

 CC-BY 4.0
CC-BY 4.0
1. Introduction
Phenanthrenes are representative π-conjugated polyaromatic compounds consisting of three ortho-fused benzenoid units. They are considered interesting materials for photoluminescent applications [1, 2]. They are known to exhibit good electrical and optical properties due to their unique photoluminescence (PL) and electroluminescence properties [3, 4, 5, 6, 7] and their ease of use in the fabrication of new sensors or nanostructured materials. Among other distinctive properties of phenanthrenes are their higher thermal stability, lower toxicity than other polyaromatic systems, and their ready functionalization, which make them very desirable for applications in materials science [8, 9, 10].
Indeed, the synthesis of phenanthrene derivatives with important properties has been attracting attention in the manufacture of new materials, opening up new fields of potential applications. Inspired by the literature, many phenanthrene derivatives have been synthesized, and their photophysical and thermal properties including their electrochemistry have been intensively studied. These derivatives offer access to various organic devices such as organic light-emitting diodes (OLEDs) [11, 12, 13, 14, 15, 16, 17, 18], solar cells [19, 20, 21], superconductors [22, 23], etc. In an independent study, Zhang and co-workers reported the synthesis of phenanthro-imidazole 1 (Figure 1) and used it for OLEDs [24]. More recently, Akula and co-workers have reported the synthesis of ruthenium complex 2, revealing good optical, electrochemical, and thermal properties, which has been used as a promising electron-transport material for organic solar cells [25].
Phenanthrene derivatives 1 and 2 as promising materials.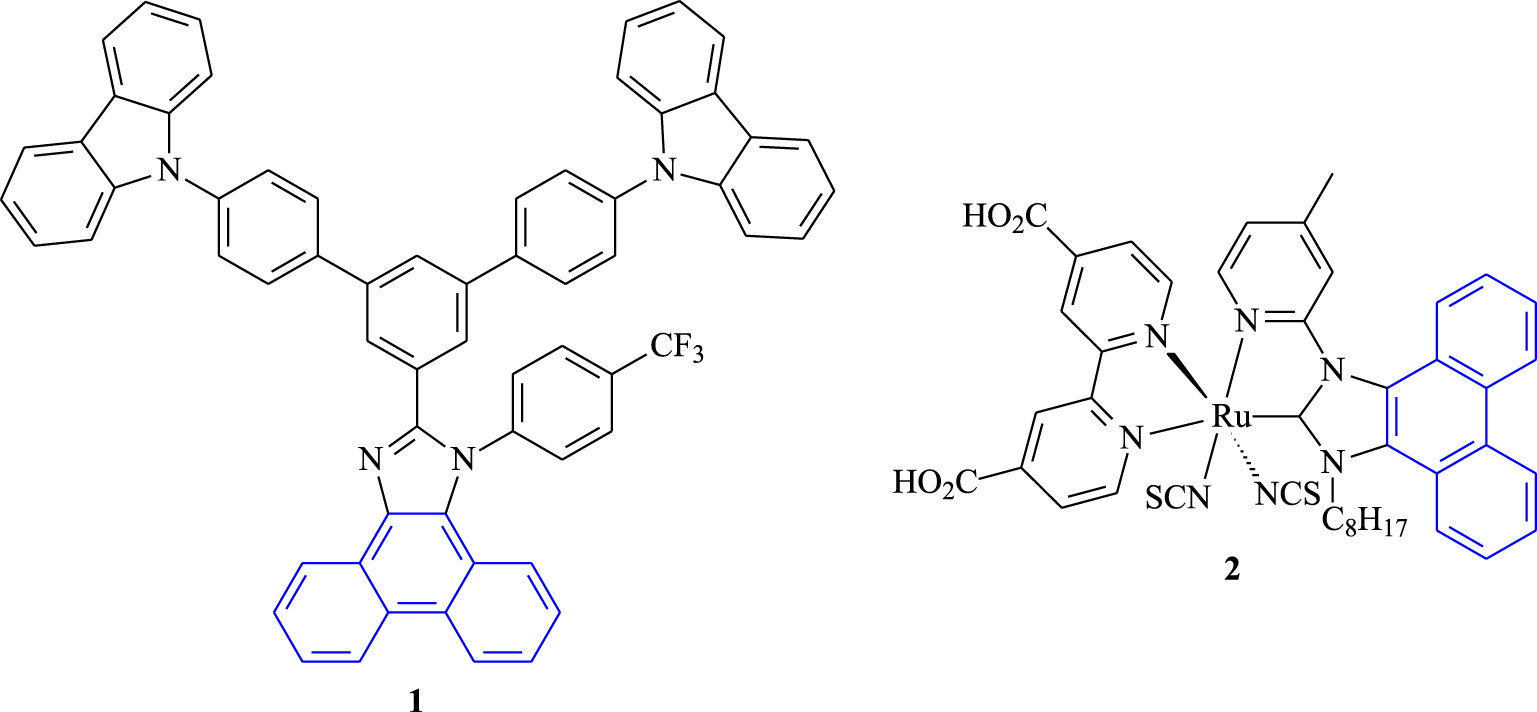
Phenanthrene derivatives are important molecules exhibiting interesting properties and have been attracting interest not only for their applications as organic materials but also for their versatile synthetic route. Up until now, various strategies have been applied for producing these π-conjugated systems. One of the most widely employed methods is the photocyclization approach, which is considered fast, economical, and practical. In addition, using this method, it is easy to obtain new electron-rich structured phenanthrenes with a wide range of characterizations and applications.
2. Experimental section
2.1. General
The majority of the experiments were performed under anhydrous conditions (dry N2 or Ar) using freshly distilled solvents. Solvents and starting materials were purchased from Aldrich. Column chromatography purifications were performed over silica gel obtained from SiliCycle chemical division (40–63 nm; 230–240 mesh). Photochemical reactions were carried out using a 150 W mercury lamp. 1H and 13C NMR spectra were recorded on a Bruker AM 300, using CDCl3 and DMSO-d6 as deuterated solvents with tetramethylsilane as the internal reference at room temperature. Chemical shifts (δ) were reported in parts per million (ppm) relatively to tetramethylsilane. Multiplicity was reported as follows: s: singlet, d: doublet, dd: doublet of doublets, and m: multiplet. UV–Vis spectroscopy was recorded on a Varian Cary 5000 spectrometer in quartz cuvettes with a path length of 1 cm. Electrochemical studies were carried out under argon atmosphere using an Eco Chemie Autolab PGSTAT 30 potentiostat for voltammetric measurements, connected to a conventional three-electrode cell. The working electrode was a platinum microdisk, the reference electrode was a saturated calomel electrode (SCE), and the counter-electrode was a platinum wire.
2.2. General procedure for the synthesis of α,β-unsaturated nitriles 2a–f
In a two-neck, 100-mL flask placed under argon, 4.5 mmol (1 eq) of p-fluorophenylacetonitrile (1), 4.5 mmol (1 eq) of aromatic aldehyde, and 30 mL of anhydrous methanol were introduced. The mixture was stirred until total dissolution of the reagents. Then 375 mg of sodium methanolate (8.3 mmol, 2 eq) was added in small portions, and the mixture was stirred at room temperature. The evolution of the reaction was monitored by TLC. The resulting product was recovered by filtration on a sintered glass, washed with cold methanol and then with distilled water, and dried.
2.2.1. (Z)-2-(p-fluorophenyl)-3-(p-methoxyphenyl) acrylonitrile (2a)
White solid, 85%; m.p. = 97–99 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 3.87 (s, 3H, OCH3), 6.97 (d, J = 9 Hz, 2H), 7.09–7.15 (m, 2H), 7.39 (s, 1H), 7.60–7.65 (m, 2H), 7.86 (d, J = 9 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ (ppm): 54.91 (OCH3), 107.22 (C), 113.95 (2CH), 115.37 (d, JC‐F = 21.8 Hz, 2CH), 117.83 (CN), 125.90 (C), 127.05 (d, JC‐F = 8.2 Hz, 2CH), 130.30 (C), 130.61 (2CH), 141.25 (CH), 161.04 (C–O), 162.48 (d, JC‐F = 251.2 Hz, CF); 19F NMR (282 MHz, CDCl3): δ (ppm): − 111.6 Hz (s, F).
2.2.2. (Z)-3-(p-bromophenyl)-2-(p-fluorophenyl) acrylonitrile (2b)
Yellow solid, 80%; m.p. = 88–90 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.12–7.17 (m, 2H), 7.39 (s, 1H), 7.59–7.67 (m, 4H), 7.73 (d, J = 8.7 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ (ppm): 110.97 (C), 115.58 (d, JC‐F = 21.9 Hz, 2CH), 117.01 (CN), 124.45 (C), 127.36 (d, JC‐F = 8.2 Hz, 2CH), 129.89 (C), 129.93 (2CH), 131.76 (2CH), 131.96 (C), 140.06 (CH), 161.22 (d, JC‐F = 249 Hz, CF); 19F NMR (282 MHz, CDCl3): δ (ppm): −111.13 Hz (s, F).
