



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Heterocyclic scaffolds represent the largest family of organic compounds and have a role in many fields of sciences such as medicinal chemistry [1, 2], biochemistry [3] or agrochemistry [4, 5]. More specifically, the isoxazole skeleton is a very important motif found in natural products or synthetic products such as antibiotics [6], analgesics [7], antirheumatic [8], anti-inflammatory [9] or antidepressant compounds [10].
Furthermore, the interest of fluorine and its methods of introduction are still gaining increasing interest as fluorination of an organic molecule significantly impacts its biological characteristics. In spite of the significant fascination for fluorine, the challenge of integrating this element into essential building blocks has postponed the examination and comprehension of its impacts as well as its utilization. Nonetheless, notable advancement has been achieved in the field over the previous decade. Considering the size of the fluorine atom and its electronegativity, the incorporation of a fluorine or a trifluoromethyl group profoundly modifies both the pharmacodynamics and pharmacokinetics of the compounds such as lipid solubility, metabolic stability or acidity [11]. Moreover, it can increase the potency of a substance’s engagement with a specific target protein.
Roughly 30% of all agrochemicals and 20% of all pharmaceuticals encompass fluorine [12] within their structures.
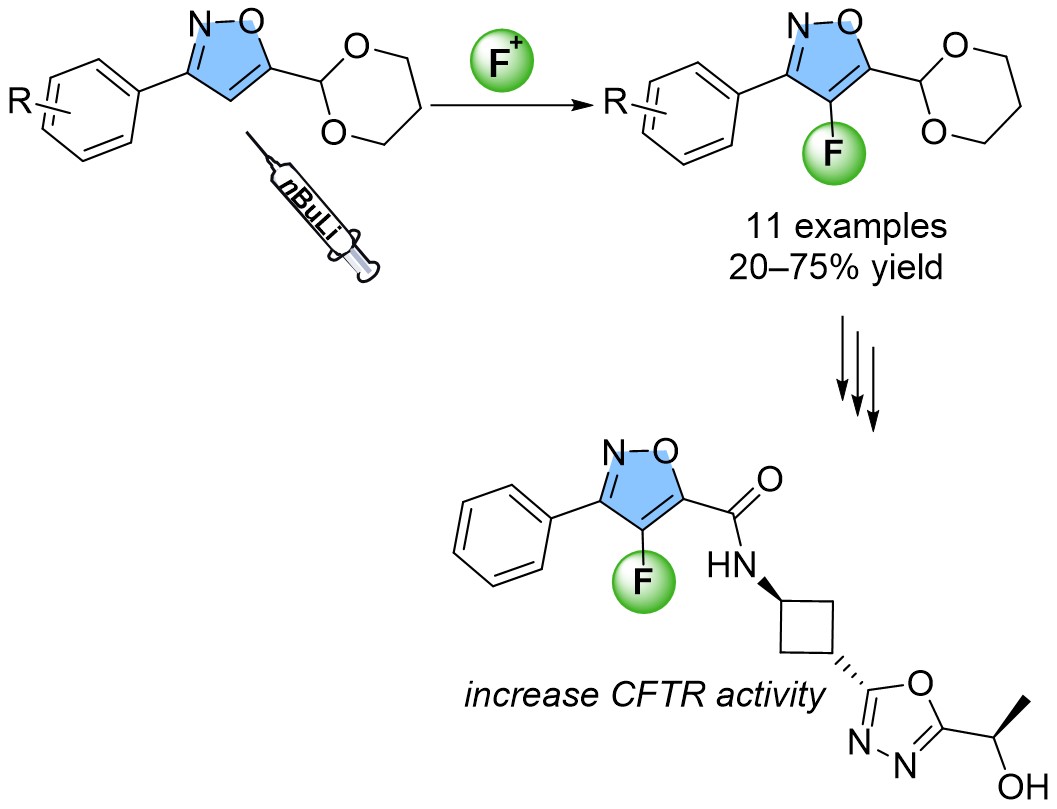
With the aim of continuing our investigations into the synthesis of fluorinated carbocycles and heterocycles [13, 14, 15, 16, 17], we envisaged to focus on a synthetic pathway for the fluorination of an isoxazole functionalized at C-5 by an acetal fragment, an area that has been relatively overlooked in the literature despite numerous molecules of interest featuring this motif (Figure 1) [18, 19, 20, 21, 22, 23]. The synthesis of 4-fluorinated 3,5-disubstituted isoxazole was reported by either condensation of 2-fluoro-1,3-diketone [24, 25] or gold-catalyzed cascade cyclization-fluorination of 2-alkynone O-methyl oximes [26] but only one method for the direct fluorination in this position was reported relying on the use of Selectfluor as the fluorinating agent and delivering the desired products in moderate chemical yields ranging from 16% to 44% (Figure 2) [27, 28]. Furthermore, the products generated during the reaction cannot undergo additional post-functionalization. In this context, the direct fluorination at the C-4 position of the 3,5-disubstituted isoxazole core, as a late-stage functionalization, was explored. With the substitution of an acetal group at the C-5 position, the resulting fluoroisoxazoles can undergo deprotection, opening-up numerous possibilities for additional functionalization.
