



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Polycyclic Aromatic Hydrocarbons (PAHs) are pollutants that are emitted naturally or produced by human activities, due to incomplete combustion or pyrolysis of organic materials (industries, road traffic, heating …) [1]. They are present in environment, but also in food, leading to a constant exposure of humans [2, 3, 4]. They are carcinogens and genotoxic substances. After entering the human body, due to their hydrophobic properties, PAHs are transported to all lipid-rich tissues. This is why a long-term exposure may lead to their accumulation in adipose tissue, liver, and kidney [4, 5]. They can also undergo different metabolic transformations resulting to the formation of reactive electrophilic intermediates able to bind cellular macromolecules such as proteins and nucleic acids. It is now well established that these pollutants are chemical hazard to human health. Long-term exposure increases the risk of a series of cancers. Moreover, an exposure during pregnancy can lead to fetal malformations, prematurity and even disorders in children and adults [6, 7, 8]. This may come from a contamination of the placenta, which ensures multiple functions directly involved in the initiation and outcome of gestation, fetal growth and possibly the initiation of parturition, and/or of the fetal tissues through a transfer of PAHs via the placenta. This is why it is highly interesting to determine PAHs in both maternal and umbilical cord bloods and also in placenta.
Today, 24 different PAHs are listed as priority pollutants: 16 by the United State Environmental Protection Agency (US-EPA) and 16 by the European Food Safety Authority (EFSA), eight of them belonging to both lists. The 24 listed PAHs belong to a wide range of Log P values reported in Figure S1 (see Supplementary Information) [9, 10, 11, 12], thus rendering their determination in biological samples particularly difficult but necessary as human contamination has multiple sources and because the EFSA PAHs are quite different from the US-EPA PAHs (higher number of aromatic cycles). Focusing on the determination of PAHs in blood, in most of the cases, only the US-EPA PAHs were targeted [13] except Pleil and coworkers who quantitated also 6 EFSA PAHs [14]. Typical blood, plasma, and serum used volumes range from 1 to 12 mL, which are quite large values, limiting the access to epidemiological cohort samples, with one exception of a study using pipette-tip solid-phase extraction (SPE) and only 200 μL of blood to determine the 16 US-EPA HAPs [15]. The most used technique for sample pretreatment consists in extracting the PAHs by liquid–liquid extraction (LLE) [6, 14, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26], but with some drawbacks such as the large volumes of used solvent and being time consuming as it involves several successive LLE steps, often three, to improve the extraction yields. Most of the time, the LLE steps were followed by SPE using C18-bonded silica, silica or silica/alumina packed in cartridge [17, 18, 19, 21, 24, 25, 26] to improve the clean-up of the sample extract by removing matrix interferents that could disturb the PAH quantitation.
SPE can also be used alone as an alternative approach to LLE to extract the PAHs from blood, plasma or serum prior to their chromatographic analysis [27, 28, 29, 30, 31]. It requires a lower volume of solvent than LLE and simple less-time consuming manipulations. Indeed, 1 mL [30] or 2 mL [28, 29, 31] of blood, plasma or serum were percolated on an apolar sorbent, C18-bonded silica when this information is given [27, 28, 29]. The SPE step was followed by an analysis with either gas chromatography coupled to mass spectrometry [27, 28, 29, 31] or liquid chromatography (LC) coupled to a fluorescence detection (FD) [30]. Unfortunately, in some of these papers, several analytical data are missing, such as the nature of the SPE sorbent [30, 31], the enrichment factors [28, 31], the extraction yields in real samples [31], and the limits of quantitation (LOQs) in the biological matrices [28, 31], whereas these data are necessary to be sure that the presented results are consistent. Moreover, only between seven and 16 PAHs were targeted and they only belong to the US-EPA list [27, 28, 29, 30, 31], even for the last study dealing with miniaturized pipette-tip SPE format involving 200 μL of sample [15].
Concerning the detected concentrations of PAHs in blood, plasma or serum, they were most often normalized with lipid content and reported in ng/g lipid. A study demonstrated that the mean lipid content in 283 maternal sera was 7.33 ± 1.86 g/L [32]. Using this value for the lipid content, it is therefore possible to convert the data reported in each study in the same unit and then to compare these estimated values, with the hypothesis that all the reported values are consistent. The highest mean values were observed by Song et al. with up to 37 μg/L of benzo[a]anthracene (BaA) in the blood of heavy-taste food consumers and a minimum mean value of 0.3 μg/L of pyrene (PYR) in the blood of Chinese non-smokers [27]. Similar values, between 0.05 and 2.7 μg/L, were determined for each of 13 US-EPA PAHs in blood plasma collected from Hong Kong residents [29]. These values are in accordance with the ones determined in sera of autopsied people in Tennessee, where the sum of the concentrations of 13 US-EPA PAHs varied between 0.1 and 12 μg/L [31]. Focusing on both maternal and cord sera, there are two Chinese studies reported values between 0.1 and 2.5 μg/L for 11 of the 16 US-EPA PAHs (the five other US-EPA PAH concentrations were below the LOQs) [28] and between 0.1 and 9.5 μg/L for the 9 targeted US-EPA PAHs [30]. This gives an idea of the PAH concentrations that one can expect in these kind of samples.
Due to the lack of consistent data of contamination level in literature, especially for the PAHs listed for food, there is still a great societal need to determine PAH concentrations in biological tissues or fluids to increase knowledge of their health effects, especially on at-risk populations such as pregnant women and their fetus. Therefore, the objective of this study is to develop a simple and low solvent consuming sample preparation step able to extract simultaneously the maximum of the 24 listed PAHs from maternal and umbilical cord sera. One of the challenges of this work is to obtain the best possible sensitivity from low volumes of sera in order to access to precious samples from epidemiological cohorts. For this, a protocol involving protein precipitation, SPE, and evaporation-redissolution before PAH analysis was developed. The key parameters of each sample pretreatment step were optimized. At last, to highlight the potential of the developed method, the extraction yields and LOQs were determined in both real spiked maternal and umbilical cord sera.
