



 CC-BY 4.0
CC-BY 4.0
1. Introduction
The importance of axially chiral biaryl compounds is rapidly expanding. The main reason is their presence in numerous biologically active natural products and therapeutic agents. They are also the privileged backbone of ligands and catalysts in asymmetric catalysis. Regarding the importance of these scaffolds, numerous strategies have been reported and among them atropo-enantioselective metallo- or organo-catalyzed cross-coupling reactions have been successfully employed to efficiently synthesize a large panel of these scaffolds (for recent reviews on the synthesis of axially chiral biaryls see [1, 2, 3, 4, 5]). Recently asymmetric C–H activation has proved to be efficient to control the chirality and more particularly to synthesize axially chiral biaryls [6, 7].
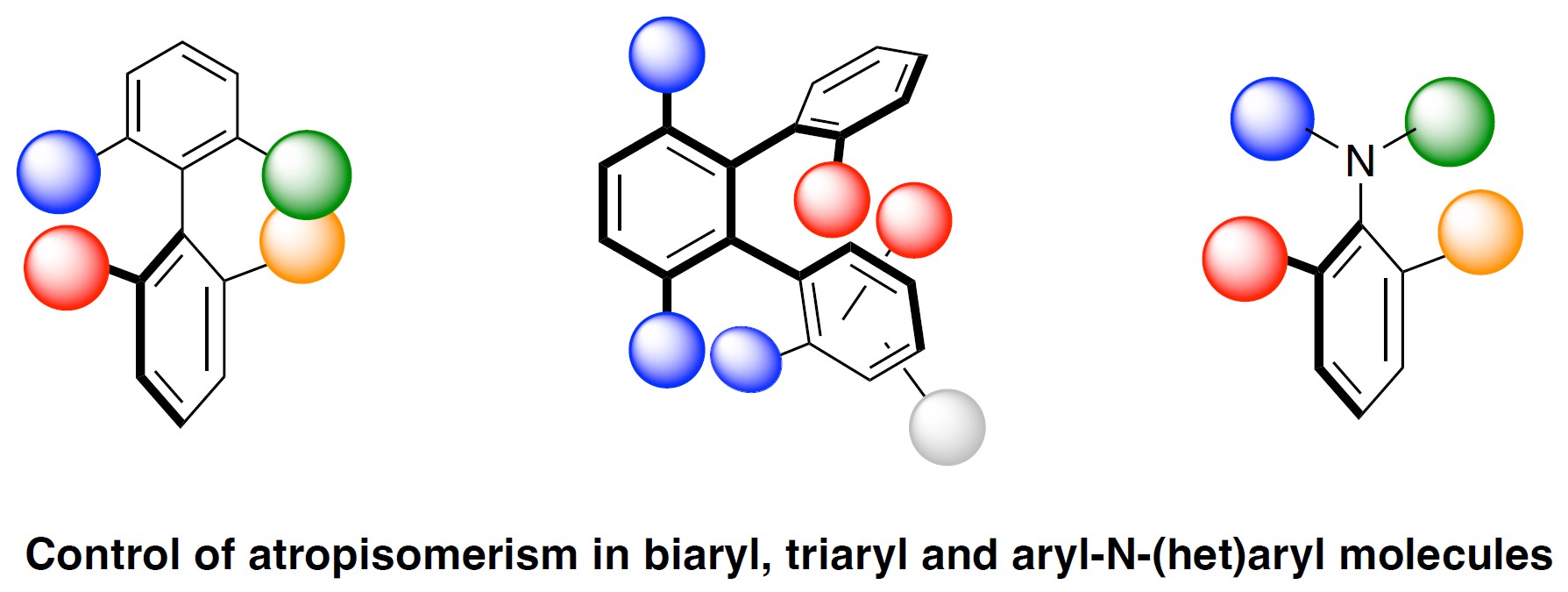
Over the past decade our laboratory has embarked on a research program dedicated to the control of atropoisomerism and we targeted at first differently substituted biaryl and triaryl skeletons bearing one or two atropisomeric axis. Our idea was to use a Pd-catalyzed atropo-diastereoselective C–H functionalization process using as chiral auxiliary an enantiopure sulfoxide, fixed on a configurationally unstable biaryl. Secondly, we turned our attention to the control of a C–N atropisomeric axis in N–Ar units via a Cu-catalyzed atroposelective aryl amination using hypervalent iodines as coupling partners (Figure 1).
Csp2-H activation: Axially chiral biaryl and triaryl compounds/Control of C–N atropisomeric axis via Ullmann-type C–N coupling reaction.
Capitalizing on our experience in total synthesis of biologically active compounds, we applied our atroposelective methods towards the synthesis of natural products as well as a new family of terphenyl ligands.
2. Results and discussion
Our first successful contribution to control atropoisomerism through a C–H activation-type protocol concerns a diastereoselective approach by using a chiral Directing Group (DG*). The chiral DG is utilized in stoechiometric amount and several criteria need to be fulfilled: the chiral DG must be (1) easy to install and not expensive (2) traceless or highly recyclable. The advantage compared to the enantioselective approach is the obtention of separable atropo-diastereomers. In addition the stereogenic center is in close proximity to both metal and substrate insuring an efficient chiral induction. We chose a sulfinyl moiety as chiral DG because we had already an extensive experience in the use of sulfoxides as chiral auxiliary (for the use of sulfoxides in Suzuki coupling reactions [8, 9, 10]). Moreover chiral sulfoxides are easily accessible on a large scale and the most important is its traceless character [11]. Sulfoxyde motif can be easily transformed via sulfoxide lithium exchange followed by electrophilic trapping with a myriad of electrophiles. Our initial hypothesis envisioned to start from a preformed biaryl, bearing the sulfinyl group in ortho position, and to introduce a substituent at the ortho position to the Ar–Ar′ linkage through a C–H functionalization process. One atropo-diastereomeric metallacycle was expected to be favored and the subsequent functionalization should thus occur in a stereoretentive manner, keeping the already fixed chirality. The removal or modification of the chiral sulfinyl group would allow the modular synthesis of axially chiral biaryls (Figure 2).