2.2.3. (Z)-4-(2-cyano-2-(p-fluorophenyl)vinyl) benzonitrile (2c)
White solid, 65%; m.p. = 208–210 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.17–7.22 (m, 2H), 7.50 (s, 1H), 7.71 (d, J = 8.1 Hz, 2H), 7.77 (d, J = 7.8 Hz, 2H), 7.97 (d, J = 7.8 Hz, 2H), 13C NMR (75 MHz, CDCl3): δ (ppm): 113.13 (C), 113.79 (C), 115.82 (d, JC‐F = 22.1 Hz, 2CH), 116.55 (CN), 117.71 (CN), 127.67 (d, JC‐F = 8.4 Hz, 2CH), 129.05 (2CH), 132.20 (2CH), 137.18 (C), 138.80 (CH), 161.55 (d, JC‐F = 252 Hz, CF); 19F NMR (282 MHz, CDCl3): δ (ppm): −109.85 (s, F).
2.2.4. (Z)-2,3-bis(p-fluorophenyl)acrylonitrile (2d)
White solid, 90%; m.p. = 173–175 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.14–7.21 (m, 4H), 7.44 (s, 1H), 7.64–7.69 (m, 2H), 7.88–7.93 (m, 2H); 13C NMR (75 MHz, CDCl3): δ (ppm): 110.00 (C), 115.53 (d, JC‐F = 21.7 Hz, 4CH), 117.19 (CN), 127.29 (d, JC‐F = 8.2 Hz, 2CH), 129.36 (C), 130.01 (C), 130.74 (d, JC‐F = 9 Hz, 2CH), 140.19 (CH), 161.13 (d, JC‐F = 248.2 Hz, CF), 161.61 (d, JC‐F = 252 Hz, CF); 19F NMR (282 MHz, CDCl3): δ (ppm): −109.47 (s, F).
2.2.5. (Z)-2-(p-fluorophenyl)-3-(3′,4′- dimethoxyphenyl)acrylonitrile (2e)
Yellow solid, 94%; m.p. = 99–101 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 3.95 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 6.91 (d, J = 8.4 Hz, 1H), 7.10–7.15 (m, 2H), 7.33 (dd, J1 = 2.1 Hz, J2 = 8.4 Hz, 1H), 7.38 (s, 1H), 7.60–7.65 (m, 2H), 7.69 (d, J = 2.1 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ (ppm): 55.49 (OCH3), 55.52 (OCH3), 107.13 (C), 110.30 (CH), 110.54 (CH), 115.41 (d, JC‐F = 21.7 Hz, 2CH), 118.01 (CN), 123.86 (CH), 126.11 (C), 127.06 (d, JC‐F = 8.2 Hz, 2CH), 130.56 (C), 141.55 (CH), 148.65 (C–O), 150.80 (C–O), 160.82 (d, JC‐F = 248.2 Hz, CF); 19F NMR (282 MHz, CDCl3): δ (ppm): −112.36 (s, F).
2.2.6. (Z)-2-(p-fluorophenyl)-3-(thiophen-2-yl)acrylonitrile (2f)
Yellow solid, 75%; m.p. = 98–100 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.12–7.19 (m, 3H), 7.56 (d, J = 5.1 Hz, 1H), 7.60–7.69 (m, 4H); 13C NMR (75 MHz, CDCl3): δ (ppm): 106.77 (C), 115.54 (d, JC‐F = 21.7 Hz, 2CH), 117.50 (CN), 127.00 (d, JC‐F = 8.2 Hz, 2CH), 127.38 (CH), 129.57 (CH), 129.63 (d, JC‐F = 3 Hz, C), 131.84 (CH), 133.59 (d, JC‐F = 1.5 Hz, CH), 137.29 (C), 160.92 (d, JC‐F = 248.2 Hz, CF); 19F NMR (282 MHz, CDCl3): δ (ppm): −111.86 Hz (s, F).
2.3. General procedure for the synthesis of phenanthrene derivatives P1–6
α,β-unsaturated nitrile 2 (1.6 mmol, 1 eq) was dissolved in 1 L of toluene. Then 40 mg of iodine (0.16 mmol, 10 mol%) was added to the solution. The mixture was vigorously stirred for approximately 3 h under UV irradiation by a 150 W mercury lamp. After completion of the reaction (CCM analysis), toluene was evaporated. The crude product was purified by flash silica gel column chromatography using cyclohexane/EtOAc (90/10) as the eluent. Spectral data and physical characteristics are given below for each phenanthrene derivative.
2.3.1. 6-fluoro-3-methoxyphenanthrene-9-carbonitrile (P1)
White solid, 78%; m.p. = 225–227 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 4.06 (s, 3H, OCH3), 7.31 (dd, J1 = 2.4 Hz, J2 = 9 Hz, 1H), 7.46 (td, J1 = 2.4 Hz, J2 = 9 Hz, 1H), 7.84–7.87 (m, 2H), 8.14 (s, 1H), 8.18 (dd, J1 = 2.4 Hz, J2 = 10.8 Hz, 1H), 8.24 (dd, J1 = 5.7 Hz, J2 = 9 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ (ppm): 55.66 (OCH3), 104.33 (CH), 106.21 (C), 108.28 (d, JC‐F = 22.5 Hz, CH); 117.11 (d, JC‐F = 23.2 Hz, CH ), 118.06 (CN), 118.56 (CH); 124.7 (C), 126.01 (C), 128.42 (d, JC‐F = 9 Hz, CH), 131.18 (CH), 131.22 (C), 132.94 (C), 134.47 (CH), 160.40 (d, JC‐F = 246.7 Hz, CF), 161.02 (C–O); 19F NMR (282 MHz, CDCl3): δ (ppm): −111.33 (s, F); IR: ν (cm-1): 3036, 2983, 2216 (CN), 1521, 1503, 1476, 1454, 1421, 1381, 1363, 1273, 1230, 1215, 1181, 1139, 1100, 1020, 952, 898, 855, 837, 808, 722, 689, 639, 563, 552, 480, 427.
2.3.2. 3-bromo-6-fluorophenanthrene-9-carbonitrile (P2)
White solid, 97%; m.p. = 247–249 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.53–7.60 (m, 1H), 7.84 (s, 2H), 8.19 (s, 1H), 8.24 (dd, J1 = 2.4 Hz, J2 = 10.2 Hz, 1H), 8.31 (dd, J1 = 5.7 Hz, J2 = 9 Hz, 1H), 8.72 (s, 1H); 13C NMR (75 MHz, CDCl3): δ (ppm): 108.50 (d, JC‐F = 23.2, CH), 109.55 (C), 117.35 (CN), 117.80 (d, JC‐F = 23.8 Hz, CH), 122.52 (C), 124.73 (C), 126.12 (CH), 128.60 (d, JC‐F = 9 Hz, CH), 128.65 (C), 130.76 (C), 130.87 (CH), 131.69 (CH), 132.43 (C), 134.04 (CH); 160.86 (d, JC‐F = 246.7 Hz, CF); 19F NMR (282 MHz, CDCl3): δ (ppm): −109.50 (s, F); IR: ν (cm-1): 3076, 2919, 2850, 2215 (CN), 1621, 1581, 1517, 1488, 1424, 1406, 1324, 1266, 1203, 1179, 1145, 1063, 1022, 917, 903, 859, 818, 807, 715, 670, 635, 535, 488, 467, 425, 411.
2.3.3. 6-fluorophenanthrene-3,9-dicarbonitrile (P3)
White solid, 92%; m.p. = 245–247 °C; 1H NMR (300 MHz, DMSO-d6): δ (ppm): 7.82–7.84 (m, 1H), 8.13–8.29 (m, 3H), 8.72 (s, 1H), 8.92–8.94 (m, 1H), 9.49 (s, 1H); 13C NMR (75 MHz, DMSO-d6): δ (ppm): 109.90 (d, JC‐F = 21 Hz, CH), 110.74 (C), 112.43 (C), 118.40 (d, JC‐F = 22.5 Hz, CH), 118.44 (CN), 118.57 (CN), 125.26 (C), 128.23 (d, JC‐F = 8.2 Hz, CH), 129.54 (CH), 129.90 (CH), 130.81 (CH), 131.13 (C), 131.88 (C), 132.00 (C), 134.67 (CH), 161.17 (d, JC‐F = 249.7 Hz, CF); 19F NMR (282 MHz, CDCl3): δ (ppm): −109.14 (s, F); IR: ν (cm-1): 3071, 2919, 2227 (CN), 2218 (CN), 1620, 1600, 1524, 1504, 1451, 1418, 1392, 1363, 1333, 1275, 1216, 1180, 1139, 1112, 1022, 952, 904, 875, 857, 838, 813, 725, 691, 644, 567, 552, 534, 488, 424.