2. Materials and methods
2.1. Chemicals
HPLC grade methanol, acetonitrile (CH3CN), propan-2-ol, and tetrahydrofuran (THF) were supplied by Carlo Erba (Val de Reuil, France). High purity water was dispensed by a milli-Q purification system (Millipore, Saint Quentin en Yvelines, France). The PAH standards were supplied by Cluzeau Info Labo (Sainte-Foy-La-Grande, France): acenaphthylene (ACY) 99%, cyclopenta[cd]pyrene (CPcdP) 99%, benzo[c]fluorene (BcF) 97%, benzo[j]fluorene (BjF) 99.7%, dibenzo[a,l]pyrene (DalP) 99.8%, dibenzo[a,e]pyrene (DaeP) 99.8%, dibenzo[a,h] pyrene (DahP) 99%, dibenzo[a,i]pyrene (DaiP) 99% at 10 mg/L in CH3CN and a standard mixture of the 16 US-EPA PAHs at 100 mg/L in CH3CN.
A stock solution mixture containing 100 μg/L of each PAH was prepared in CH3CN and stored at 4 °C until further use. An aqueous solution containing 10% (w/v) sodium dodecyl sulfate (SDS) was purchased from Thermofisher Scientific (Villebon-Courtaboeuf, France). A synthetic serum was prepared from Earle’s balanced salt solution containing albumin at 25 g/L and spiked with a mixture of 16 representative PAHs at 4 μg/L for each PAH in CH3CN.
2.2. Apparatus and analytical conditions
The LC-diode array detector (DAD)/FD analyses were performed using an Agilent 1200 series system (Agilent Technologies, Massy, France) that was controlled by the Chemstation software. The separation was performed on a Pursuit PAH column (100 × 2.1 mm, 3 μm, Agilent Technologies) maintained at 35 °C with a column oven (Croco-cil, Interchim, Montluçon, France). Samples were analyzed using a linear gradient elution with water (A) and a mixture of CH3OH:CH3CN (6:4, v/v) (B). The gradient started at 50% of B and increased to 90% of B in 27 min, held for 13 min, and increased to 100% of B in 1 min, held for 10 min, and finally returned to initial composition within 1 min and let 10 min to equilibrate the system. The flow-rate was set at 0.2 mL/min and the injection volume was 5 or 10 μL. ACY, CPcdP, BjF, and indeno[1,2,3-cd]pyrene (IcdP) were quantified by UV, at 230 nm for both ACY and CPcdP, 240 nm for BjF, and 300 nm for IcdP. For the other PAHs, a time program of the excitation and emission wavelengths was performed and they were quantified with FD. The wavelengths are reported in Table 1.
LOQs (S/N = 10) in μg/L obtained with fluorescence and UV detections and estimated by injecting 10 μL of a solution of PAHs at 1, 5, and 20 μg/L in CH3CN:water (1:1, v/v)
| Compounds | 𝜆UV (nm) | 𝜆ex, 𝜆em (nm) | LOQFD (μg/L) | LOQUV (μg/L) |
|---|---|---|---|---|
| NAPH | 220 | 269, 327 | 1.2 | 2.2 |
| ACY | 230 | - | - | 2.1 |
| ACE | 230 | 227, 315 | 0.5 | 2.3 |
| FLO | 260 | 227, 315 | 1.1 | 13.2 |
| PHE | 250 | 250, 364 | 0.4 | 1.5 |
| ANT | 250 | 250, 364 | 0.9 | 0.7 |
| FLT | 230 | 234, 454 | 0.9 | 4.8 |
| PYR | 240 | 237, 388 | 0.2 | 0.9 |
| BcF | 230 | 237, 388 | 0.3 | 8.0 |
| CPcdP | 230 | - | - | 6.8 |
| BaA | 275 | 265, 376 | 1.5 | 8.7 |
| CHR | 270 | 265, 376 | 0.1 | 6.3 |
| 5MCHR | 270 | 265, 376 | 0.2 | 5.5 |
| BjF | 240 | - | - | 4.9 |
| BbF | 260 | 290, 440 | 0.3 | 4.6 |
| BkF | 240 | 290, 440 | 0.1 | 5.7 |
| BaP | 260 | 290, 440 | 0.2 | 4.5 |
| DahA | 295 | 292, 428 | 0.3 | 3.6 |
| DalP | 315 | 292, 428 | 0.4 | 6.7 |
| BghiP | 285 | 292, 428 | 0.3 | 15.9 |
| IcdP | 300 | - | - | 9.3 |
| DaeP | 310 | 280, 404 | 0.8 | 5.7 |
| DaiP | 240 | 390, 439 | 0.4 | 6.2 |
| DahP | 310 | 260, 456 | 0.4 | 2.0 |
2.3. SPE procedure
The SPE cartridges were Hypersep (25 mg of trifunctional octadecyl non-end-capped silica, 40–63 μm, 60 Å, 520 m2/g, 21–23% carbon, ThermoFisher Scientific), Versaplate (25 mg of trifunctional octadecyl end-capped silica, 40 μm, 60 Å, 500 m2/g, 17.4% carbon, Agilent Technologies) and BondElut SPEC (30 mg of trifunctional octadecyl non-end-capped silica, 70 Å, 220 m2/g, 7% carbon, Agilent Technologies). The first SPE developments were carried out by percolating ultra-pure water spiked at 5 μg/L with PAHs on the Hypersep cartridges. Except when it is mentioned, the protocol was as follows: the cartridge was conditioned with 300 μL each of THF, CH3OH, and H2O:CH3CN (1:1, v/v). After percolating 100 or 200 μL of a H2O:CH3CN (1:1, v/v) mixture spiked with PAHs, a washing step with 100 μL of H2O:CH3CN (1:1, v/v) was carried out and the cartridge was next dried. Finally, the PAHs were eluted with 300 μL of THF. The elution fraction was diluted with water to obtain a THF:H2O mixture (1:1, v/v) before injection in LC-DAD/FD system or subjected to evaporation.
2.4. Optimization of the evaporation step
To enrich the SPE elution fraction, the optimization of an evaporation step was carried out by evaporating 200 μL of THF spiked with 5 μg/L of 20 PAHs to dryness or partially when 10 or 40 μL of an aromatic solvent (xylene, trimethylbenzene, or toluene) were added to THF. For both approaches, after the evaporation step, the extract was recovered in CH3CN before its injection in LC-DAD/FD (5 μL).