Axially chiral C–H functionalization of a biaryl moiety.
Thus, the hypothesis was validated by designing a Fujiwara–Moritani reaction with methyl acrylates or styrenes. We were delighted to obtain the corresponding coupling products in high yields and high atropo-diastereoselectivities [12, 13, 14]. Remarkably, the reaction is very tolerant towards many functional groups. Progressively, similar C–H acetoxylation in the presence of AcOH as well as the C–H iodination in the presence of N-iodosuccinimide (NIS) were developed, delivering the atropostable biaryls in excellent yields and stereoselectivities (Scheme 1) [15].
Atropodiastereoselective oxidative Heck, acetoxylation and iodination.
To rationalize these experimental results, a phenomenon of dynamic kinetic asymmetric transformation has been proposed and corroborated by DFT calculations. It was thus shown that the rotational barrier between the two substrates S1 and S2 is much higher than between the two palladacycles C1 and C2 (a difference of 10 kcal/mol) (Figure 3). Accordingly, the dynamic character of the overall transformation arises from a possible rotation around the chiral axis once the metallacyclic intermediates are formed whereas the substrate remains atropostable under the reaction conditions.
Conventional DFT studies in gazeous phase without solvatation.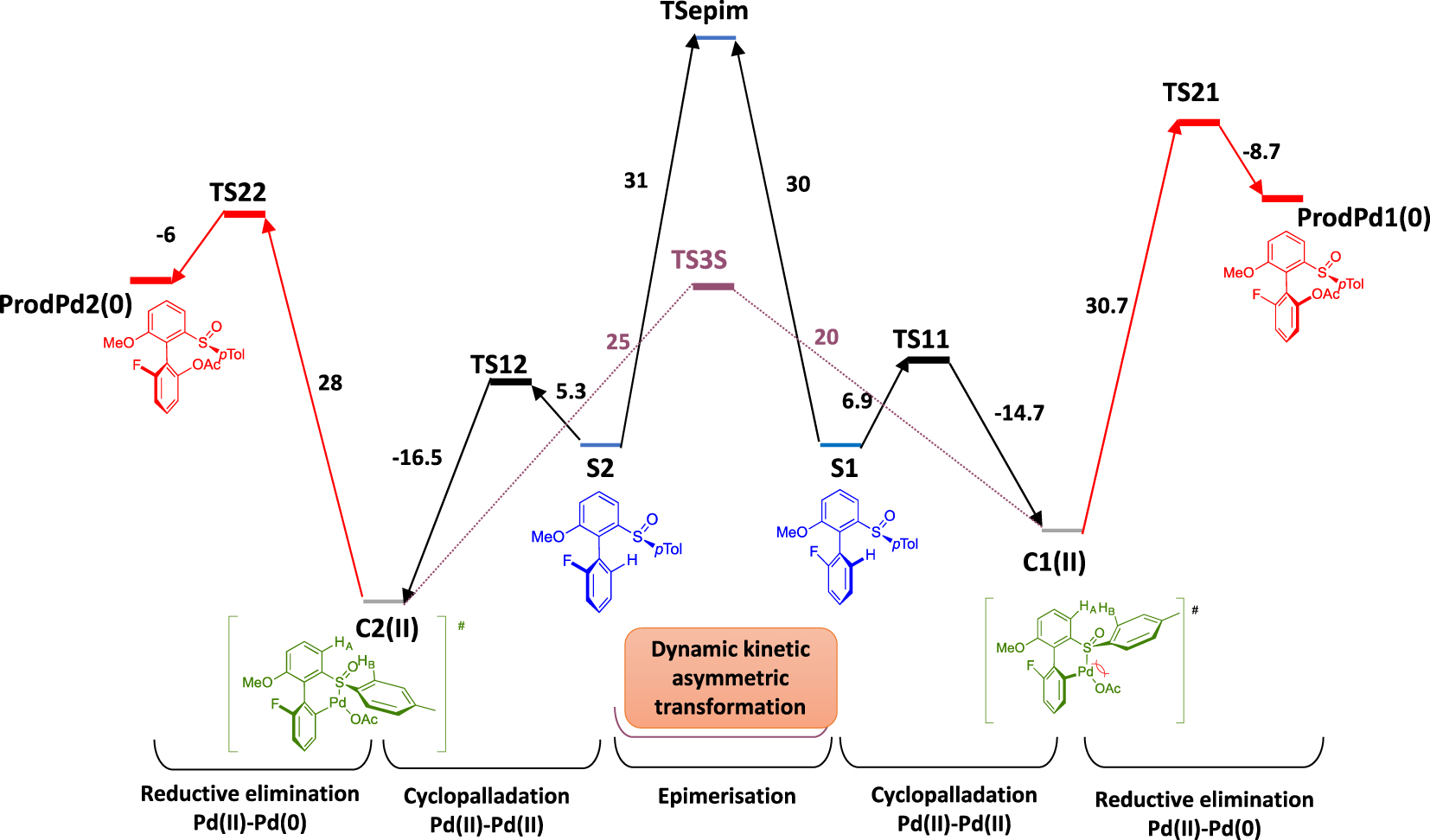
To demonstrate the broad synthetic utility of this atroposelective method, sulfinyl/Li exchange was performed at −78 °C to avoid the epimerisation of the atropisomeric axis and quenching with different electrophiles allows the introduction of different functional groups with a total retention of the axial chirality (Scheme 2).