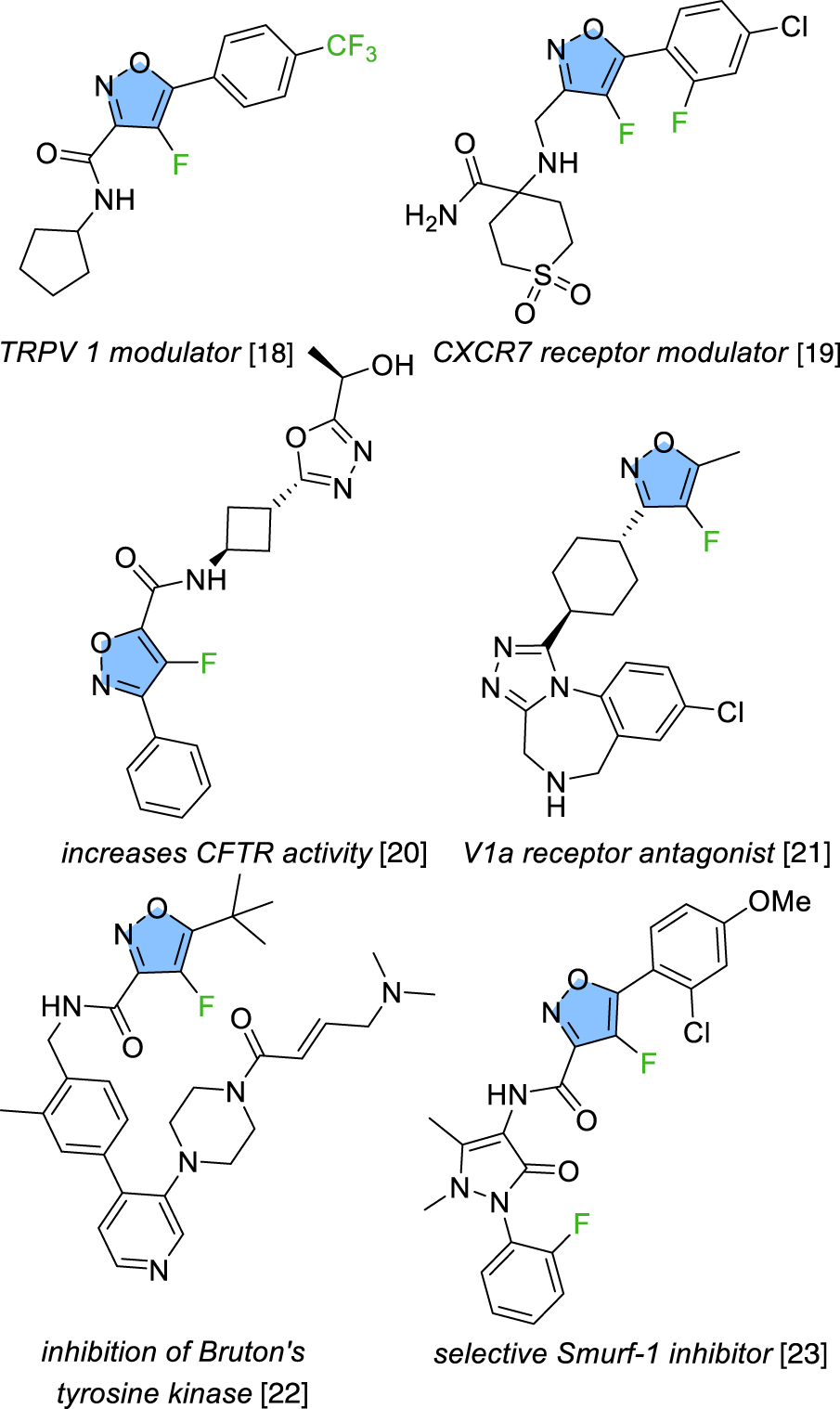
Examples of bioactive molecules containing a 4-fluorinated isoxazole core.