2.5. Optimization of the protein precipitation procedure with a design of experiment
The optimization of the protein precipitation parameters was performed with a design of experiment (DOE). The DOE was constructed with two levels for the percentage of CH3CN and heating and three levels for the percentage of SDS. The DOE was realized with an Earle’s balanced salt solution containing albumin at 25 g/L (synthetic serum) and spiked at 4 μg/L with 16 PAHs (acenaphthene (ACE), fluorene (FLO), phenanthrene (PHE), anthracene (ANT), fluoranthene (FLT), PYR, BcF, chrysene (CHR), 5-methylchrysene (5MCHR), benzo[b]fluoranthene (BbF), benzo[k]fluoranthene (BkF), benzo[a]pyrene (BaP), dibenzo[a,h]anthracene (DahA), DalP, DaeP, and DahP) to prevent from wasting precious sera samples. For these experiments, only 16 PAHs among the 24 were selected in order to cover the whole range of hydrophobicity, except the less hydrophobic compounds, naphthalene (NAPH) and acenaphthylene (ACY) because of their higher LOQs. All samples were introduced in 2 mL Protein Lobind tubes (Eppendorf, Montesson, France). The denaturation and the precipitation of the albumin were carried out by adding an aqueous solution containing 10% of SDS (w/v) to achieve a final concentration of 0.1 or 0.2%. Some samples were next heated at 60 °C during 25 min. Then, different volumes of CH3CN were added to obtain a final content of 50 or 70% (v/v) and samples were vortexed. Finally, a centrifugation at 4500 rpm at 4 °C during 30 min was carried out and the supernatants were collected. Before SPE, as the samples already contained 700 μL of CH3CN, a dilution with water to achieve a final content of 50% of CH3CN in the sample was implemented. After conditioning the SPE cartridge, 400 μL of the diluted supernatant were percolated. A washing step with 100 μL of H2O:CH3CN (1:1, v/v) was done and the cartridge was dried. Finally, the PAHs were eluted with 300 μL of THF and a partial evaporation with the addition of 40 μL of toluene and a recovered volume of 35 μL of CH3CN was performed before injection in LC-DAD/FD system (5 μL). The measured responses were the PAH extraction recoveries. The data treatment was performed using JMP 10.0 (S.A.S Institute Inc, Cary, NC, USA) software.
2.6. Extraction of PAHs from maternal and umbilical cord sera
2.6.1. Sample collection and storage
Biological samples were obtained following informed written patient consent and approval from our local ethics committee (CPP 2015-Mai-13909). Umbilical cord and maternal blood were collected with harmonized procedures from patients delivered in Obstetric Units of Paris’ Hospitals. Sera were obtained after blood centrifugation at 3000 rpm during 15 min and stored at −20 °C in Protein Lobind tubes.
2.6.2. Sera pretreatment procedure
100 μL of maternal or umbilical cord serum were spiked at 10 μg/L with each PAH except at 20 μg⋅L−1 for CPcdP and IcdP. A solution containing 10% of SDS was next added to achieve a final concentration of 0.2% (w/v). Sample was vortexed and 225 μL of CH3CN were added. A centrifugation at 4500 rpm during 30 min at 4 °C was carried out. 300 μL of supernatant were then collected and 120 μL of water were added to obtain a final content of 50% of CH3CN (v/v). 400 μL of this sample were percolated on SPE cartridge. A washing step with 100 μL of CH3CN:H2O (1:1, v/v), a drying and an elution with 300 μL of THF were implemented. 40 μL of toluene were added to the elution fraction that was next vortexed and evaporated. The final extract was suspended in 100 μL of CH3CN:H2O (1:1, v/v) before injection in LC-DAD/FD system (10 μL).
3. Results and discussions
3.1. Limits of quantitation of the LC-DAD/FD analysis
The analysis of the 24 PAHs by LC-DAD/FD was carried out with an Agilent Pursuit PAH (100 × 2.1 mm, 3 μm) column containing a polymeric C18-bonded silica stationary phase, chosen for its shape selectivity towards PAHs [33]. Considering the wide polarity range of the 24 targeted PAHs with log P values from 3.3 up to 7.7 (see Figure S1) [9, 10, 11, 12], different temperatures (30 and 35 °C), mobile phases (CH3CN and water, CH3OH and water, CH3OH plus CH3CN and water), and gradients were tested to find optimal conditions implementing a ternary gradient with water, CH3CN, and CH3OH (see Figure S2).
The FD detection schedule was optimized by studying the excitation and emission fluorescence spectra of each PAH. Nevertheless, some compromises were necessary as it is only possible to change the wavelengths when the time gap between two peaks is sufficient. Table 1 presents the LOQ values (defined as the concentration level that gives a signal to noise ratio of 10) determined with the optimized fluorescence schedule. They ranged from 0.1 to 1.5 μg/L for an injection volume of 10 μL. It is worthwhile to notice that no values are given for four PAHs (ACY, CPcdP, BjF, and IcdP) in FD. Indeed, ACY and CPcdP do not fluoresce while BjF have a poor fluorescence. Concerning IcdP, its quantification in UV was preferred because the resolution was insufficient to change the excitation and emission wavelengths in order to better detect it in FD. The LOQs were also determined in UV, at the maximal absorption wavelength of each PAH (see Table 1), and are comprised between 0.7 and 15.9 μg/L. As expected, except for ACY, CPcdP, BjF, IcdP and ANT, these LOQ values are significantly better in FD than in UV, by factors of 1.8 to 63. This is why FD was used for quantitation of all these PAHs. For ANT, LOQs in LC-UV and -FD are very close and FD was chosen in order to exploit its specificity for the analysis of real samples. It can also be noticed that the obtained LOQs for these standard solutions are close to the expected concentrations in sera already determined in literature thus indicating that the sample pretreatment method does not necessitate to provide a high enrichment factor. This should favor the possibility of using reduced sample volumes, which is often required by cohort leader.
3.2. Development of the SPE procedure
In order to determine PAHs at trace level in sera that are complex biological samples, a purification and extraction by SPE on C18-bonded silica-based sorbent was optimized in order to remove as much as possible matrix components.