Traceless character of the sulfinyl group.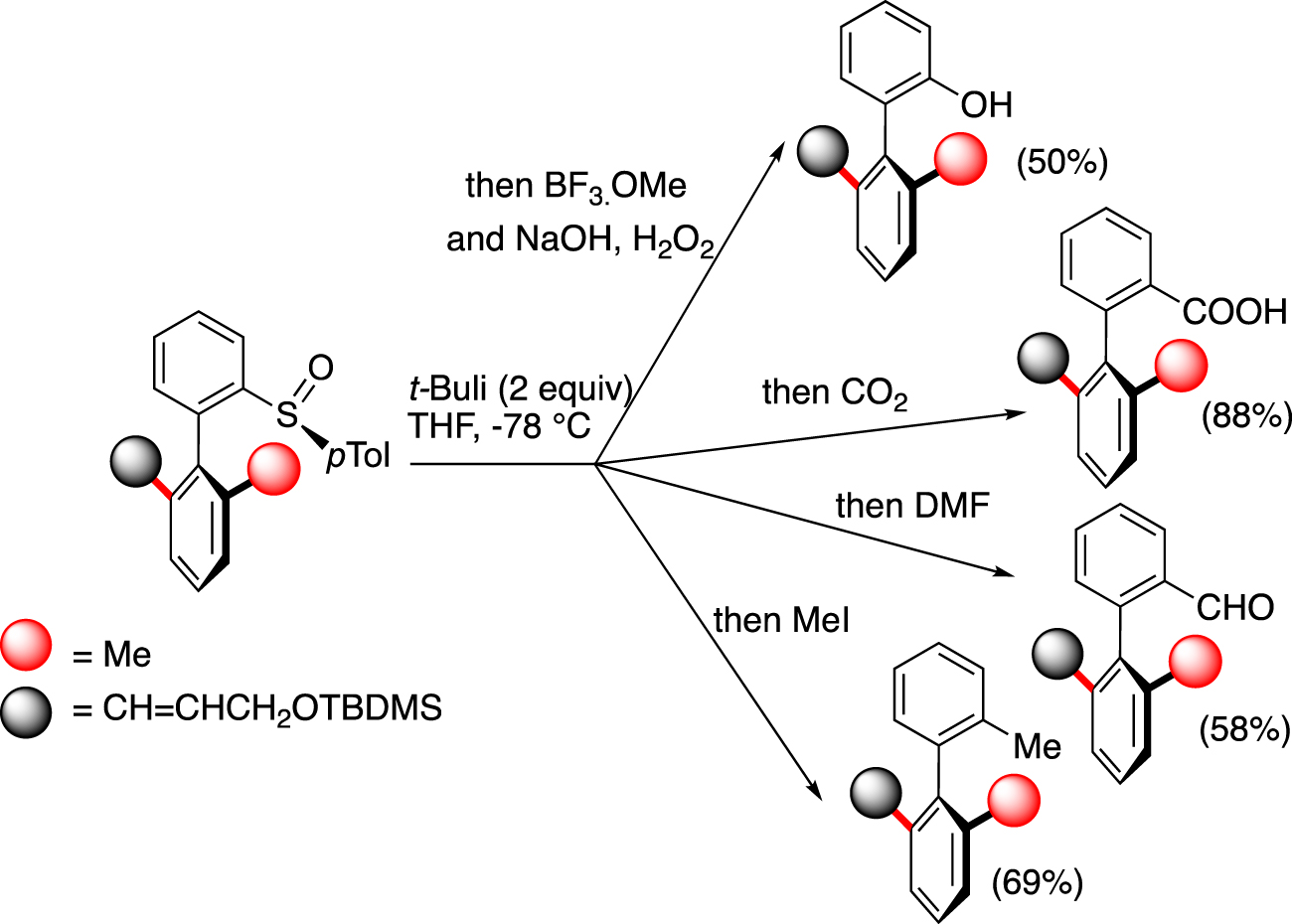
We applied this strategy to the formal synthesis of the biaryl subunit of (-)steganone that has been often used to illustrate the efficiency of a new atroposelective method. Starting from bromobenzodioxole we performed the most expedient synthesis in 10 steps with a 42.3% overall yield and with a total atropostereoselectivity (Scheme 3) [16].
Formal synthesis of the biaryl subunit of (-)steganone.
Moreover, in collaboration with Speicher et al., we applied our method towards the first total synthesis of atropopure isoriccardin C. An atropostereoselective macrocyclization through an oxidative Heck type C–H activation process allowed complete control of the atroposelectivity (de > 98%) and the traceless character of the sulfinyl moiety in ortho position of the biaryl enabled the introduction of an OH group (Scheme 4) [17].
Total synthesis of atropopure isoriccardin C.
Following this work, we were interested in the synthesis of doubly atropoisomeric, ortho-orientated dissymmetrical terphenyls that are truly appealing for the design of chiral ligands. Following this idea we explored the C–H arylation of a biaryl bearing in the ortho position the chiral sulfinyl DG with ortho-substituted aryl iodides. After an extensive optimization of the reaction conditions, we succeeded in obtaining the terphenyl in moderate to good yields but with excellent atropo-diastereoselectivity. What is remarkable in this reaction is the control of the two contiguous chiral axes in one step. An X-ray crystal structure highlighted a unique architecture of these scaffolds, featuring a cavity comparable to the one of a seashell well designed for the coordination with metal in organometallic catalysis (Scheme 5) [18].