2.3.4. 3,6-difluorophenanthrene-9-carbonitrile (P4)
Yellow solid, 88%; m.p. = 217–219 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.44–7.55 (m, 2H), 7.92 (dd, J1 = 6 Hz, J2 = 8.7 Hz, 1H), 8.11–8.17 (m, 3H), 8.26 (dd, J1 = 5.7 Hz, J2 = 8.7 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ (ppm): 107.94 (d, JC‐F = 23.2 Hz, CH), 108.07 (d, JC‐F = 22.5 Hz, CH), 116.97 (CN), 117.00 (d, JC‐F = 23.2 Hz, CH), 117.27 (d, JC‐F = 23.5 Hz, CH), 125.24 (2C), 126.35 (2C), 128.08 (d, JC‐F = 8.2 Hz, CH), 131.42 (d, JC‐F = 9.7 Hz, CH), 133.55 (CH), 160.15 (d, JC‐F = 249 Hz, CF), 161.20 (d, JC‐F = 249.7 Hz, CF); 19F NMR (282 MHz, CDCl3): δ (ppm): −107.70 (s, F); −110.03 (s, F); IR: ν (cm-1): 3076, 2983, 2215 (CN), 1620, 1597, 1523, 1507, 1472, 1449, 1426, 1387, 1364, 1274, 1229, 1218, 1202, 1172, 1142, 1104, 1032, 1020, 967, 956, 902, 857, 849, 838, 815, 728, 693, 644, 606, 564, 553, 482, 463, 429.
2.3.5. 6-fluoro-2,3-dimethoxyphenanthrene-9-carbonitrile (P5)
White solid, 70%; m.p. = 237–239 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 4.08 (s, 3H, OCH3), 4.15 (s, 3H, OCH3), 7.24 (s, 1H), 7.44 (td, J1 = 1.2 Hz, J2 = 9 Hz, 1H, H7), 7.78 (s, 1H), 8.09 (s, 1H), 8.10 (dd, J1 = 1.2 Hz, J2 = 11.4 Hz, 1H, H5), 8.23 (dd, J1 = 6 Hz, J2 = 8.7 Hz, 1H, H8); 13C NMR (75 MHz, CDCl3): δ (ppm): 56.17 (OCH3), 56.21 (OCH3), 103.17 (CH), 106.71 (C), 107.60 (d, JC‐F = 22.5 Hz, CH), 108.63 (CH), 116.20 (d, JC‐F = 24 Hz, CH), 118.16 (CN), 125.17 (C), 125.37 (C), 126.51 (C), 128.45 (d, J = 9 Hz, CH), 130.96 (d, JC‐F = 9 Hz, C), 133.53 (CH), 150.49 (C–O), 151.87 (C–O), 160.35 (d, JC‐F = 246 Hz, CF); 19F NMR (282 MHz, CDCl3): −111.41 (s, F); IR: ν (cm-1): 3082, 3006, 2925, 2838, 2215 (CN), 1616, 1525, 1509, 1472, 1418, 1373, 1259, 1213, 1160, 1115, 1037, 1024, 995, 908, 899, 853, 836, 817, 807, 767, 758, 718, 646, 614, 586, 497, 478, 423, 413.
2.3.6. 6-fluoronaphto[2,1-b]thiophene-9-carbonitrile (P6)
White solid, 65%; m.p. = 224–226 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.36 (td, J1 = 2.4 Hz, J2 = 9 Hz, 1H, H6), 7.78 (d, J = 5.4 Hz, 1H, H2 or H3), 7.85 (d, J = 5.4 Hz, 1H, H3 or H2), 7.88 (dd, J1 = 2.4 Hz, J2 = 9.6 Hz, 1H, H4), 8.23 (s, 1H, H9), 8.25 (dd, J1 = 5.4 Hz, J2 = 9 Hz, 1H, H7); 13C NMR (75 MHz, CDCl3): δ (ppm): 106.12 (C), 108.17 (d, JC‐F = 22.5 Hz, CH), 116.41 (d, JC‐F = 24.7 Hz, CH), 117.43 (CN), 121.83 (CH), 125.49 (C), 126.82 (CH), 128.05 (d, JC‐F = 9 Hz, CH), 129.51 (d, JC‐F = 9.7 Hz, C), 130.61 (CH), 135.71 (C), 138.28 (C), 159.71 (d, JC‐F = 249 Hz, CF); 19F NMR (282 MHz, CDCl3): δ (ppm): −110.46 (s, F); IR: ν (cm-1): 3111, 3084, 2215 (CN), 1622, 1510, 1464, 1423, 1371, 1324, 1303, 1261, 1224, 1203, 1188, 1146, 1099, 1051, 968, 884, 858, 847, 811, 769, 728, 708, 667, 615, 564, 485, 459.
3. Results and discussion
In this work, the synthetic approach followed for the construction of tricyclic architectures was based on the oxidative photocyclization reaction of suitably functionalized olefins. First, p-fluorophenylacetonitrile (1) was reacted with different commercially available aromatic aldehydes according to a Knoevenagel reaction, producing α,β-unsaturated nitriles 2a–f with (Z)-configuration in 65–94% yields (Scheme 1 and Table 1). The resulting nitriles are well soluble in different organic solvents including toluene, acetone, dichloromethane, chloroform, and ethyl acetate.
Synthesis of α,β-unsaturated nitriles 2a–f.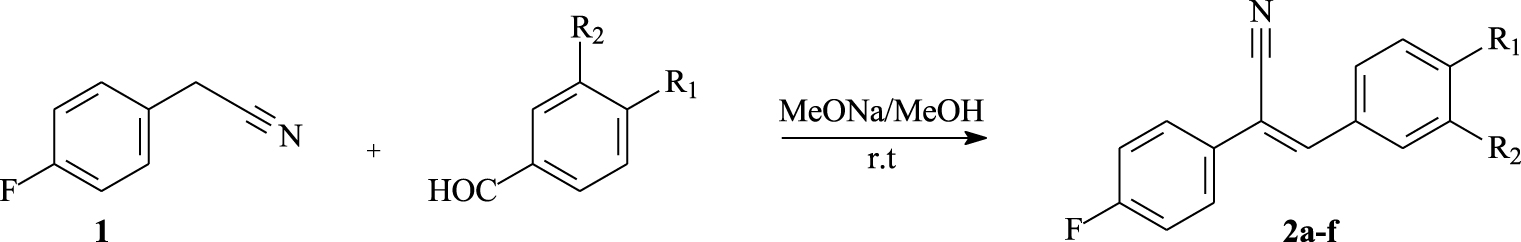
Chemical yields of α,β-unsaturated nitriles 2a–e
| Compound | R1 | R2 | Aspect | Yield (%)a |
|---|---|---|---|---|
| 2a | OMe | H | White solid | 85 |
| 2b | Br | H | Yellow solid | 80 |
| 2c | CN | H | White solid | 65 |
| 2d | F | H | White solid | 90 |
| 2e | OMe | OMe | Yellow solid | 94 |
a Isolated yields.
Chemical yields of phenanthrene derivatives P1–5
| Compound | R1 | R2 | Aspect | Yield (%)a |
|---|---|---|---|---|
| P1 | OMe | H | White solid | 78 |
| P2 | Br | H | White solid | 97 |
| P3 | CN | H | White solid | 92 |
| P4 | F | H | Yellow solid | 88 |
| P5 | OMe | OMe | White solid | 70 |
a Isolated yields.
p-Fluorophenylacetonitrile (1) was also reacted with 2-thiophene carbaldehyde under the same conditions. This provided 2f in 75% yield as a yellow solid (Scheme 2).