3.2.1. Choice of the percolation solvent
The solubility of some PAHs is very low in aqueous solutions, particularly for the high molecular weight PAHs. This can induce their adsorption on the vial or cartridge walls and consequently their loss and then the underestimation of their concentration in samples. The strategy to avoid their adsorption consists in adding some organic solvent in the sample. Moreover, the treatment of serum samples as envisaged in this study, generally required a first precipitation step usually carried out in organic solvent that will also prevent the risk of loss of PAHs by adsorption to proteins. Therefore, the critical parameters are the solvent nature and its proportion to add in the sample without reducing too much the breakthrough volume of the lowest molecular weight PAHs. Preliminary experiments were performed by percolating water containing 30% of CH3OH, CH3CN or isopropanol and spiked with CHR, BaP, and DaiP, i.e. three PAHs selected to be representative of some of the most hydrophobic targeted ones. For the washing step, an CH3CN:H2O (1:1, v/v) mixture was chosen to remove some potential interferents that could be present latter in real samples since it has a slightly higher eluent strength than the percolation media. A volume of 100 μL of this solution was introduced to limit the risk of reaching the breakthrough volume of the most polar PAHs. For the elution step, one has to select a solvent able to disrupt the interactions between the analytes and the sorbent with a volume as low as possible to either lead to some enrichment or to limit the time of the potential subsequent evaporation step. In literature, THF [34] or mixtures of hexane:CH2Cl2 (1/1) [28, 29] and CH3CN:butyl chloride (55/45) [31] or ethyl acetate [30] were used. THF was selected, with an elution volume of 300 μL. Figure 1 shows the resulting average extraction recoveries obtained with this SPE protocol, percolating 100 μL of hydro-organic mixtures (7:3, v/v) spiked at 5 μg/L with the three PAHs on 25 mg of a C18-bonded silica (Hypersep). It appears that the DaiP concentration in the elution fraction was below the LOQ whereas it was not the case with CH3CN and isopropanol. CH3OH appears as too polar to ensure the solubilization of this highly hydrophobic PAH (log P(DaiP) = 7.3), leading to its adsorption on the vial or cartridge walls. Therefore, CH3OH was removed for the end of the study.
Effect of the nature and the volume of organic solvent (30% or 50%) introduced in spiked water on the extraction recoveries. SPE procedure (Hypersep cartridge): percolation of 100 μL of water containing 30 or 50% of the organic studied solvent and spiked with 5 μg/L of each PAH, washing with 100 μL of CH3CN:H2O (1:1, v/v), and elution with 300 μL of THF, dilution of the THF extract with water (THF:H2O, v:v, 1/1) (n = 3). * not quantifiable PAH.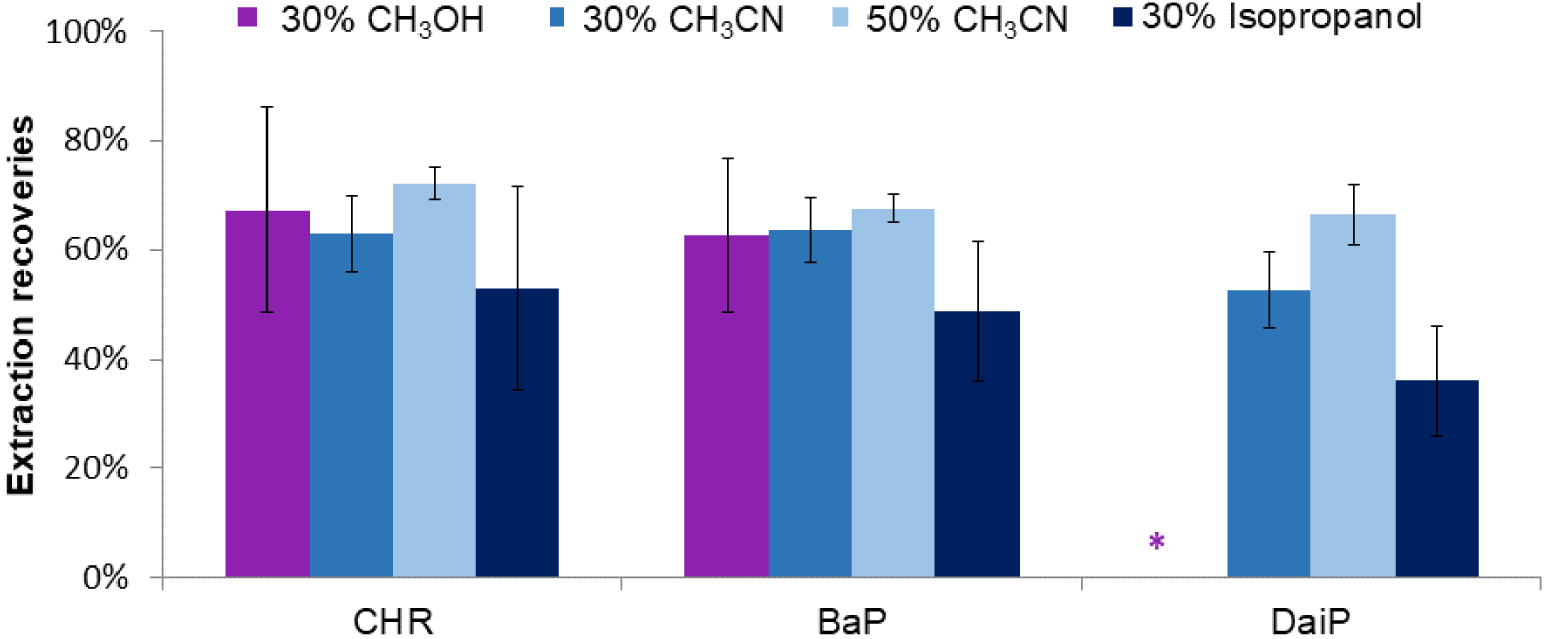
An ANOVA test (𝛼 = 5%) demonstrated that the recoveries of the three PAHs obtained after percolating them in 30% of CH3CN or isopropanol were not significantly different. Nevertheless, CH3CN led to lower standard deviation (SD) values than isopropanol and was thus preferred. Its proportion in the sample was next increased up to 50% to favor the PAH solubility, especially for the most hydrophobic ones. It appears in Figure 1 that the obtained extraction recoveries did not significantly change for CHR and BaP (ANOVA test, 𝛼 = 5%), but was increased for DaiP. Therefore, for the rest of the development of the SPE step, a content of 50% of CH3CN was added in the percolation media.