Synthesis of terphenyls with two chiral axes.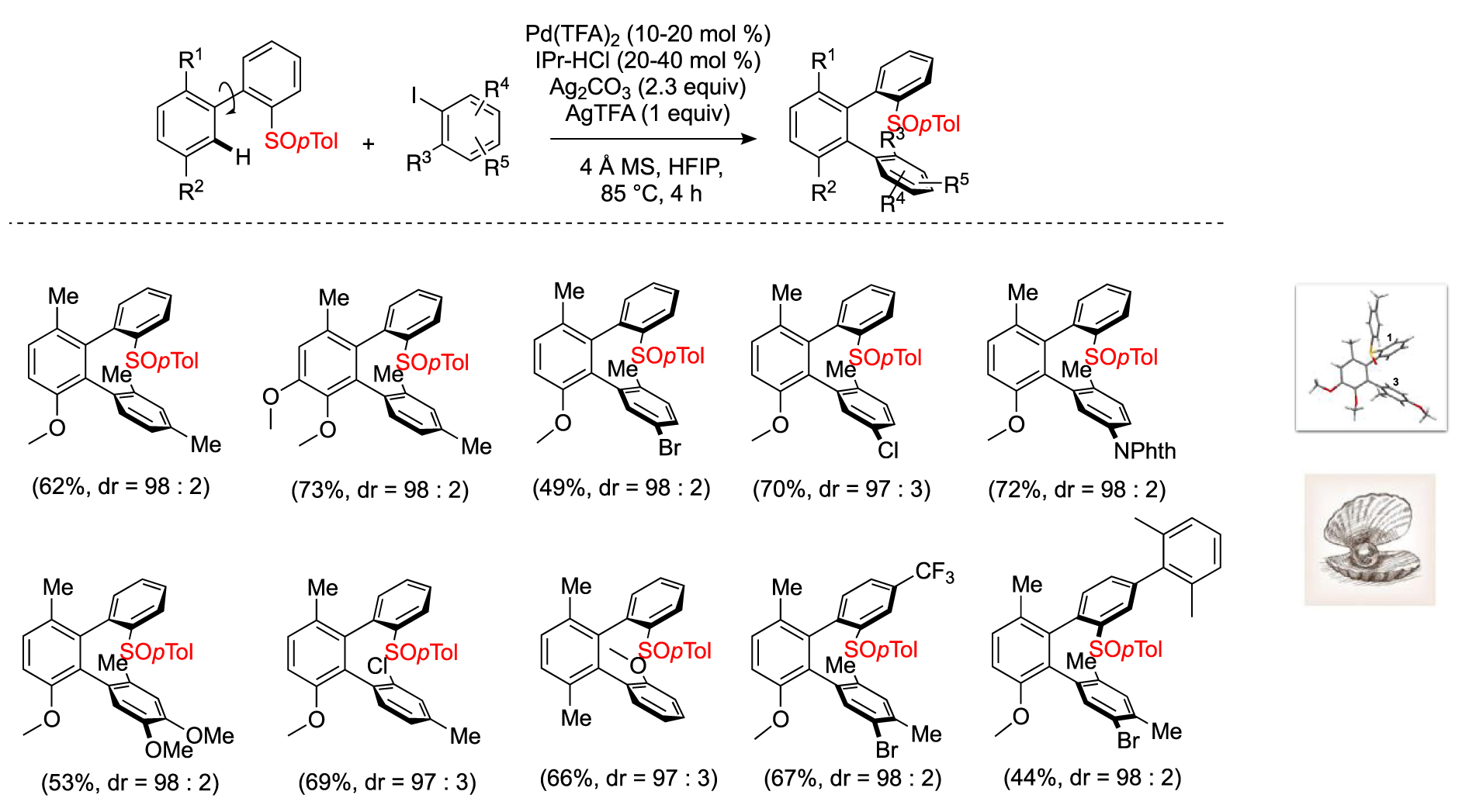
Of particular interest is the atropoenantiopure terphenyl scaffold A, bearing the sulfinyl group and the bromine which can be obtained in good yield (49%), with total atroposelectivity (>98:2) and on a gram scale in one day, this compound can be transformed by simple chemoselective lithium exchange and then quenched by various electrophiles to give access to a large variety of ligands. On the one hand, we performed the double exchange with Li base giving access to diphosphines BiaxPhos (Scheme 6) and, on the other hand, we developed an elegant two-step sequential chemoselective functionalization to install different coordinating groups like two different phosphines BiaxPhos* (Scheme 7) [19].