Synthesis of compound 2f.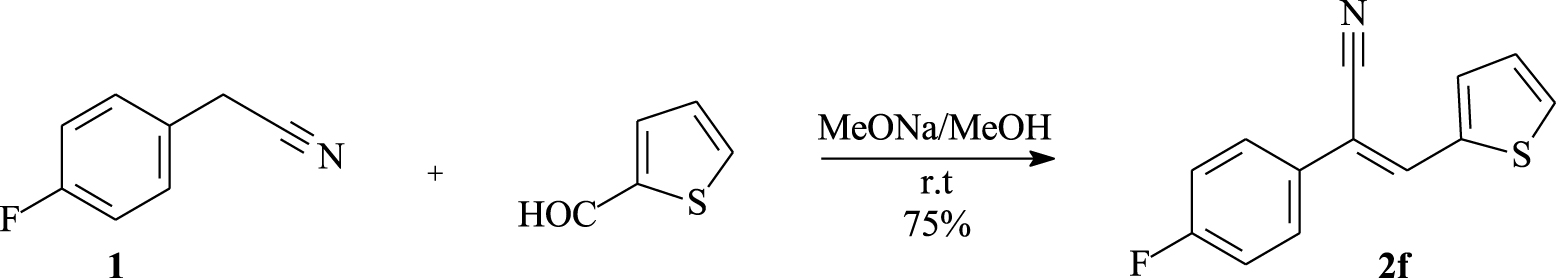
Photocyclization of α,β-unsaturated nitriles into phenanthrenes P1–5.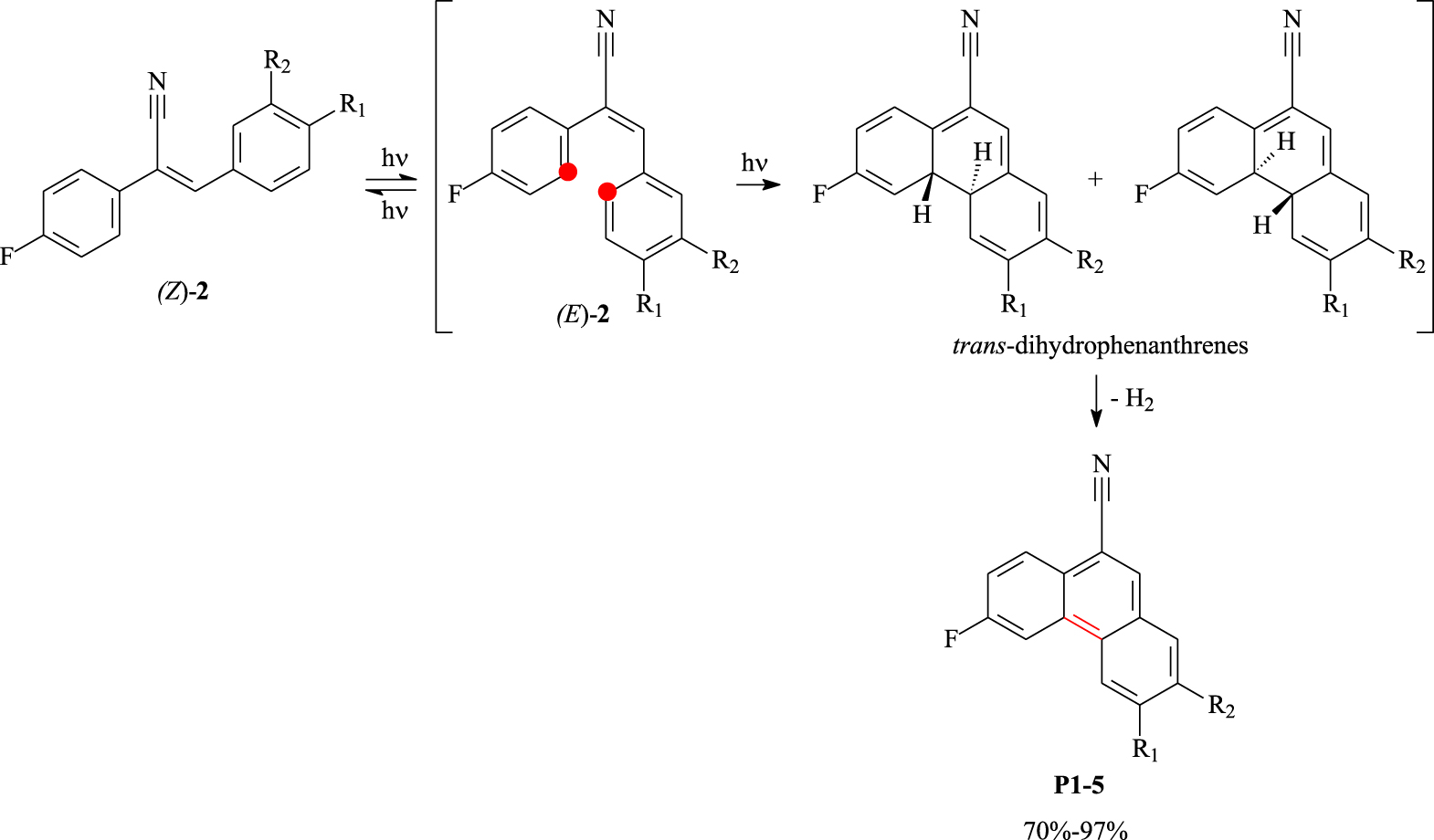
Fluorinated α,β-unsaturated nitriles 2a–f were then irradiated with a 150 W mercury lamp, through a Pyrex glass jacket, in toluene for approximately 3 h in the presence of a catalytic amount of iodine as an oxidizing agent. Unfortunately, this reaction conducted on a 500 mg scale per run provided only the desired fluorinated phenanthrene derivatives P1–5 in 70–97% yields (Scheme 3 and Table 2). It is worth noting that the photo-irradiation conditions are compatible with the different functionalities grafted on the target tricyclic compounds. No photodimer was observed during the photocyclization step. The whole photocyclodehydrogenation reaction proceeded via isomerization of (Z)-2 to (E)-2 followed by intramolecular electrocyclization leading to trans-dihydrophenanthrenes, which were oxidized to phenanthrene derivatives.
The phenanthrene derivatives were characterized by NMR spectroscopy. For example, the proton NMR spectrum of compound P5 (Figure 2) displayed three characteristic singlets at 7.24, 7.78, and 8.09 ppm, which were attributed to protons H1, H4, and H10. A triplet of doublets (J1 = 1.2 Hz, J2 = 9 Hz) was observed at δ = 7.44 ppm, which is typical of proton H7.
1H NMR spectrum (CDCl3, 300 MHz, 298 K) of compound P5.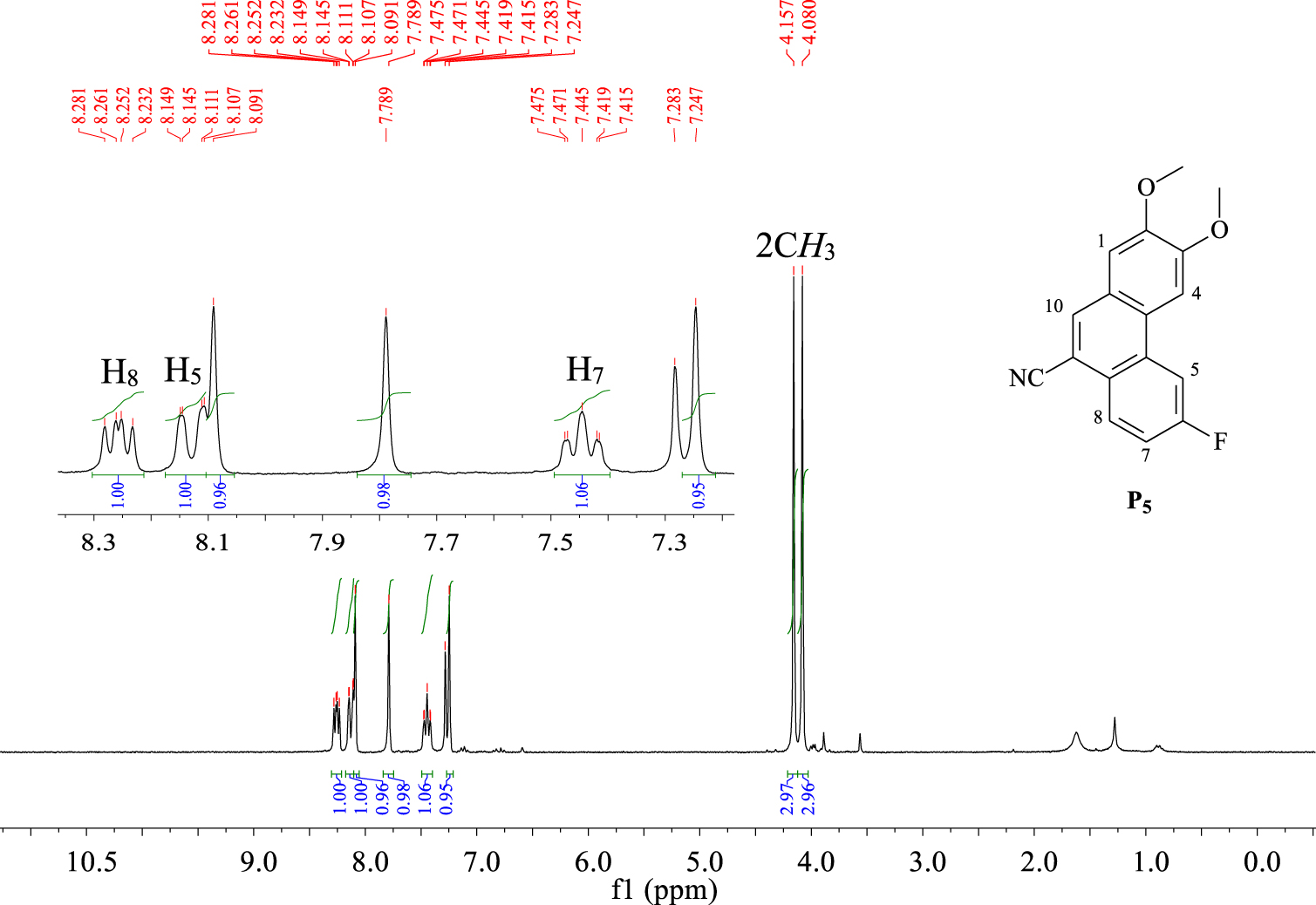
We investigated the 1H–1H COSY NMR spectrum of P5 (Figure 3) to provide more structural information. Two doublets of doublets at 8.10 ppm (J1 = 1.2 Hz, J2 = 11.4 Hz) and 8.23 ppm (J1 = 6 Hz, J2 = 8.7 Hz) are attributed to H5 and H8, respectively, which couple with H7.