3.2.2. Choice of the eluting solvent
Another solvent with a higher elution strength (𝜀0), CH3CN, was evaluated for the desorption of the retained PAHs on the cartridge. The experiments were performed with a spiked CH3CN:H2O (1:1, v/v) mixture solution and the same washing conditions described in Section 3.2.1 (n = 3). It was observed that CH3CN did not allow the elution of DaiP, and led to not significantly different extraction recoveries for CHR, and lower ones (47%) for BaP (ANOVA test, 𝛼 = 5%) compared to THF (68%). Therefore, THF was the best choice as eluting solvent and the results were in good agreements with the 𝜀0 values of CH3CN (𝜀0 = 3.1) and THF (𝜀0 = 3.7), which give a measure of their elution strength on a C18-based sorbent [35].
3.2.3. Comparison of three different SPE sorbents
In order to select a C18-based sorbent favoring the PAH extraction and leading to the best recoveries, three SPE cartridges containing 25 or 30 mg of C18-bonded silica from different suppliers that mainly differ by their carbon content were evaluated (see Section 2.3) by applying the optimized conditions. For these experiments, nine PAHs among the 24 were selected in order to cover the whole range of hydrophobicity, except the less hydrophobic compounds, NAPH and ACY, because of their higher LOQ (see Table 1). After conditioning the SPE sorbents, 200 μL of H2O:CH3CN (1:1, v/v) spiked with the nine PAHs were percolated. After washing and drying, PAHs were eluted with 300 μL of THF. Results are reported in Figure 2. High extraction recoveries between 65% and 98% were obtained. An ANOVA test (𝛼 = 5%) demonstrated that the recoveries obtained on the three sorbents for each PAH were not significantly different. The single exception was for 5MCHR, which has a significantly higher extraction recovery with the Hypersep cartridge. Therefore, the Hypersep cartridge was preferred for the end of the study.
Comparison of the extraction recoveries obtained using three different SPE sorbents. SPE procedure: percolation of 100 μL of CH3CN:H2O sample (1:1, v/v) spiked with 5 μg/L of each PAH, washing with 100 μL of CH3CN:H2O (1:1, v/v) and elution with 300 μL of THF (n = 3).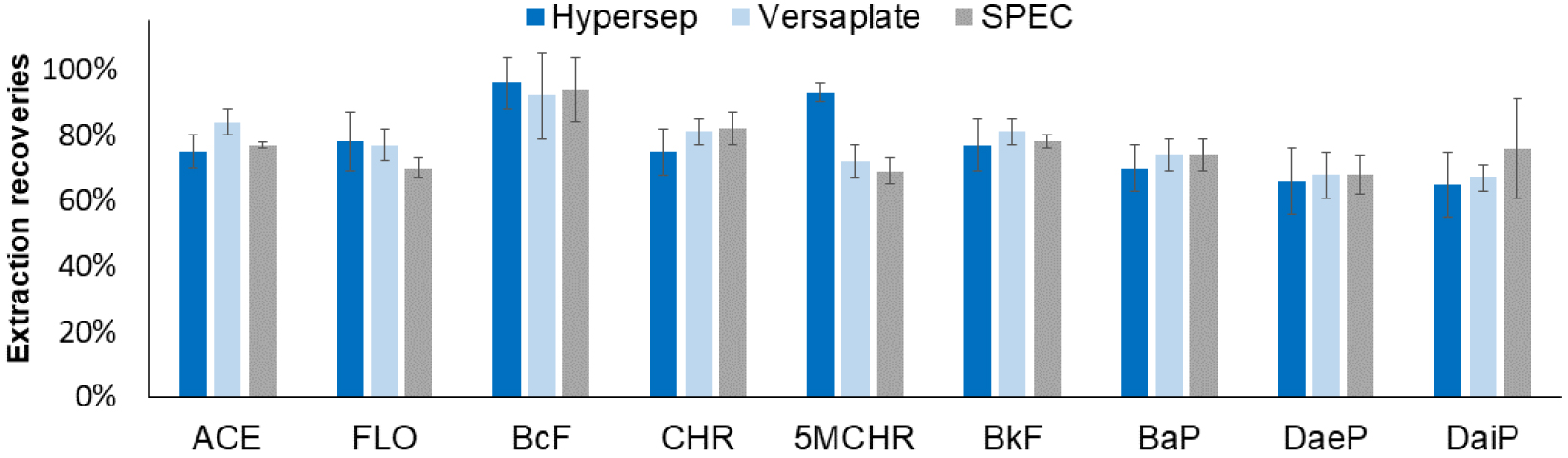
3.2.4. Study of the evaporation step
At this stage of the study, the SPE elution fraction had never been evaporated before its LC analysis. The introduction of an evaporation step was next evaluated to improve the enrichment factor and the method sensitivity, but this step can also induce some loss of the most volatile PAHs. To evaluate this risk, 200 μL of THF was spiked with 20 PAHs and was evaporated to dryness and further reconstituted in 50 μL of CH3CN before analysis. Results are reported in Figure 3. This evaporation step led to the partial loss of the five most volatile PAHs (NAPH, ACE, FLO, PHE, and ANT). Their recoveries ranged from 15% (NAPH) to 54% (ANT) with SD values up to 20%. For the other PAHs, the losses were lower and the recoveries were higher than 70%. To improve the recoveries of the most volatile PAHs, a small volume of an aromatic solvent having a low volatility and able to develop π–π interactions with PAHs was added in the extract before starting the evaporation and the evaporation was stopped before a complete dryness in order to trap all PAHs in this small residual volume of the aromatic solvent, this volume being controlled thanks to the shape of the used vials. For these experiments, three aromatic solvents were selected: toluene (boiling point (BP): 110.6 °C, vapor pressure (VP): 0.29 kPa), xylene (BP: 144.4 °C, VP: 0.8 kPa), and trimethylbenzene (BP: 175 °C, VP: 0.18 kPa). First, 10 μL of each aromatic solvent was added in 200 μL of THF spiked at 5 μg/L with each PAH. The evaporation was stopped before dryness (last drop) and the extract was diluted with 50 μL of CH3CN before analysis. Figure 3 presents the obtained results.