Synthesis of diphosphines BiaxPhos bearing the same phosphorus moiety.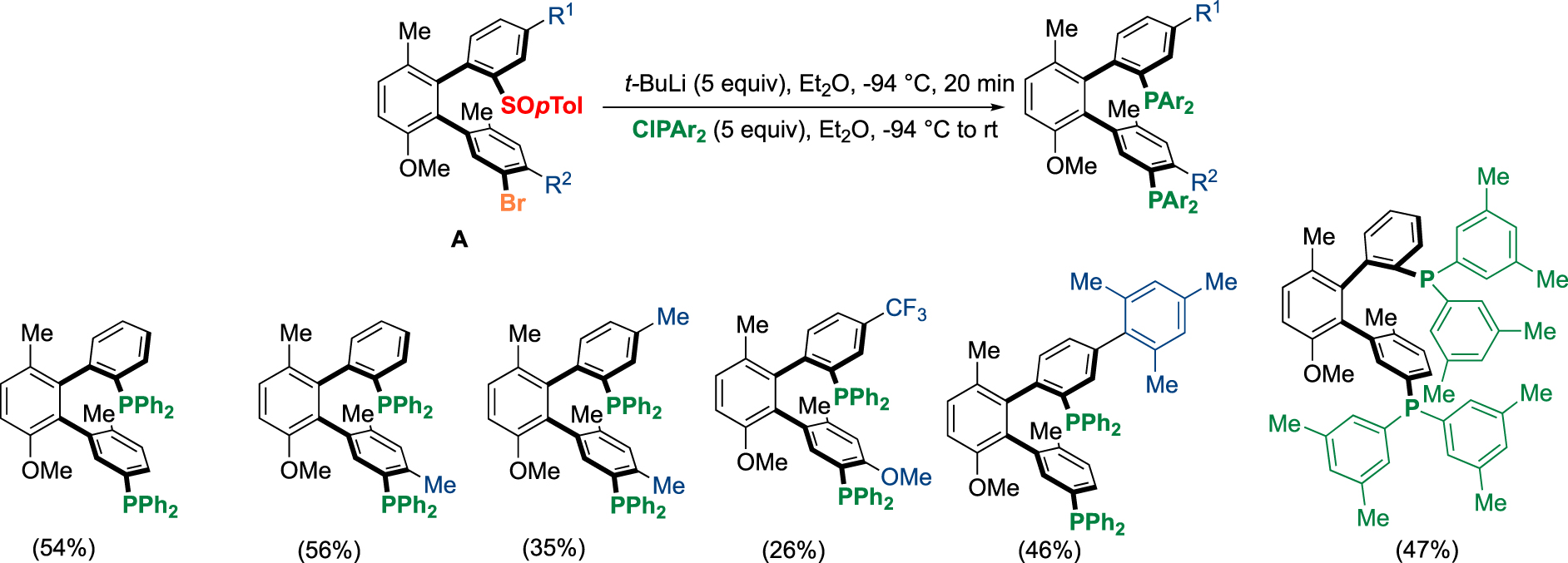
Synthesis of diphosphines BiaxPhos* bearing different phosphorus moieties.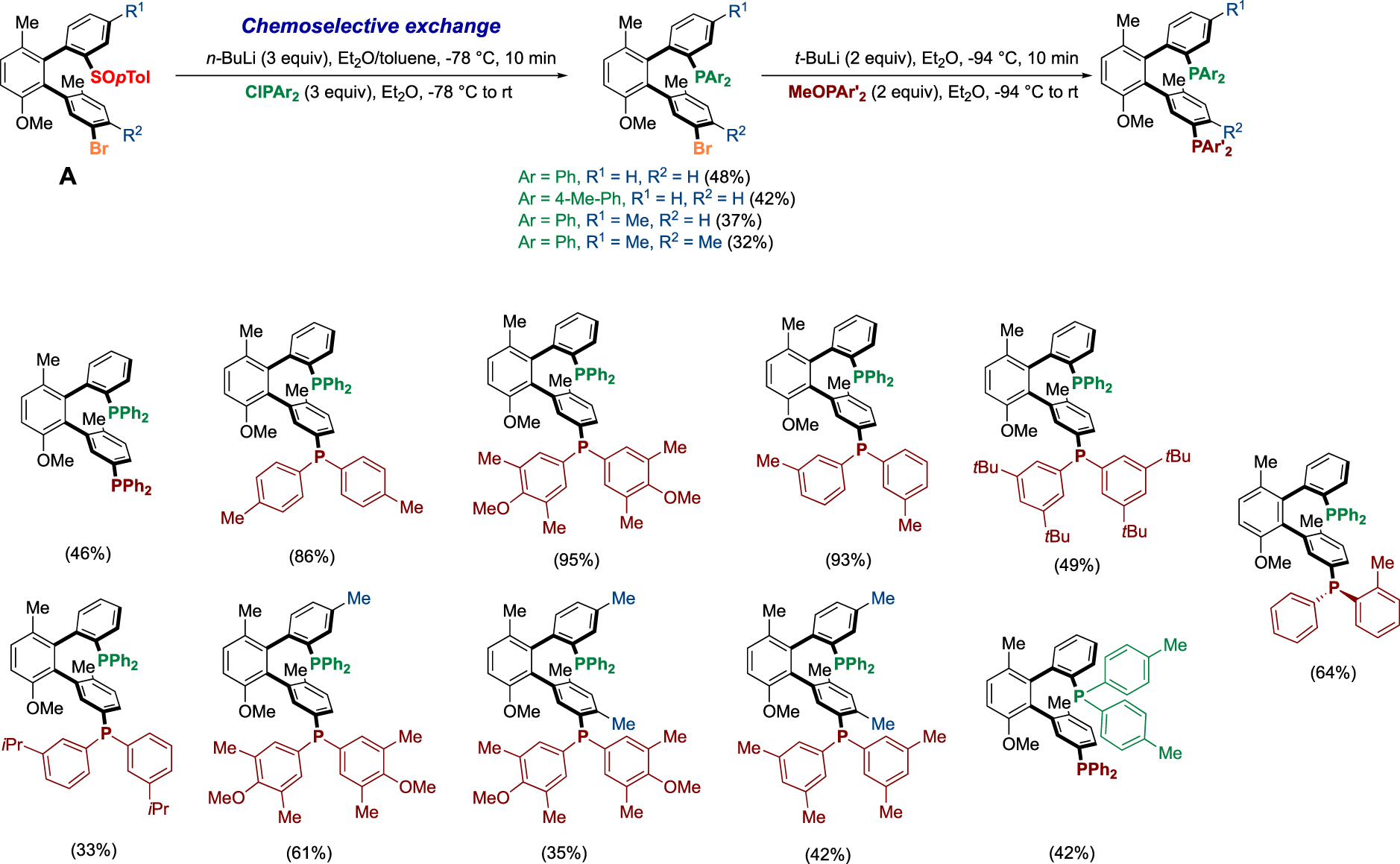
With the new ligands in hand, their potential in asymmetric catalysis, focusing mainly on the Ir-catalyzed hydrogenation of imines, was explored [20]. After optimization of the reaction conditions and catalyst screening, the desired chiral amines were isolated in high yields and excellent enantioselectivities under a remarkably low hydrogen pressure (Scheme 8) [19].