1H–1H COSY NMR spectrum (CDCl3, 300 MHz, 298 K) of compound P5.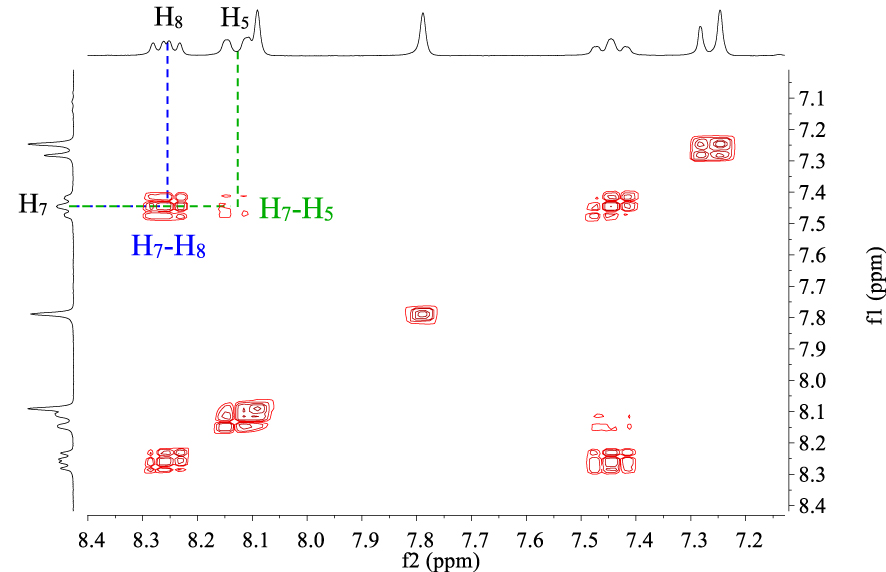
The photo-irradiation of the α,β-unsaturated nitrile 2f, conducted under the same conditions, provided 5-fluoronaphto[2,1-b]thiophene-8-carbonitrile P6 as a white solid in 65% yield (Scheme 4).
Synthesis of 5-fluoronaphto[2,1-b]thiophene-8-carbonitrile P6.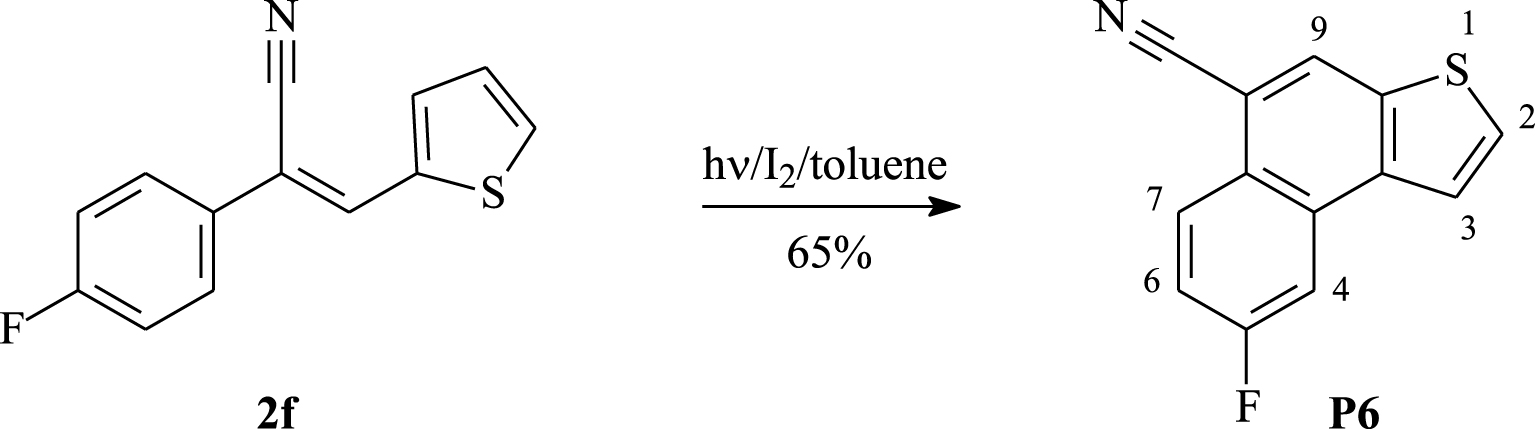
The proton NMR spectrum (Figure 4) of compound P6 shows a triplet of doublets at 7.36 ppm (J1 = 2.4 Hz, J2 = 9 Hz) attributed to proton H6. Two doublets at 7.78 ppm (J = 5.4 Hz) and 7.85 ppm (J = 5.4 Hz) relative to H2 and H3 and a singlet at 8.23 ppm, which is typical of proton H9, are observed.
1H NMR spectrum (CDCl3, 300 MHz, 298 K) of compound P6.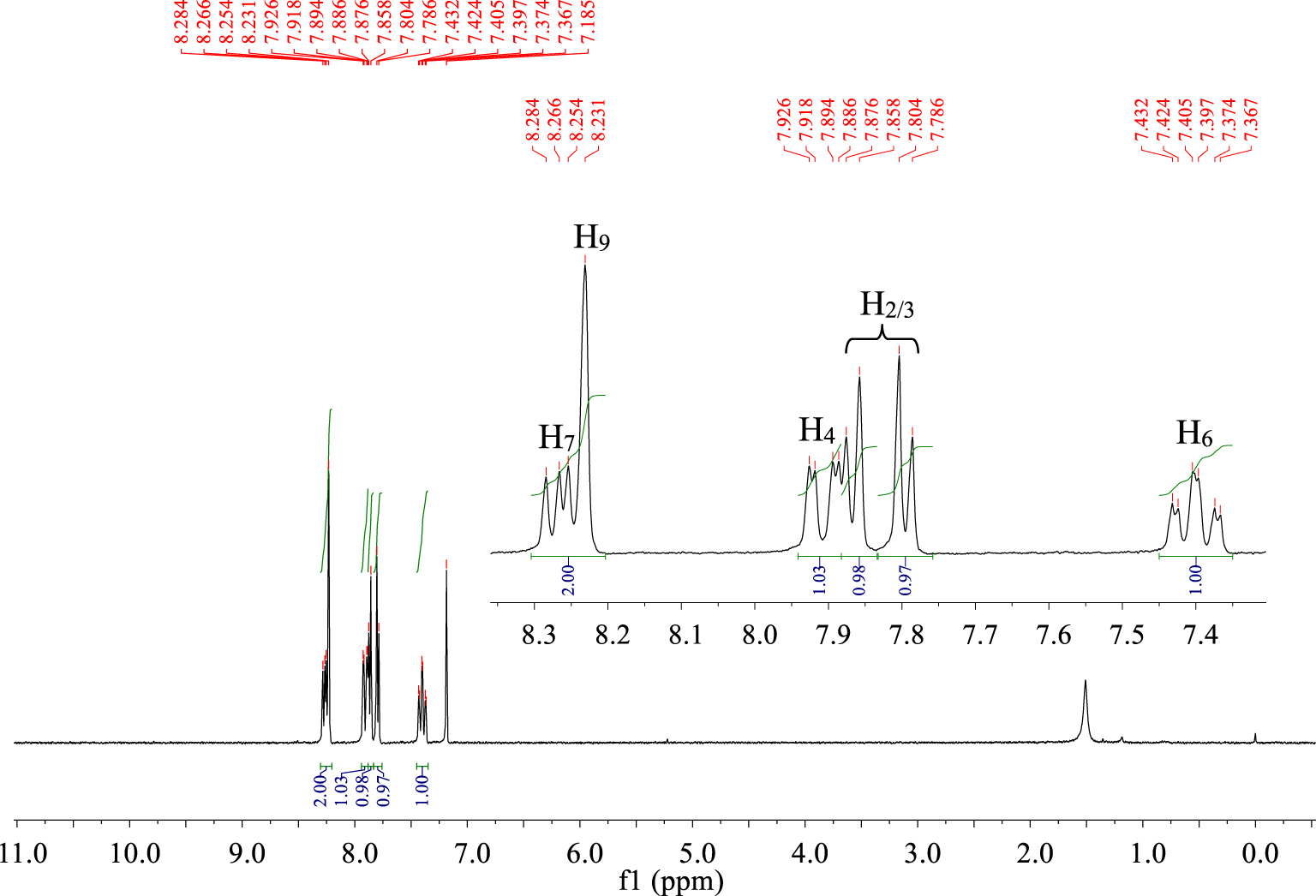
By using 1H-1H COSY NMR spectroscopy, it is possible to differentiate the peaks for protons H4 and H7. In Figure 5, the H4 signal is located at 7.88 ppm as a doublet of doublets (J1 = 2.4 Hz, J2 = 9.6 Hz), while the H7 signal is overlapped with the H9 proton signal and is found at 8.23 ppm (J1 = 5.4 Hz, J2 = 9 Hz).