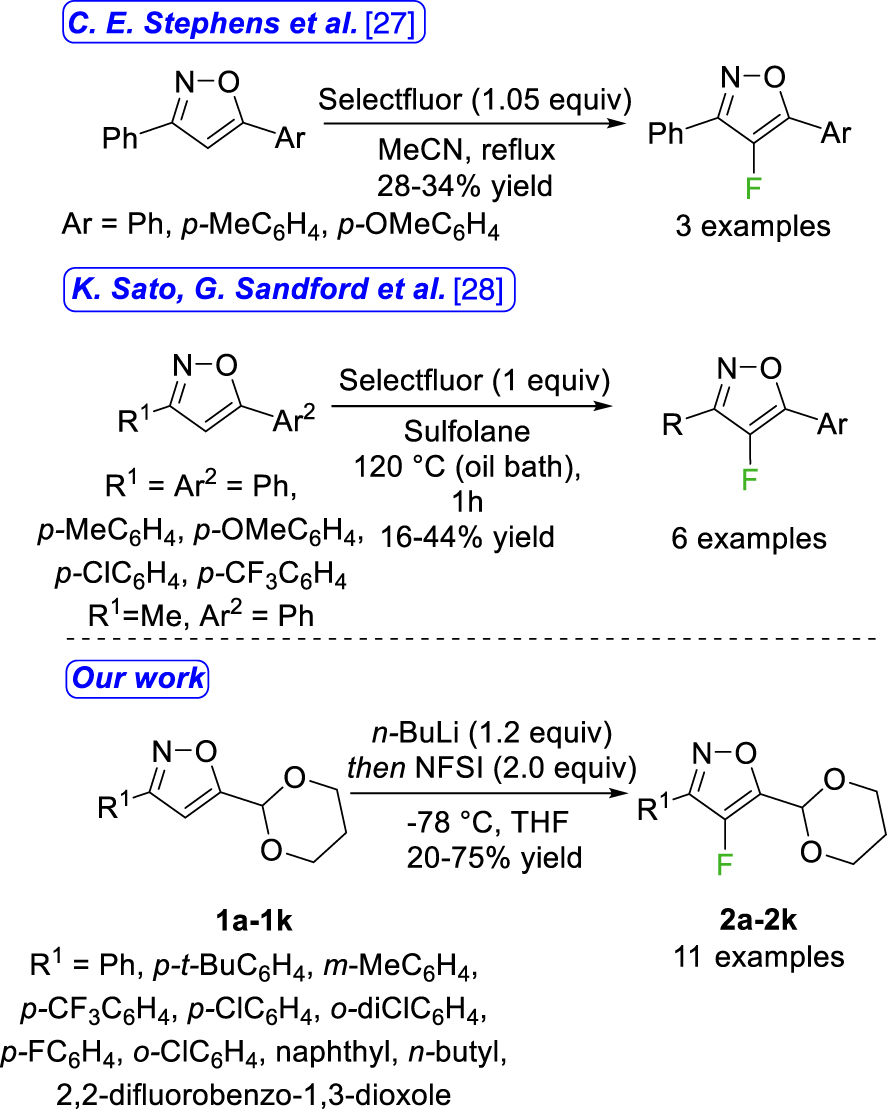
Synthesis of a 4-fluorinated 3,5-disubstituted isoxazoles by direct fluorination.