Ir-catalyzed asymmetric hydrogenation of imines with BiaxPhos* ligands.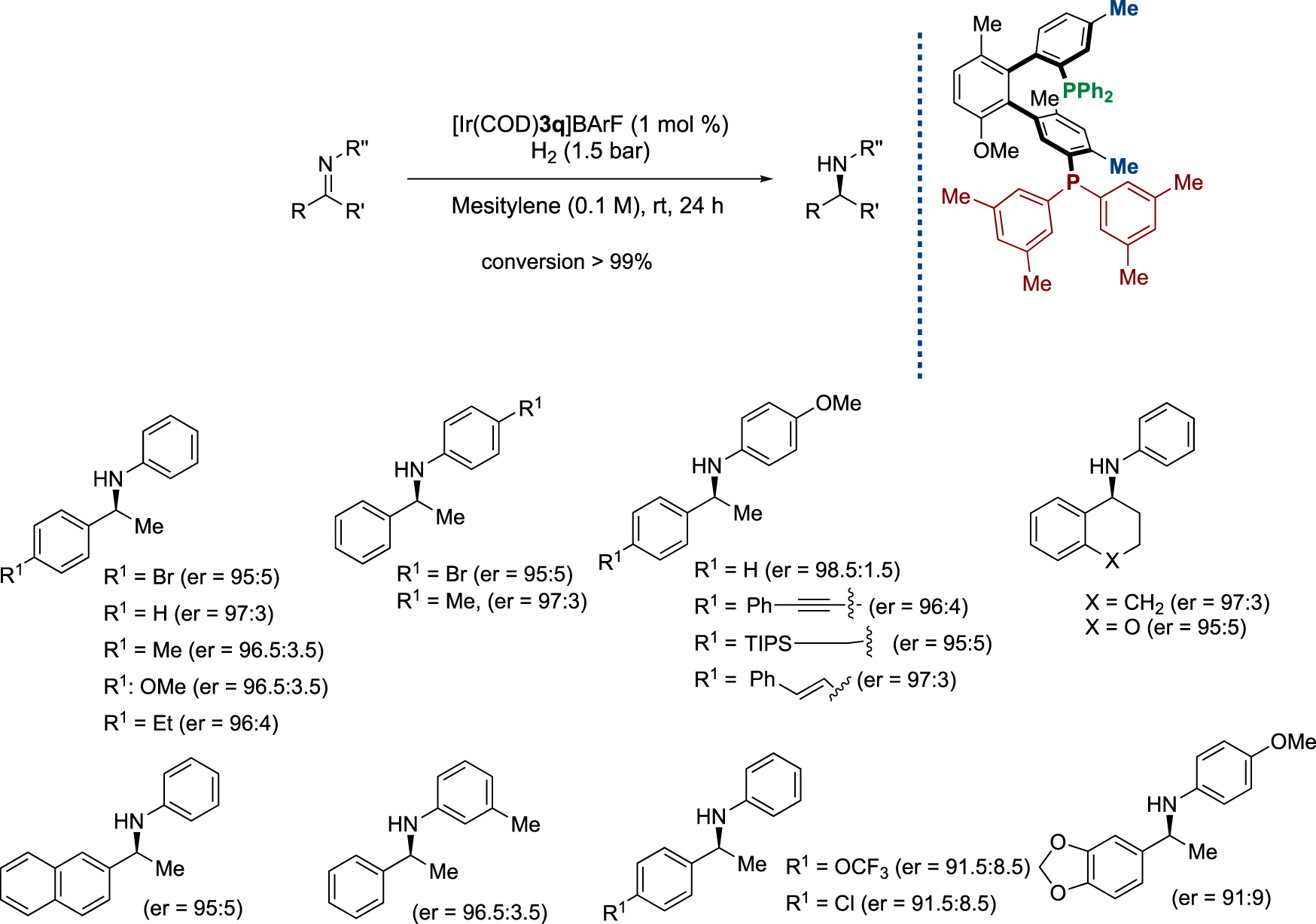
Intrigued by the growing interest of compounds with C–N atropoisomeric axes in medicinal chemistry with unique kinase inhibitor activities [21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31], we turned our attention to their atropo-stereoselective synthesis. However, the stereoselective control of the C–N axially chiral compounds remains a big challenge due to their low configurational stability compared to the well-developed C–C atropisomers and also due to the harsh reaction conditions generally required for the C–N coupling reaction. The extremely challenging character of these transformations is unambiguously illustrated by the lack of literature precedents on asymmetric Ullmann and Buchwald–Hartwig couplings.
Following such an ambitious goal, our idea was to use highly electrophilic coupling partners such as hypervalent iodines to perform the Cu-catalyzed C–N Ullmann-type coupling at low temperature to ensure the configurational stability of the C–N atropoisomer formed. Besides, the sterecontrol could be reached either with enantiomerically pure, stereogenic iodanes bearing a sulfinyl moiety allowing an atropo-diastereoselective C–N coupling with different N-coupling partners or using chiral ligands compatible with an atropo-enantioselective C–N coupling (Figure 4).
Cu-catalyzed atropo-stereoselective C–N coupling reaction.