1H-1H COSY NMR spectrum (CDCl3, 300 MHz, 298 K) of compound P6.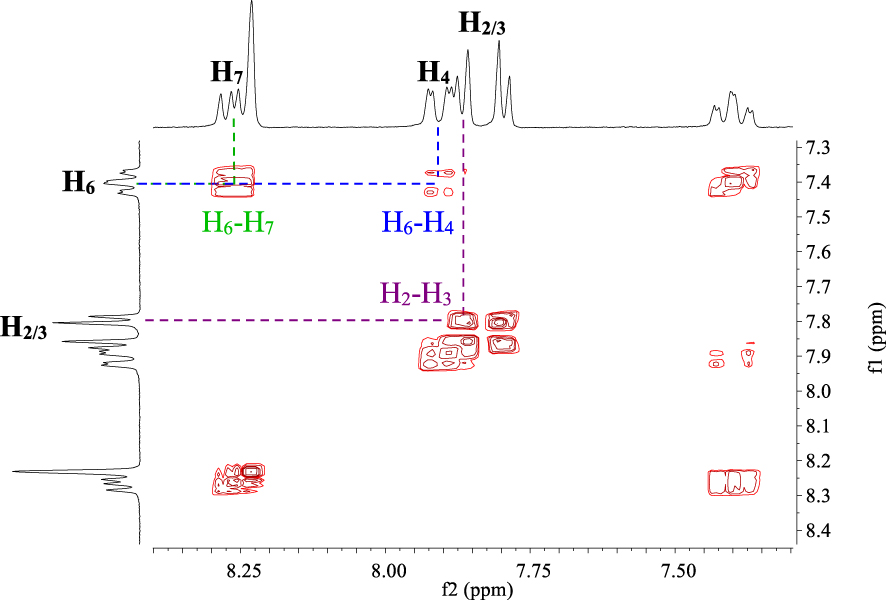
UV–Vis spectra of the synthesized α,β-unsaturated nitriles 2a–f were recorded in chloroform solutions (C ≈ 3 × 10−5 mol⋅L−1) at room temperature. As seen in Figure 6, the UV–Vis spectra of 2a–f display several absorption bands below 275 nm (256, 263, and 270 nm) with slightly different absorption coefficients (64000–82000 M−1⋅cm−1) as indicated in Table 3. They are accompanied by one broad absorption band in the low-energy region (Δ𝜆 > 65 nm). These absorption bands are characteristic of π–π∗ and n–π∗ transitions. By examining the low-energy region, it is clear that 2d shows the strongest absorption band at 315 nm (𝜀 = 3.89 × 104 M−1⋅cm−1), 2c displays a weaker band at 322 nm (𝜀 = 1.21 × 104 M−1⋅cm−1), and 2e exhibits the longest band at 349 nm (the molar extinction coefficient is more than twice as high as that for 2c). Moving from 2a to 2e, the addition of a methoxy group induces a bathochromic shift of 12 nm for the low-energy band (centered at 337 nm) with a slight decrease in the absorption intensity (Δ𝜀 ≈ 2400 M−1⋅cm−1). From 2d to 2b, replacing the fluorine atom by a bromine atom results in a significant decrease in the absorption intensity for the band located between 277 and 367 nm. It appears that the α,β-unsaturated nitriles with electron donor substituents (e.g., methoxy group) on the benzene ring (2a and 2e) or with thiophene nuclei (2f) shift the low-energy ( >300 nm) absorption maxima to higher wavelengths. The α,β-unsaturated nitriles with electron-withdrawing substituents (e.g. cyano, fluoro, and bromo groups) (2b, 2c, and 2d) shift the low-energy ( >300 nm) absorption maxima to lower wavelengths. The electron-donating substituents lead to a bathochromic shift of the absorption maxima at longer wavelengths between 377 and 400 nm. Electron-withdrawing substituents lead to hypsochromic shifts. The optical band gap energy (Eg‐op) of each α,β-unsaturated nitrile has been estimated according to the empirical formula
| (1) |
Electronic UV–Vis absorption spectra of α,β-unsaturated nitriles 2a–f in chloroform solutions (C ≈ 3 × 10−5 M).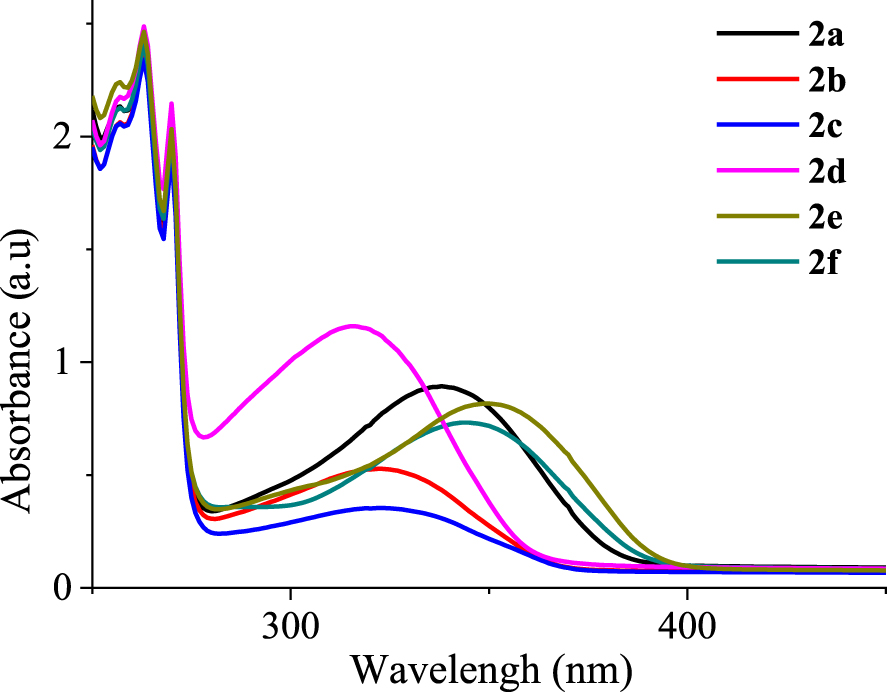
Electronic UV–Vis absorption properties of the α,β-unsaturated nitriles 2a–f in chloroform solutions
| Compound | 𝜆abs a/nm (𝜀/104 M−1⋅cm−1)b | 𝜆onset (nm) | Eg‐op c (eV) |
|---|---|---|---|
| 2a | 256 (7.11), 263 (8.02), 270 (6.58), 337 (2.99) | 395 | 3.13 |
| 2b | 256 (6.88), 263 (7.87), 270 (6.46), 321 (1.71) | 377 | 3.28 |
| 2c | 256 (6.88), 263 (7.84), 270 (6.46), 322 (1.21) | 377 | 3.28 |
| 2d | 256 (7.26), 263 (8.26), 270 (7.11), 315 (3.89) | 374 | 3.31 |
| 2e | 256 (7.45), 263 (8.26), 270 (6.77), 349 (2.75) | 398 | 3.11 |
| 2f | 256 (7.11), 263 (8.02), 270 (6.58), 343 (2.46) | 395 | 3.13 |
a Absorption maxima measured in CHCl3 solution (3 × 10−5 mol⋅L−1) at room temperature.
b Calculated using the Beer–Lambert law (𝜀 = A∕lC), where A = absorbance at 𝜆abs, l = path length (1 cm), and C = concentration in mol⋅L−1.
c The optical band gap (Eg‐op) was estimated from the onset point of the absorption spectrum: Eg‐op = 1240∕𝜆onset.