2. Results and discussion
A range of 3,5-disubstituted isoxazoles 1a-1k were prepared from diversely substituted chloroximes and bromovinyl acetals, according to a three-step procedure developed previously [29]. Having those substrates in hand, our study started with a series of optimization experiments on 5-(1,3-dioxan-2-yl)-3-phenylisoxazole 1a (Table 1). The first experiment required 3.5 equiv of base and N-fluorobenzenesulfonimide (NFSI) to afford the fluorinated isoxazole 2a in 47% yield for a conversion of 63% at −78 °C (Table 1, entry 1). Other commercially available electrophilic fluorinated agent such as Selectfluor or N-fluoropyridinium triflate were tested but no conversion was observed under these conditions. Reducing the quantity of base and fluorinating agent to 1.2 and 2.0 equiv respectively led to better conversions (72–74%) and higher yields in 2a (60–62%) (Table 1, entries 2–3). Subsequently, the impact of the starting material concentration in THF on the reaction outcome was examined. The concentration was gradually increased step by step from 0.1 mol/L to 1.0 mol/L (Table 1, entries 4–9), leading to an optimized yield of 75% at 0.4 mol/L (Table 1, entry 6). In fact, for a concentration higher than 0.6 mol/L, the reaction medium reaches such a viscosity that stirring stops after addition of the electophile.
Optimisation of the reaction conditionsa
| Entry | Base | Solvent | x (equiv) | c (mol/L) | T | t (h) | Conversionb (%) | Yieldc (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | n-BuLi | THF | 3.5d | 0.05 | −78 °C | 20 | 63 | 47 |
| 2 | n-BuLi | THF | 2.5e | 0.05 | −78 °C | 20 | 72 | 62 |
| 3 | n-BuLi | THF | 2.0 | 0.05 | −78 °C | 2 | 74 | 60 |
| 4 | n-BuLi | THF | 2.0 | 0.1 | −78 °C | 2 | 78 | 61 |
| 5 | n-BuLi | THF | 2.0 | 0.2 | −78 °C | 2 | 82 | 73 |
| 6 | n-BuLi | THF | 2.0 | 0.4 | −78 °C | 1 | 85 | 75 |
| 7 | n-BuLi | THF | 2.0 | 0.6 | −78 °C | 0.5 | 89 | 71 |
| 8 | n-BuLi | THF | 2.0 | 0.8 | −78 °C | 0.5 | 91 | 70 |
| 9 | n-BuLi | THF | 2.0 | 1.0 | −78 °C | 0.5 | 90 | 70 |
| 10 | n-BuLi | THF | 2.0 | 0.2 | −40 °C | 1 | 100 | 0f |
| 11 | n-BuLi | Et2O | 2.0 | 0.2 | −78 °C | 3 | 20 | nd |
| 12 | n-BuLi | MeTHF | 2.0 | 0.2 | −78 °C | 1 | 58 | nd |
| 13 | LiHMDS | THF | 2.0 | 0.4 | −78 °C | 0.5 | 0 | nd |
| 14 | LiHMDS | THF | 2.0 | 0.4 | 0 °C | 0.5 | 0 | nd |
| 15 | LiTMP | THF | 2.0 | 0.3 | −78 °C | 0.5 | 100 | 0f |
| 16 | LDA | THF | 2.0 | 0.1 | −78 °C | 1.5 | 81 | 67 |
| 17 | NaH | THF | 2.0 | 0.1 | 0 °C | 2 | 0 | nd |
aReaction conditions: (1a, 0.43 mmol, 1.0 equiv), nBuLi (0.52 mmol, 1.2 equiv), NFSI (0.86 mmol, 2.0 equiv), THF (0.4 M, 1.1 mL), −78 °C to rt, under argon.
bAfter NMR 1H analysis of the crude reaction mixture.
cAfter isolation by column chromatography.
d3.5 equiv of base were used.
e1.5 equiv of base were used.
fOnly degradation was observed.
Carrying out the reaction at −40 °C to achieve a full conversion led to complete degradation of the starting material (Table 1, entry 10). Other solvents were tested such as Et2O or 2-MeTHF, but lower conversions were observed (20% and 58% respectively, Table 1, entries 11 and 12). The influence of the base was also studied. The starting material was fully recovered when treated with LiHMDS at −78 °C and 0 °C or with NaH at 0 °C (Table 1, entries 13, 14 and 17). Unfortunately, LiTMP led to a complete degradation of the isoxazole (Table 1, entry 15). Using LDA gave an encouraging result of 81% conversion and 67% yield in 2a but was still inferior when n-BuLi was used (Table 1, entries 16 vs 6).