Effect of the nature and the volume of the aromatic solvent added in 200 μl of THF and spiked with 5 μg/L of each PAH on extraction recoveries. The extract was recovered in 50 μL of CH3CN before analysis (n = 3). * not quantifiable PAHs.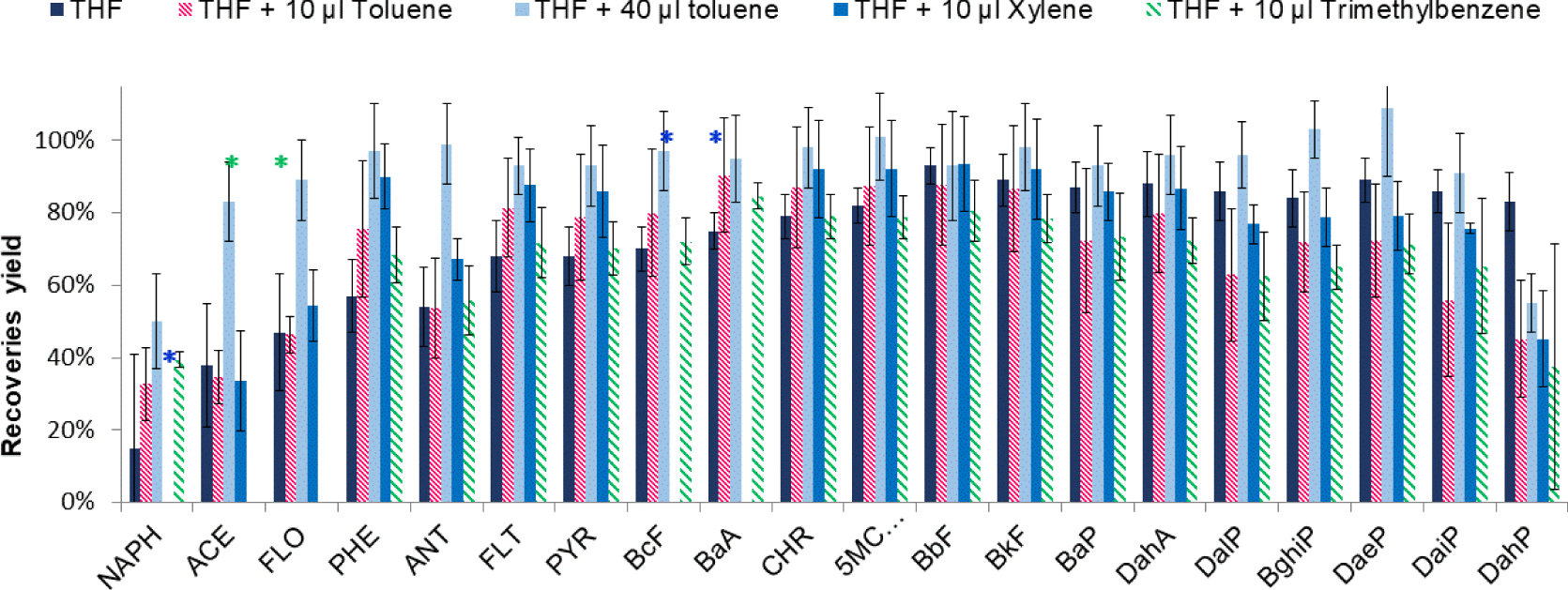
With xylene, the quantitation of NAPH, BcF, and BaA was not possible because of the presence of co-eluting peaks in the chromatogram due to xylene and its impurities. There was a similar problem with trimethylbenzene, which prevents from the quantitation of ACE and FLO. Therefore, toluene was selected. Nevertheless, 10 μL of toluene were not sufficient to ensure high recoveries for NAPH, ACE, FLO, and ANT. Then, the effect of the addition of 40 μL of toluene was evaluated. In that case, the recoveries for the five most volatile PAHs were improved: about 50% for NAPH, 60% for DahP, and above 80% for all the other PAHs. Only DahP has a lower recovery than without the addition of an aromatic solvent. The addition of 40 μL of toluene in the SPE extract before its evaporation was then selected to limit the loss of most of the PAHs.
Then, different volumes of CH3CN (50, 100, and 150 μL) were tested for the dilution of the extracts after evaporation and before injection in LC. According to the ANOVA test (𝛼 = 5%), the extraction yields were similar for all the tested volumes except for DahP (data not shown) for which a volume of 150 μL of CH3CN gave higher extraction recoveries (87%). Therefore, 150 μL of CH3CN was selected for the optimization of the next step.
3.2.5. Optimization of the elution volume
As described previously, two elution solvents were tested, CH3CN and THF, and THF was preferred. At this stage, 300 μL of THF were used for the elution but it is important to ensure that this volume is sufficient to elute the most hydrophobic PAHs from the SPE cartridge. This is why an increase in the elution volume was studied. Three volumes of THF were tested: 300, 400 and 500 μL and Figure 4 presents the obtained recoveries. An ANOVA test (𝛼 = 5%) demonstrated that they are not significantly different except for NAPH and IcdP. NAPH is the most polar and the most volatile PAH and its recovery decreases when the elution volume increases. This cannot be due to a problem of desorption from the SPE cartridge but rather to the subsequent evaporation step that takes a longer time, leading to an increasing lost of this volatile compound. For IcdP, there is a decrease in the recovery with the increase in the elution volume. This unexpected result for this highly hydrophobic compound may be also due to the evaporation step. Indeed, with a higher elution volume to evaporate, the compound may adsorb on the entire walls of the vials and may become more difficult to recover within a resuspension volume of 150 μL of CH3CN even after vortexing. This is why a volume of 300 μL of THF was finally selected.