The photocyclization of the α,β-unsaturated nitriles into the corresponding phenanthrene derivatives leads to significant modifications in their absorption spectra by the appearance of new bands between 275 and 400 nm. The photophysical properties of compounds P1–6 are then examined in chloroform solutions (C ≈ 3 × 10−5 mol⋅L−1) at room temperature. These are displayed in Figure 7. The absorption spectra of these compounds show similar features including three intense bands located between 250 and 275 nm (𝜀 ∼ 69200–93900 M−1⋅cm−1) and a shoulder peak around 275 nm followed by several less intense bands up to 380 nm (Table 4). The weaker absorption bands are observed in the low-energy region with intensities in the range 5100–18000 M−1⋅cm−1. Moving from P1 to P5, the presence of a second methoxy group induces a bathochromic shift of approximately 2–5 nm for the absorption maxima at 275–375 nm with a significant decrease in intensities for the bands at 316 and 327 nm in the range 14300–16100 M−1⋅cm−1. Compared to P4, the bromine atom in P2 induces a bathochromic shift of approximately 3–8 nm for the bands at 299, 311, 338, and 354 nm. The absorption bands of P2 below 316 nm are less intense than those of P4, but above 316 nm, they become slightly more intense. The optical band gap energy (Eg‐op) of each α,β-unsaturated nitrile is calculated, and the results are summarized in Table 4.
Electronic UV–Vis absorption spectra of phenanthrene derivatives P1–6 in chloroform solutions (C ≈ 3 × 10−5 M).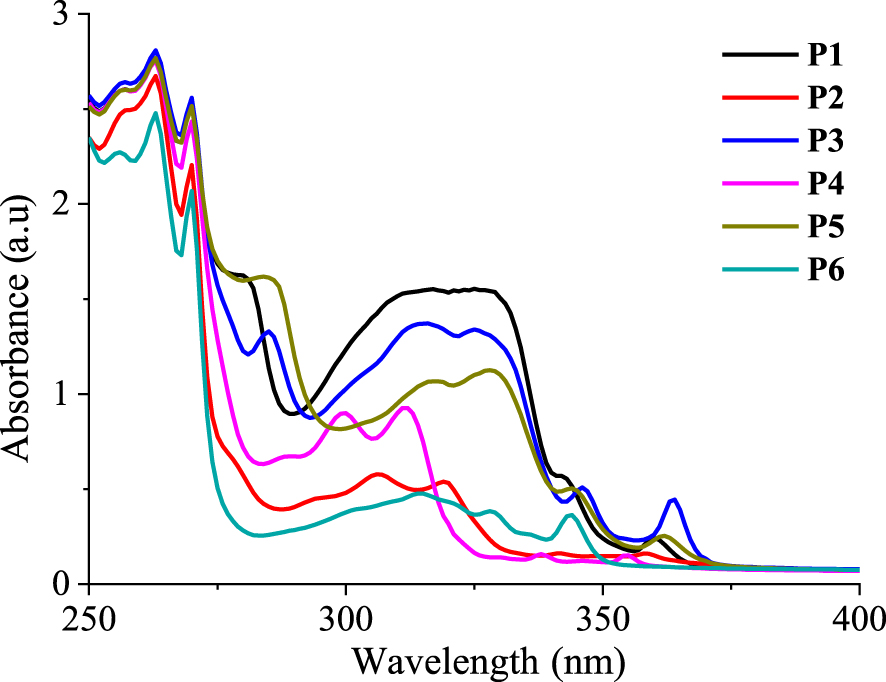
Electronic UV–Vis absorption properties of phenanthrene derivatives P1–6 in chloroform solutions
| Compound | 𝜆abs a/nm (𝜀/104 M−1⋅cm−1)b | 𝜆onset (nm) | Eg‐op d (eV) |
|---|---|---|---|
| P1 | 256 (8.79), 263 (9.39), 270 (8.55), 280 (5.44), 315 (5.19), 326 (5.19), 342 (1.8), 360c (0.80) | 371 | 3.34 |
| P2 | 257 (8.34), 263 (8.97), 270 (7.35), 306 (1.96), 319 (1.82), 341 (0.56), 358c (0.54) | 378 | 3.28 |
| P3 | 256 (8.79), 263 (9.39), 270 (8.55), 285 (4.46), 315 (4.61), 325 (4.51), 345 (1.72), 363c (1.48) | 378 | 3.28 |
| P4 | 256 (8.66), 263 (9.19), 270 (8.07), 288 (2.27), 299 (3.02), 311 (3.09), 338, (0.53), 353c (0.51) | 358 | 3.46 |
| P5 | 256 (8.68), 263 (9.24), 269 (8.37), 284 (5.42), 317 (3.58), 328 (3.76), 344 (1.71), 362c (0.86) | 378 | 3.28 |
| P6 | 256 (7.59), 263 (8.27), 270 (6.92), 301 (1.31), 314 (1.58), 328 (1.28), 343c (1.21) | 355 | 3.49 |
a Absorption maxima measured in chloroform (C ≈ 3 × 10−5 mol⋅L−1) at room temperature.
b Calculated using the Beer–Lambert law (𝜀 = A∕lC), where A = absorbance at 𝜆abs, l = path length (1 cm), and C = concentration in mol⋅L−1.
c First absorption maxima (𝜆1abs).
d The optical band gap (Eg‐op) was estimated from the onset point of the absorption spectrum: Eg‐op = 1240∕𝜆onset.
Photoluminescence properties of phenanthrene derivatives P1–6 in chloroform. Stokes shifts are calculated in wavelength and wavenumber units
| Compound | Photoluminescence | Stokes shift | ||
|---|---|---|---|---|
| 𝜆ems a (nm) | FWHMd (nm) | –𝜆1abs (nm) | Δ𝜈 (cm−1) | |
| P1 | 361, 379c, 399, 417b | 49 | 19 | 1392 |
| P2 | 359, 376c, 397, 415b | 30 | 18 | 1337 |
| P3 | 437c, 450b | 31 | 74 | 4664 |
| P4 | 353c, 369, 375, 392, 415 | 47 | 0 | 0 |
| P5 | 366c, 383, 400b | 36 | 5 | 378 |
| P6 | 348, 362c, 381, 398b | 25 | 19 | 1530 |
a Emission measured in chloroform (C ≈ 10−5 mol⋅L−1) at room temperature; fluorescence excitation at 350 nm.
b Shoulder peak.
c Maximum emission ().
d Spectrum full width at half maximum.
The PL spectra of phenanthrene derivatives P1–6 are recorded in chloroform solutions (C ∼ 10−5 mol⋅L−1) at room temperature (Figure 8). Compounds P1–5 exhibit structured emissions displaying several bands at different intensities (Table 5). The difluorinated compound P4 shows an maximum emission at 353 nm followed by three other bands (369, 375, and 392 nm) that are less intense and a shoulder peak at 415 nm. The phenanthrene-like derivative P6 exhibits an emission with three bands at 348, 362, and 381 nm and one shoulder peak at 398 nm. The maximum emission of P6 (𝜆ems = 362 nm) is shifted to the highest wavelengths with 7 nm, in comparison to that of P4 (𝜆ems = 353 nm). The emission spectrum of P2 shows the same signature as that of P4, including three emission bands (359, 376, and 397 nm) and a shoulder peak (415 nm) with enhanced intensities and are shifted to the highest wavelengths with 11–14 nm. Unlike the previous compounds, P5 shows two emission bands at 366 and 383 nm followed by a shoulder peak located at 400 nm, the first of which is the most intense. P1 exhibits two emission bands at 361 and 399 nm with almost the same intensities and a shoulder peak at 417 nm. Finally, the emission spectrum of compound P3 is totally different. It is shifted more to the highest wavelengths (red-shifted) than the others since it displays an emission at 437 nm and a shoulder peak at 450 nm. The destructured profile as well as the bathochromic shift of this emission illustrates the intense character of the charge transfer within the molecule P3 in the excited state.