If we consider the atropo-diastereoselective C–N coupling occurring via the chiral transfer from the iodanes (sulfoxide-substituted iodanes were prepared by C–H functionalization of the arylsulfoxide with Koser’s reagent), we studied the C–N coupling with indolines first as standard substrates, and we obtained the desired coupling products at room temperature in excellent yield and high atropopurity. However, in the first two examples, we noticed a slow racemization of the product at room temperature. Therefore, to increase the atropostability of the coupling products, substituted indolines at the C7 position or iodanes ortho-substituted by a methyl, instead of a methoxy, were used as coupling partners to obtain atropostable products [32].
Remarkably, the reaction tolerates many functional groups including chlorine, bromine, methoxy, benzyloxy but also boronic ester compatible for further functionalization (Scheme 9).
Cu-catalyzed Ullmann-type C–N coupling using a chiral sulfinyl directing group.
Subsequently, an atropo-enantioselective version of this C–N coupling was explored. Drawing inspiration from Gaunt et al. and Mc Millan et al. who investigated asymmetric copper-catalyzed coupling reaction with hypervalent iodines, the BOX-type ligand was preselected as the chirality source. We rapidly established that a free amide as DG on the iodine substrate was important to reach high enantioselectivity whereas the optimization of the BOX ligand revealed that the presence of benzyl groups as well as gem-dimethyl group was essential. Optimization of the reactions conditions indicated that the addition of a Lewis acid such as BF3⋅OEt2 warrants significantly increased yields [33].
The results, in most of the examples, show excellent enantioselectivities with moderate to good yields. As previously, a substituent in the C7 position of the indoline is necessary to guarantee atropostability of the newly formed axis. The presence of Cl-, Br-, F-substituents is well tolerated either on the indoline or the iodane.
The X-ray crystal structure which shows the absolute aR-configuration and a hydrogen bonding between the N atom of indoline and the free NH2 is in agreement with the importance of the free amide as DG (Scheme 10).
Cu-catalyzed Ullmann-type C–N coupling using a BOX ligand.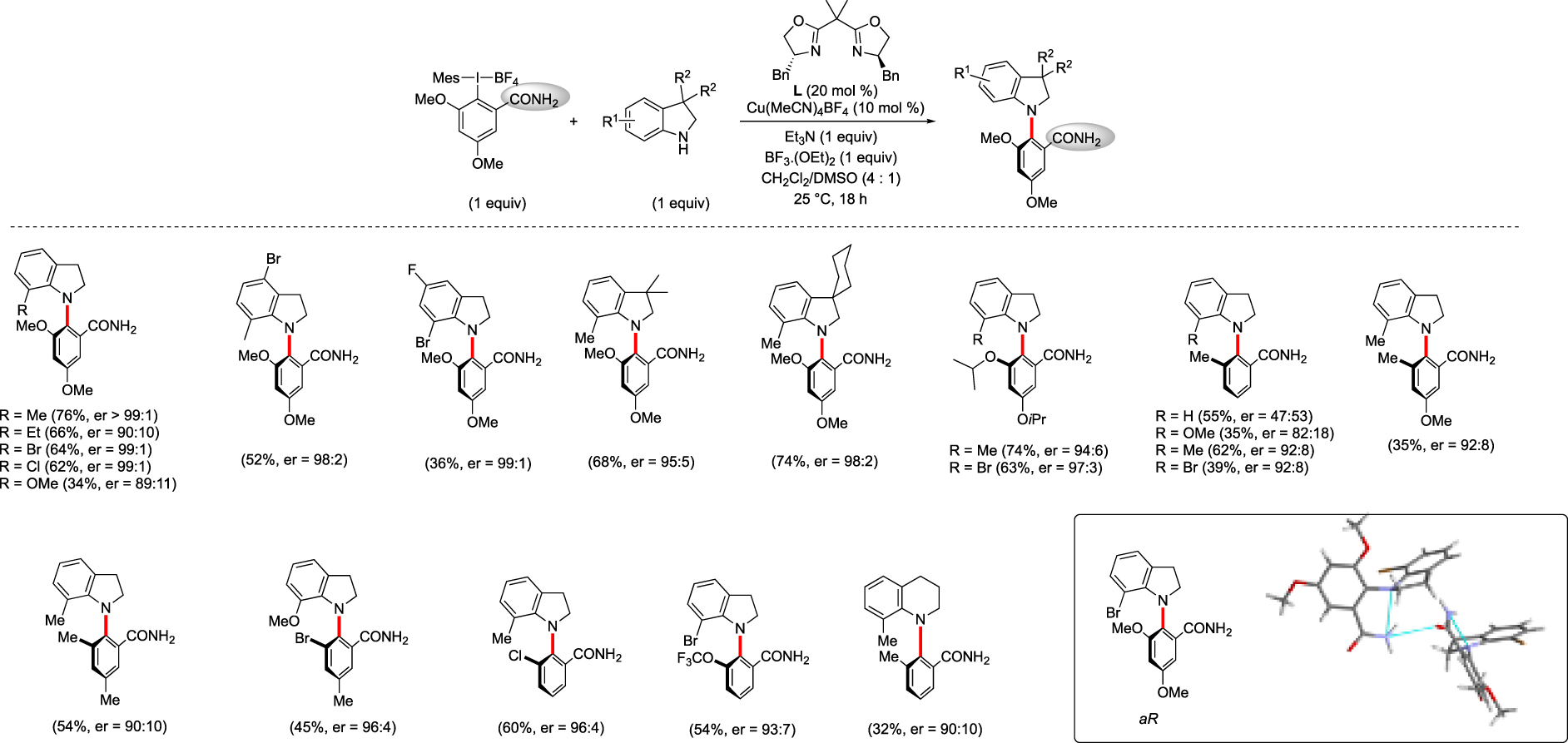
In order to highlight the synthetic potential of this method, the post-modifications of the coupling products were studied. Thanks to the traceless character of the sulfoxide, various functional groups could be introduced such as, for example, an aldehyde. The halogenated substrates allow mild Stille- and Suzuki-coupling under micellar conditions, thus overcoming the racemization issues. Oxydation of indoline to indole works very well with sulfinyl or amide DG. Moreover, indoline-2,3-dione, a privileged synthetic intermediate of oxindole scaffolds could also be obtained by oxydation with t-butylhydroperoxide in presence of copper. The amide DG can be transformed in two steps into a carboxylic acid with a complete atropostability (Figure 5).