Optimization of the SPE elution volume. SPE procedure (Hypersep cartridge): percolation of 200 μL of CH3CN:H2O (1:1, v/v) spiked with 5 μg/L of each PAH except ACY, CPcdP, BjF, and IcdP at 50 μg/L, washing with 100 μL of CH3CN:H2O (1:1, v/v), elution with THF, and partial evaporation (addition of 40 μL of toluene in the elution fraction). The extract was recovered with 150 μL of CH3CN before analysis (n = 3).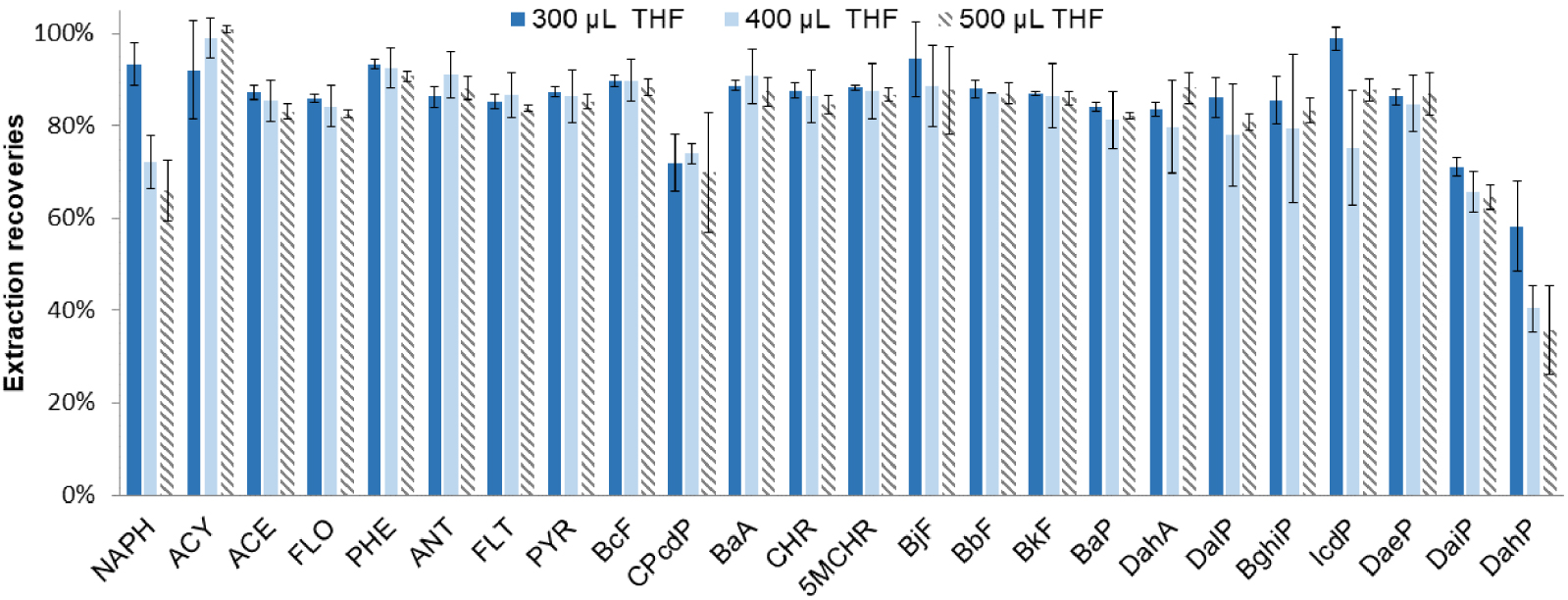
3.2.6. Improvement of the enrichment factor
At this stage of the study, almost all the parameters of the pretreatment steps were optimized except the sample volume and the final volume of solvent to recover compounds after the evaporation just before analysis. Both are key parameters for the recoveries and enrichment factors of the SPE procedure, that have to be as high as possible to detect the PAHs present at trace levels in real samples. The loading volume was first increased from 200 to 400 μL. The obtained extraction recoveries are presented in Figure 5. For the first four less hydrophobic PAHs, a dramatic decrease was observed. Indeed, a loading volume of 400 μL followed by a washing volume of 100 μL may exceed their breakthrough volumes. For the other PAHs, the obtained extraction recoveries were quite similar and SD values were between 2 and 17% for both conditions. As the aim of this study was to quantify the 24 listed PAHs at trace levels, a compromise had to be made and a loading volume of 400 μL was chosen to favor the enrichment factor.
Optimization of the SPE percolation volume. SPE procedure (Hypersep cartridge): percolation of 200 μL or 400 μL of CH3CN:H2O sample (1:1, v/v) spiked with 5 μg/L of each PAH except for ACY, CPcdP, BjF, and IcdP at 50 μg/L, washing with 100 μL of CH3CN:H2O (1:1, v/v), elution with 300 μL of THF and partial evaporation (addition of 40 μL of toluene in the elution fraction). The extract was recovered with 150 μL of CH3CN before analysis (n = 3).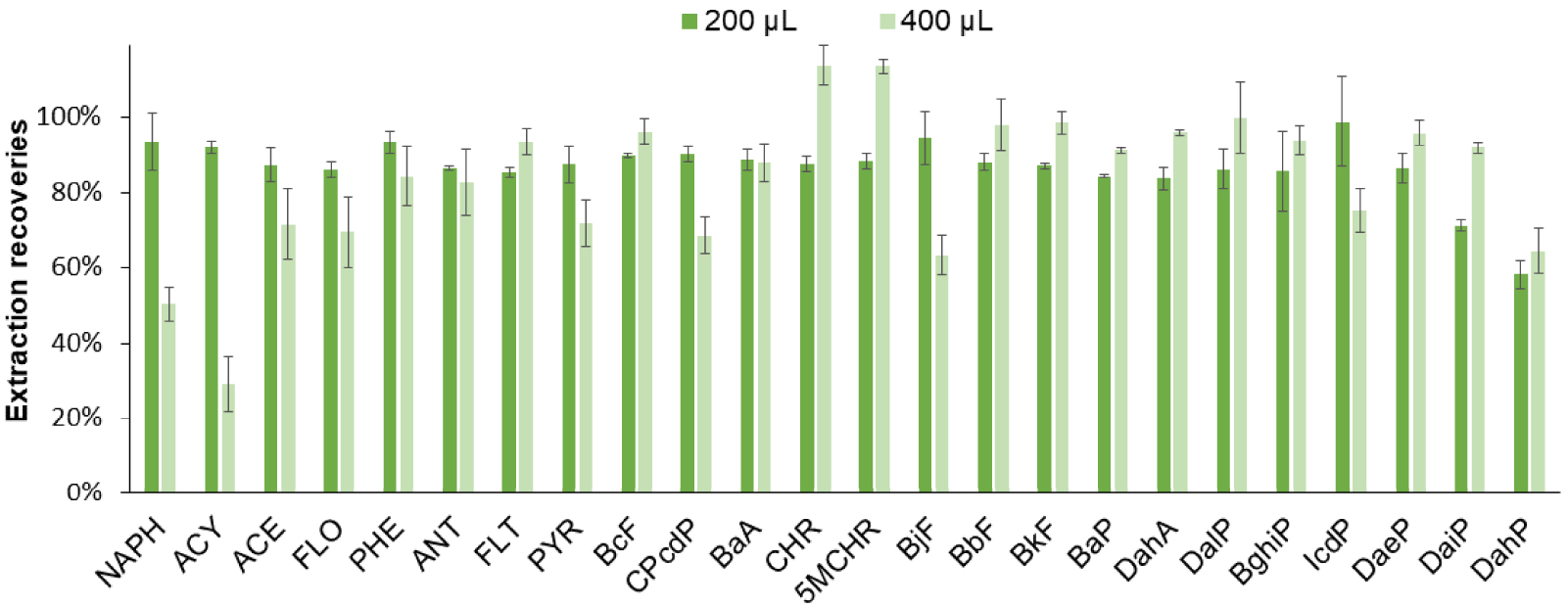
The volume needed for recovering all the PAHs after the evaporation and before analysis was finally studied. For this, three volumes of CH3CN were tested: 35, 100, and 150 μl. According to the results presented in Figure 6 and the ANOVA test (𝛼 = 5%), the extraction recoveries were not significantly different. To improve LOQs, the injection volume of 5 μL used for all the injection achieved for