Normalized emission spectra (𝜆exc = 350 nm) of phenanthrene derivatives P1–6 measured in chloroform solutions (C ∼ 10−5 mol⋅L−1).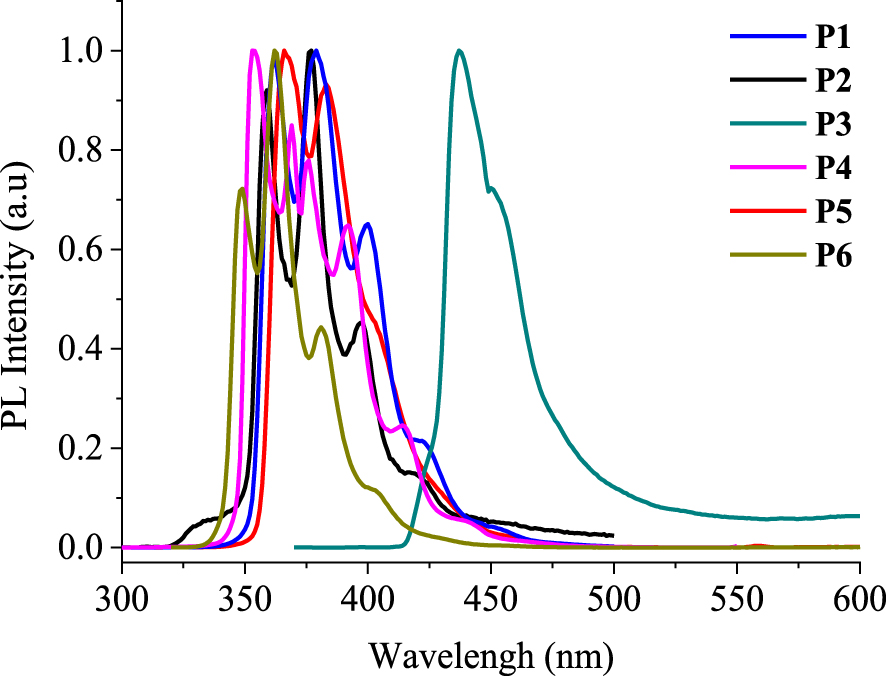
Cyclic voltammograms of P1–6 registered in 0.1 M nBu4NPF6/CH2Cl2 at a scan rate of 50 mV/s.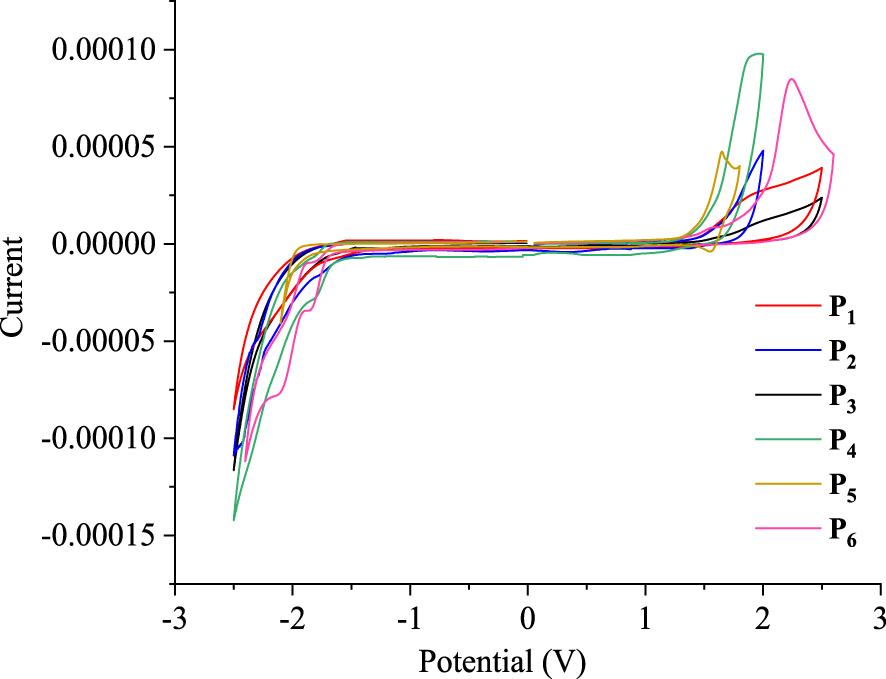
The electrochemical behaviors of the phenanthrene derivatives P1–6 are evaluated using cyclic voltammetry (CV) in dichloromethane with nBu4NPF6 (0.1 M) as the supporting electrolyte versus the SCE. As shown in Figure 9, the cyclic voltammograms, recorded at a scanning rate of 50 mV/s, indicate that compounds P1–6 exhibit irreversible anodic and cathodic peaks. Analysis of responses in CV allowed the determination of the oxidation potentials, which were found to be 1.42, 1.48, 1.46, 1.55, 1.28, and 1.64 V, versus the SCE. In addition, the reduction potentials were −1.67, −1.60, −1.59, −1.63, −1.75, and −1.67 V for P1–6, respectively.
Electrochemical onset potentials and electronic energy levels of P1–6
| Compound | Vonset‐ox (V) | Vonset‐red (V) | EHOMO (eV) | ELUMO (eV) | Eg‐el (eV) |
|---|---|---|---|---|---|
| P1 | 1.42 | − 1.67 | − 5.72 | − 2.63 | 3.09 |
| P2 | 1.48 | − 1.60 | − 5.78 | − 2.70 | 3.08 |
| P3 | 1.46 | − 1.59 | − 5.76 | − 2.71 | 3.05 |
| P4 | 1.55 | − 1.63 | − 5.85 | − 2.67 | 3.18 |
| P5 | 1.28 | − 1.75 | − 5.58 | − 2.55 | 3.03 |
| P6 | 1.64 | − 1.67 | − 5.94 | − 2.63 | 3.31 |
An empirical method [26, 27] was then utilized to examine the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels as well as the electrochemical band gap (Eg‐el) as follows:
- EHOMO (IP, ionization potential) = −(Vonset‐ox − VFOC + 4.8)
- ELUMO (EA, electron affinity) = −(Vonset‐red − VFOC + 4.8)
- Eg‐el = (ELUMO − EHOMO) eV.
Here the value 4.8 represents the energy level of the ferrocene/ferrocenium couple, VFOC corresponds to the ferrocene half-wave potential (0.50 V), Vonset‐ox is the oxidation onset, and Vonset‐red is the material reduction onset, all measured versus Ag/AgCl. As shown in Table 6, the calculated electrochemical band gap (Eg‐el) varies from 3.03 to 3.31 eV.
Comparison of the HOMO–LUMO energy levels of P1–6 and those of BCP, indium tin oxide (ITO), and aluminum.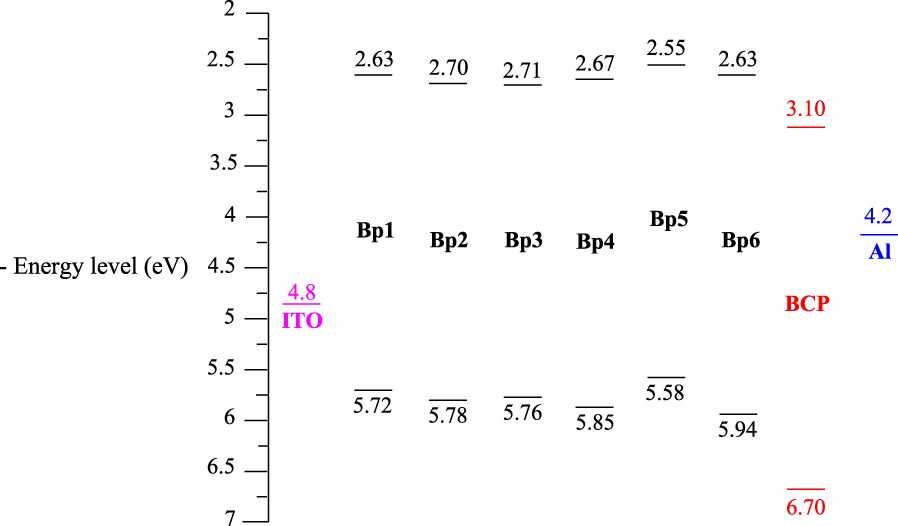
The difference between the electrochemical and optical band gaps is attributed to the interface barrier between the electrode and each phenanthrene derivative [28, 29] and the exciton binding energy. Interestingly, the energy band gap values of the tricyclic units P1–6 appear to be less than the electrochemical band gap (Eg‐el = 3.46 eV) of a benzo[ghi]perylene derivative bearing two electron-withdrawing methoxy carbonyl groups, which has been elaborated and used recently for the construction of field-effect transistors [30].
The LUMO levels estimated for systems P1–6 are as good as that of bathocuproine (BCP), which is extensively used as an electron-injection, hole-blocking layer in OLEDs [31]. It prohibits excitons diffusing toward the Al electrode where they would otherwise be quenched. However, the quick crystallization of BCP reduces the device performance [32, 33]. For this reason, it is better to replace BCP by another material in such devices. In fact, P1–6 might be suitable candidates to play the role of BCP by serving as hole-transport as well as electron-transport layers (Figure 10).
4. Conclusion
In this work, we prepared new functional fluorophenanthrene derivatives through a short synthetic approach under mild conditions by using commercially available and very low cost starting materials. The absorption and the PL properties of the target phenanthrenes were experimentally evaluated in solutions, and significant results were noted. Phenanthrenes P1, 2, and 4-6 reveal a blue emission. P3 shows a red-shifted emission due to an impressive intramolecular charge transfer. The electrochemical behavior of these tricyclic systems was also evaluated, demonstrating a notable charge transfer interaction owing to their π-conjugated electronic system. These properties suggest the application of these compounds as candidates for OLED-like and electroluminescent devices.
Acknowledgment
The authors extend their appreciation to the deanship of scientific research at University of Tabuk for funding this work through research group number RGP S-1440-0315.