Postfunctionalization of the C–N atropoenriched coupling products.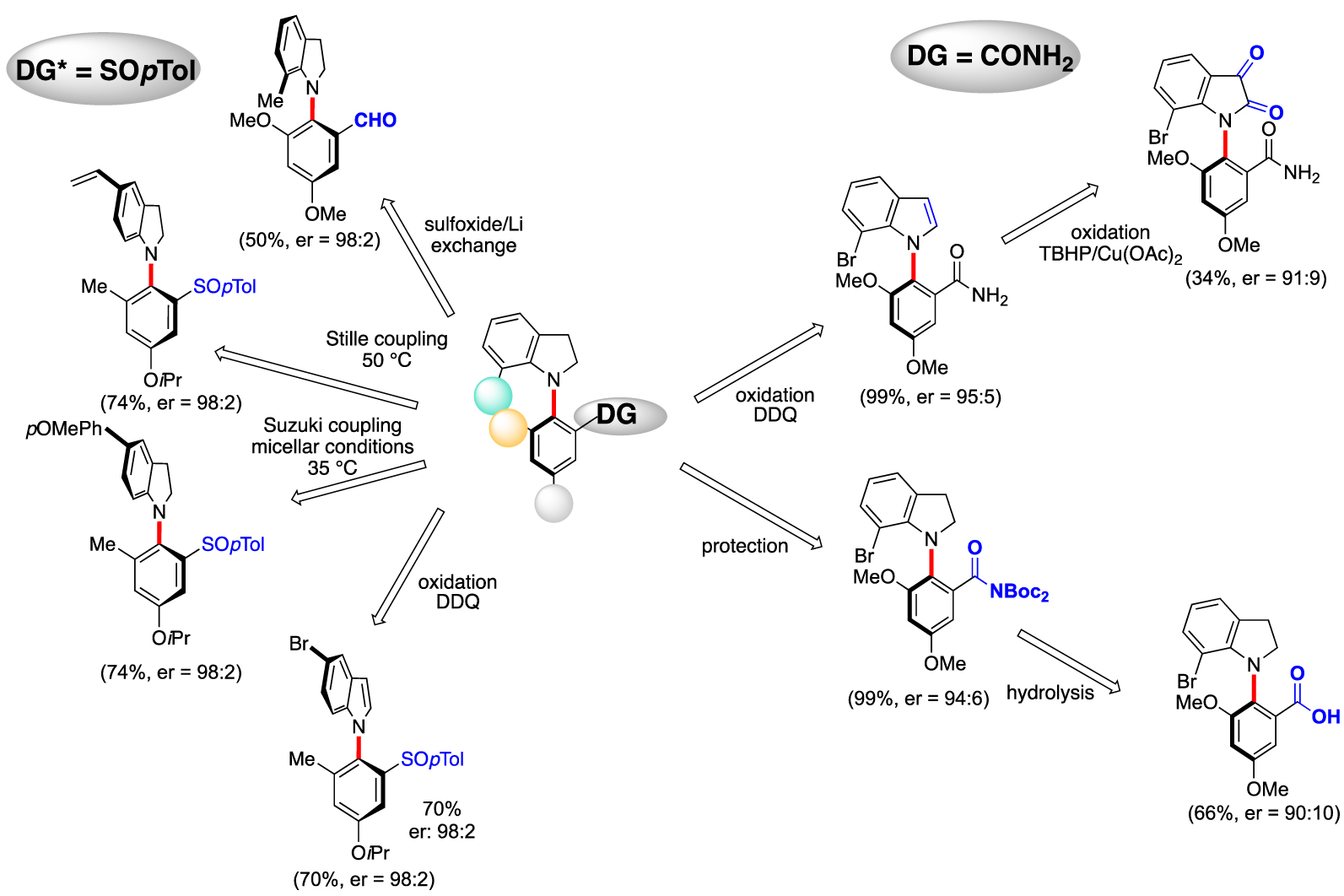
In conclusion, this account summarizes our recent efforts to control atropoisomerism in both biaryl and aryl-N-(het)aryl molecules. The general methods described such as the C–H functionalization of the biarylsulfoxide afforded complex molecules such as biologically active natural products: steganone and isoriccardin C. In parallel, the challenging C–H arylation gives access to unprecendented terphenyl ligands with two atropisomeric axes that have been demonstrated to be highly efficient in Ir-catalyzed asymmetric hydrogenation. Their original scaffold as well as their modularity are appealing candidates for asymmetric catalysis in the future.
In addition, the use of hypervalent iodines in Ullmann type C–N coupling at room temperature solved the problem of the weak atropostability of the C–N axis leading to C–N axially chiral compounds in high stereoselectivities. This first example of atroposelective N-arylation, using hypervalent iodines, is opening the way to synthesize different C–N axially chiral products with different N-coupling partners.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.