Having these optimized conditions in hand, we further explored the scope and limitations of the reaction with 3,5-disubstituted isoxazoles previously synthesized. This process allowed us to obtain the corresponding fluorinated isoxazoles bearing various electron-donating or -withdrawing substituents in the para-position of the aryl ring such as t-butyl- 2b, chlorine 2c, fluorine 2d or trifluoromethyl 2e groups in yields between 59–70%. Compounds 2g and 2h having a 3-chloro-4-fluorophenyl or a 2,6-dichlorophenyl substituent were obtained in 44% and 21% yield, respectively (Figure 3). Having a methyl in meta-position (1f) led to 75% isolated yield of the corresponding fluorinated product 2f. To extend the feasibility of our process, a bulky group such as a naphthyl or a 2,2-difluorobenzodioxole was also investigated and afforded the products 2i and 2j in 20% and 60% yields, respectively. The reaction was also suitable for isoxazoles bearing a non-aromatic substituent at the C-3 position and compound 2k, bearing a n-butyl chain, was synthesized in 25% yield.
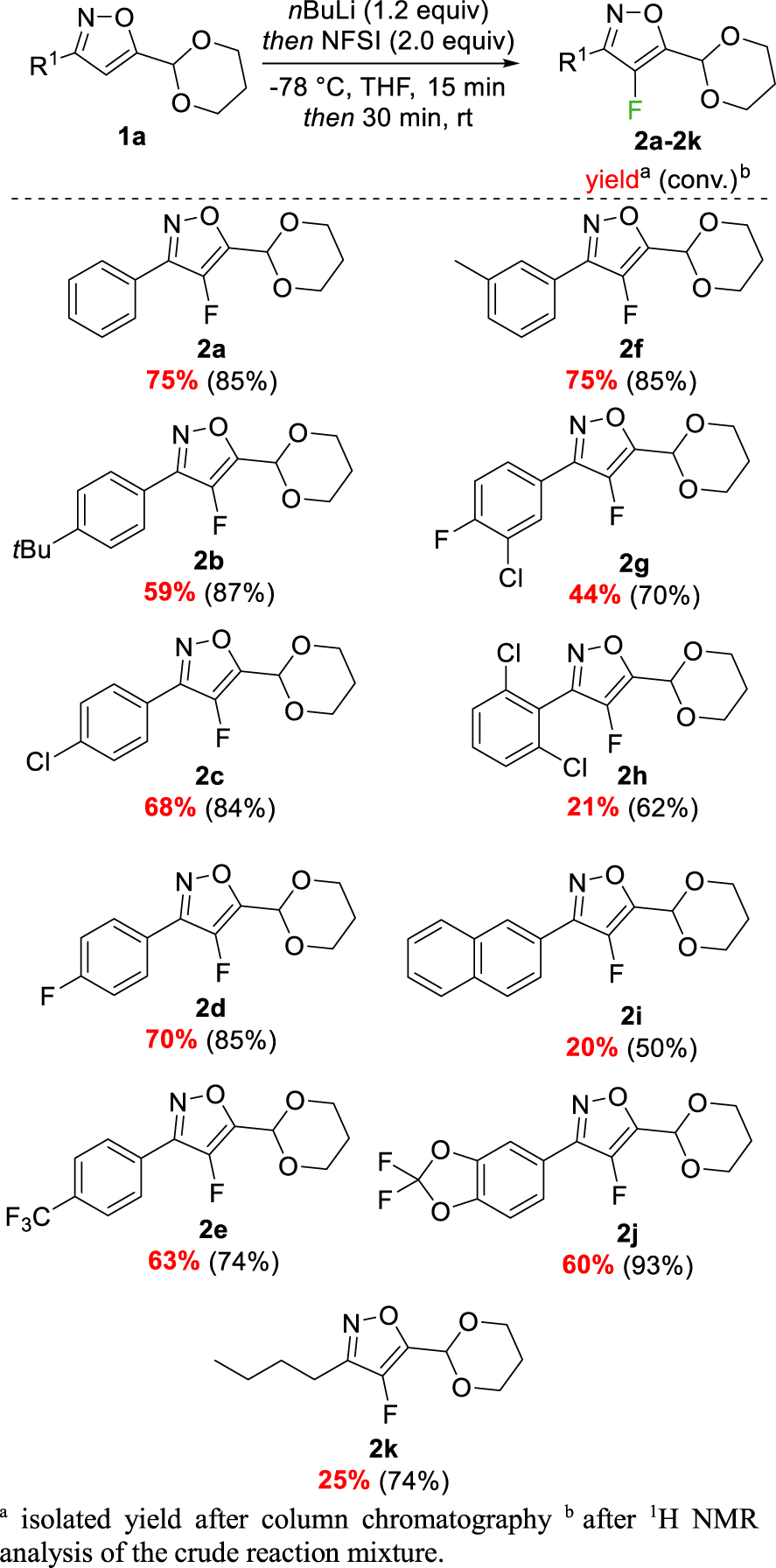
Scope of the fluorination.
