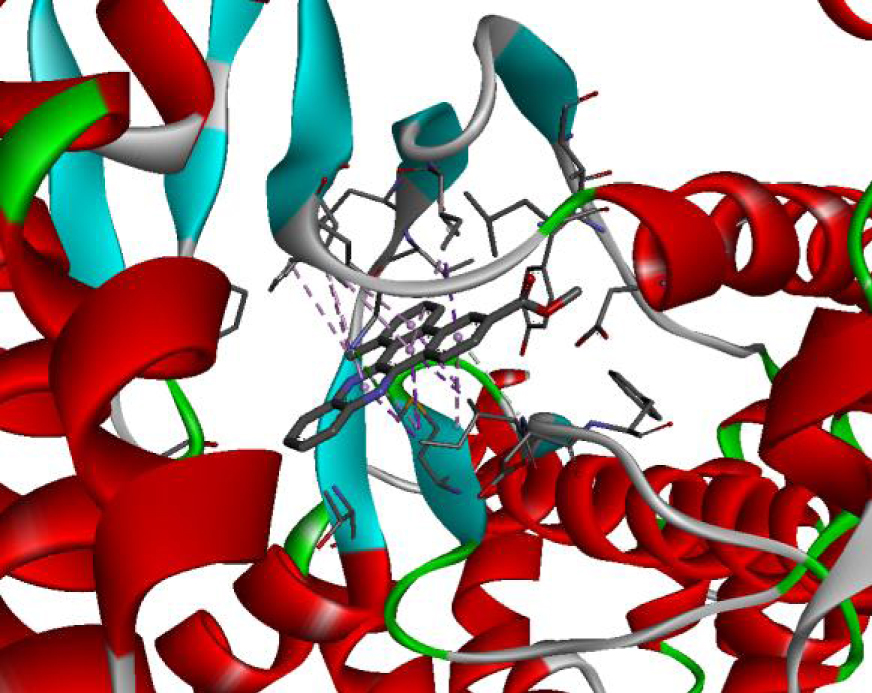 |
| 4U5J | 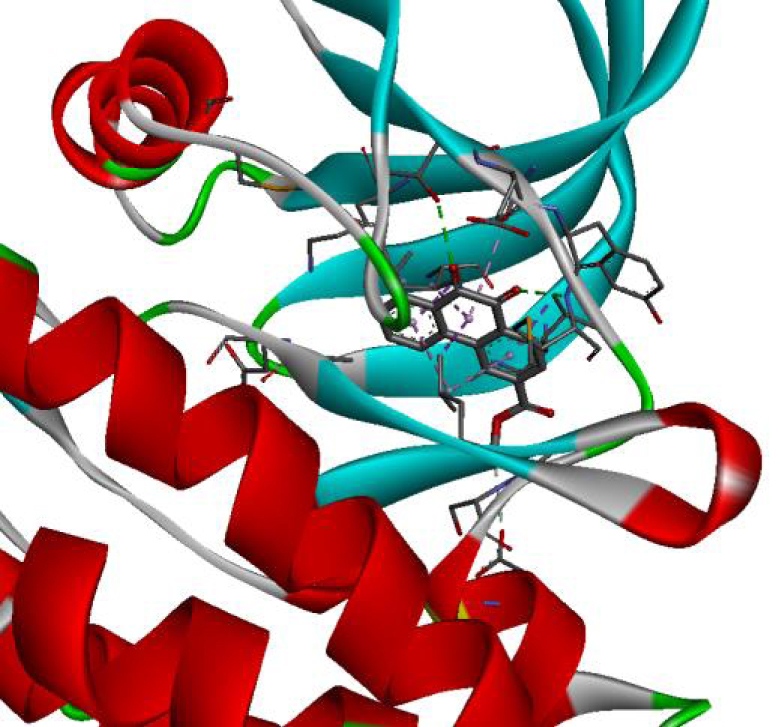 | 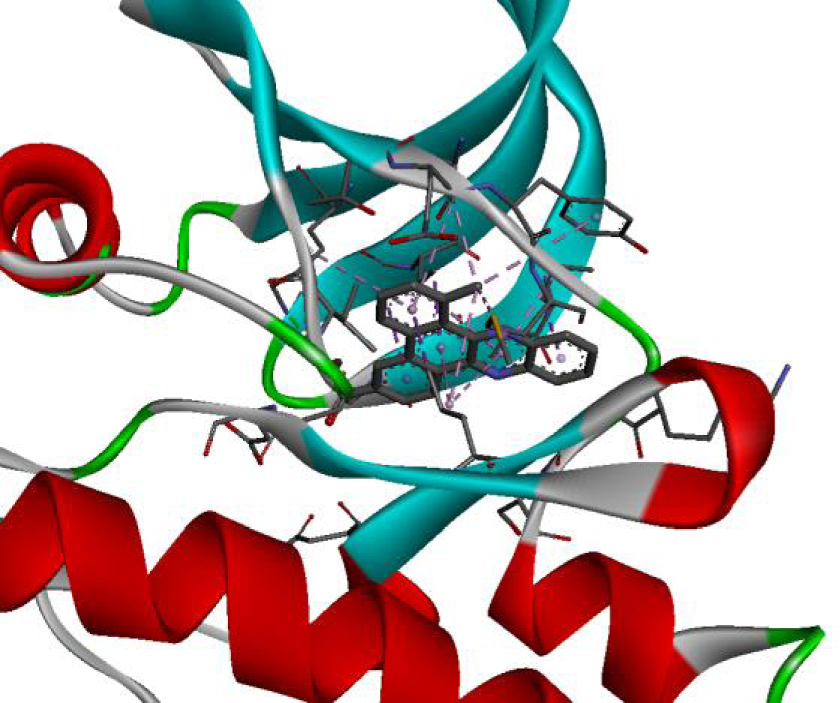 |
| 6N2J | 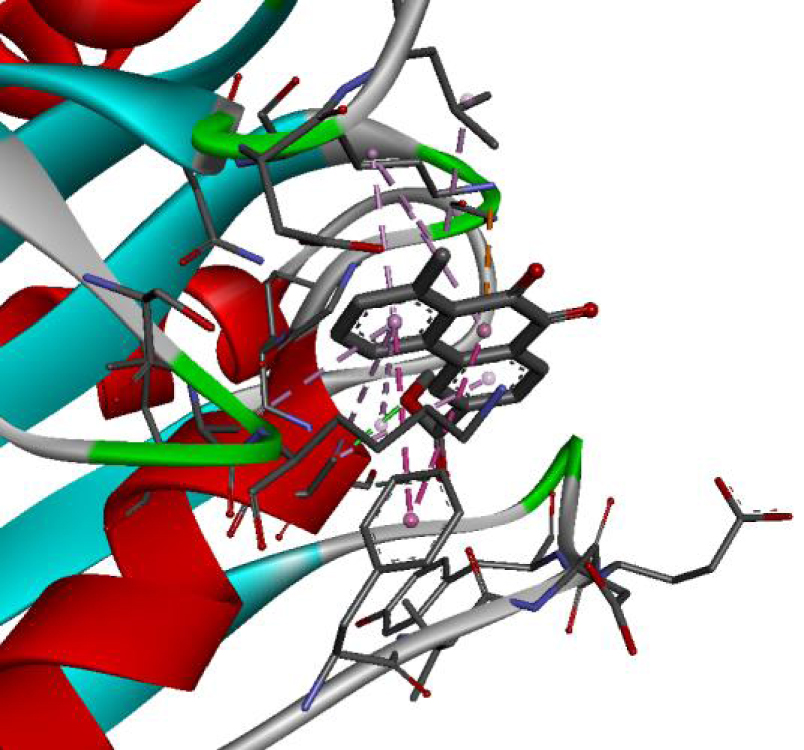 | 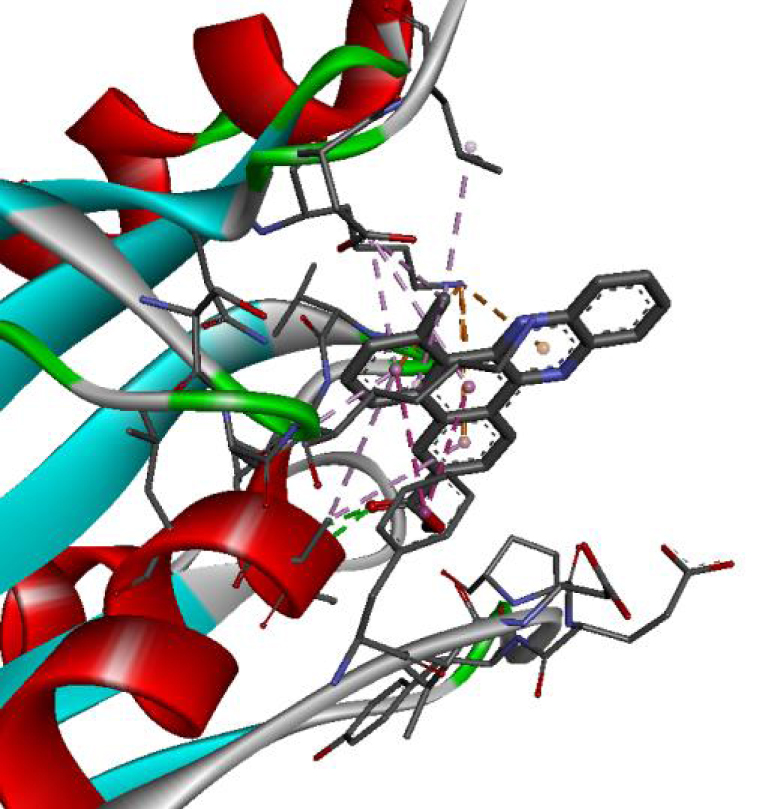 |
Based on affinity, stability, and the study of different interactions, we can ensure that B-Raf proto-oncogene serine/threonine-protein kinase is the potential target for these molecules. To better confirm these results, we have compared the types and numbers of bonds of our molecules D-1 and D-2 with those of the native ligand PLX4720 (Figure 3).
It can be seen from the comparison of the interactions between the potential target and the studied molecules as well as the reference molecule that most of the amino acids that interact with the reference molecule also interact with our studied molecules. These interactions are essentially hydrophobic bonds, hydrogen bonds, and π-interactions (Figure 4).
3. Conclusion
Following the study of H. Guédouar and co-workers that evaluated the antiproliferative activity of phenanthrene derivatives against the Caco-2 cancer cell line, this study proposed two molecules that exhibited the best activity against this cancerous cell. For understanding the mode of action of these molecules to propose a potential therapeutic target in the two types of studied cancers, we established molecular docking against eight vital cancer targets. As a result, molecular docking results confirmed that tricyclic molecules D-1 and D-2 show significant activity. Both of the studied compounds were found to display low binding energies and the best affinity is noted in the protein B-Raf proto-oncogene serine/threonine-protein kinase, which is an important target in both types of studied cancers. Moreover, the comparison of the types and the modes of interactions between these molecules and the reference ligand, which is an inhibitor of this protein, shows remarkable similarity in the binding of amino acids in the types of interactions, which suggests that molecules D-1 and D-2 are ligands that can inhibit this protein.