Furthermore, our goal was to obtain a range of fluorinated isoxazoles that could be subsequently modified after deprotection of the acetal fragment at the C-5 position. In this context, the acetal was deprotected in acidic media (HCl 1N, 5 equiv), in a mixture of water/acetone (1/1) for 4 days at 90 °C affording the corresponding aldehyde 3a in 65% yield (Figure 4).
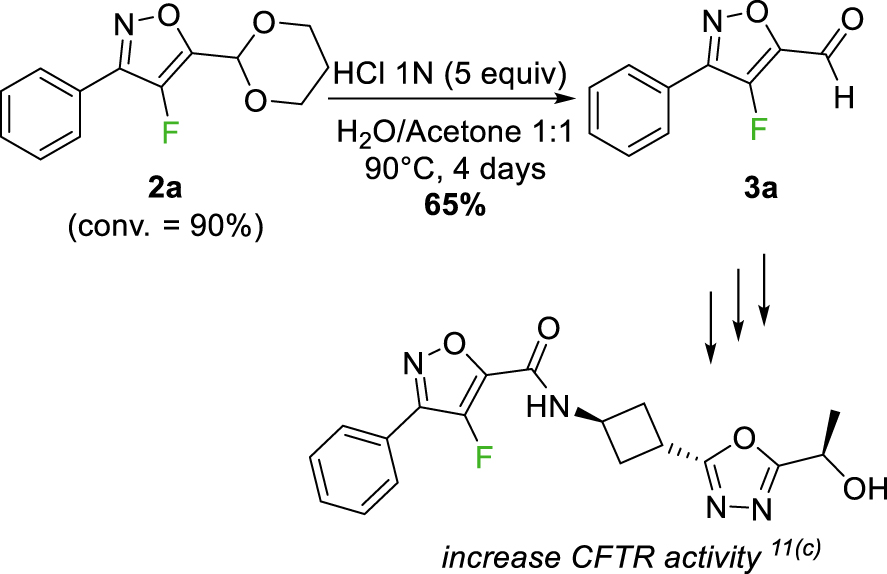
Formal synthesis of a bioactive compound that increases CFTR activity.
Isoxazole 3a is an intermediate in the preparation of a bioactive compound that increases Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) activity [15]. Cystic fibrosis is a result of genetic mutations in the CFTR gene, responsible for encoding a chloride channel that spans multiple cell membranes in epithelial tissues. The loss of a functional CFTR channel leads to a reduced lung function and increases mucus viscosity resulting in chronic infection and inflammation [30]. Besides respiratory impairment, CFTR also affects the regular operation of additional organs such as the pancreas, intestine, or gall bladder.
The effectiveness of this fluorination process was supported by a scale-up experiment conducted under the defined standard conditions using 5-(1,3-dioxan-2-yl)-3-phenylisoxazole 1a (Figure 5) to obtain the desired fluorinated product 2a in 61% yield.

Scale-up experiment of the fluorination of 1a.
3. Conclusion
In conclusion, we succeeded in synthesizing a range of new 4-fluorinated 3,5-disubstituted isoxazoles bearing various substituents on the aromatic ring at the C-3 position in 20–75% yields. This process was also tolerant to bulkier substituents such as a naphthyl or a difluorodioxobenzene and even a non-aromatic substituent such as a n-butyl fragment. The objective of this project was also to obtain a substrate that could be subsequently functionalized. This was achieved through the deprotection of the acetal, resulting in a much more advantageous aldehyde for subsequent post-functionalization. The usefulness of this method was demonstrated through the formal synthesis of a bioactive compound that increases CFTR activity.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Acknowledgement
We thank Dr. C. Fosse (Chimie ParisTech - PSL) for HRMS analysis.
Vous devez vous connecter pour continuer.
S'authentifier