the development of the SPE procedure was increased to 10 μL, which prevents from the use of 35 μL as volume use to dilute the final extract after the evaporation step. Therefore, a volume of 100 μL was selected. However, an injection volume of 10 μL of PAHs in CH3CN induced a peak deformation for the first eluted PAHs. Therefore, the 100 μL of CH3CN was replaced by the addition of 50 μL of CH3CN for resuspension followed by 50 μL of H2O. Finally, the optimized SPE protocol with spiked pure media, at this stage, was: percolation of 400 μL of H2O:CH3CN (1:1, v/v), washing with 100 μL of H2O:CH3CN (1:1, v/v), drying, elution with 300 μL of THF, addition of 40 μL of toluene in the elution fraction, partial evaporation (last drop), and dilution of the extract with 100 μL of H2O:CH3CN (1:1, v/v) before analysis.
Optimization of the resuspension volume after evaporation before LC analysis. SPE procedure (Hypersep cartridge): percolation of 400 μL of CH3CN:H2O sample (1:1, v/v) spiked with 5 μg/L of each PAH except for ACY, CPcdP, BjF, and IcdP at 50 μg/L, washing with 100 μL of CH3CN:H2O (1:1, v/v), elution with 300 μL of THF, and partial evaporation (addition of 40 μL of toluene in the elution fraction). The extract was recovered with 35, 100 or 150 μL of CH3CN (n = 3).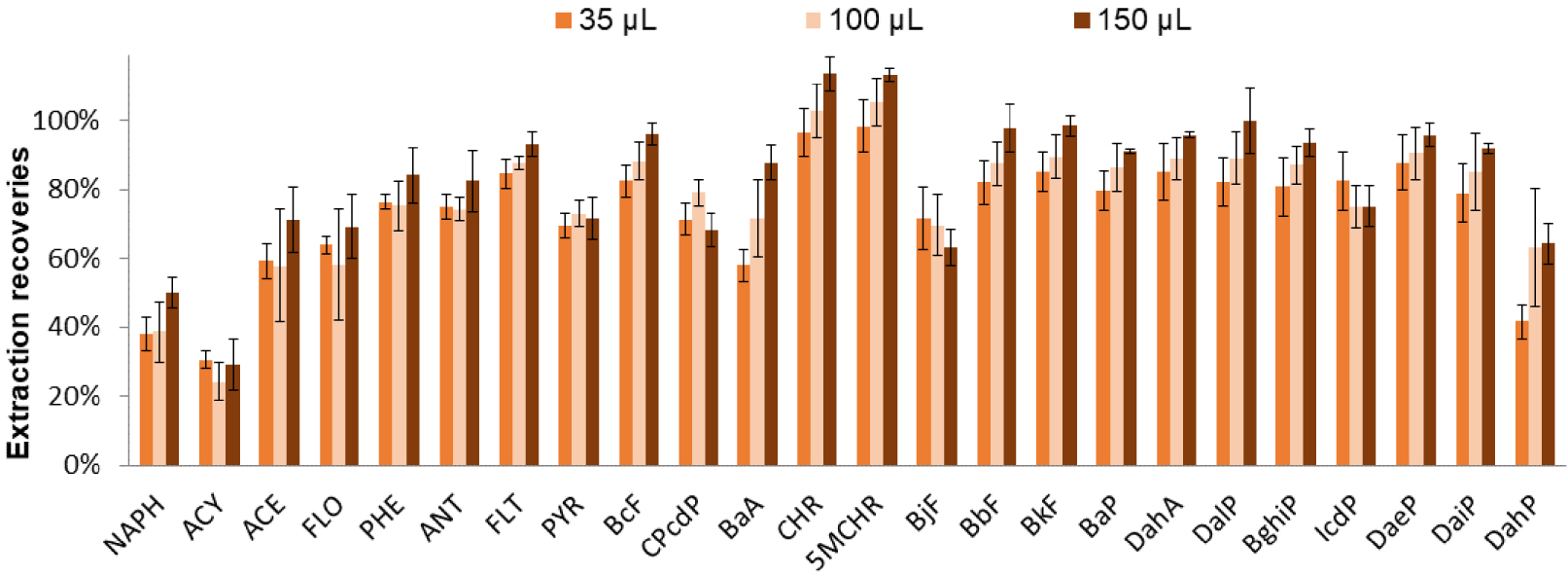
3.3. Optimization of the protein precipitation procedure by a DOE
Previous results were obtained for spiked pure media. Nevertheless, the analysis of real sera is targeted. Therefore, it is necessary to proceed first to the precipitation of the proteins contained in the sera minimizing PAHs losses that may occur due to their potential interactions with the serum proteins. The traditional approach involves the addition of an organic solvent, but also of a surfactant, such as SDS [36, 37, 38, 39]. The CH3CN percentage, SDS concentration, and temperature may affect these interactions. A DOE-based approach was selected to study the potential effects and interactions of these three factors and determine the optimum precipitation conditions.
According to literature and the conventional protocols used for protein precipitation, the low and high levels for the CH3CN content was fixed at 50 and 70% (v/v), respectively [36, 37], the three levels of SDS were fixed at 0, 0.1, and 0.2% (v/w) [38], and the precipitation was carried out at 25 °C (rt) for 2 min or at 60 °C during 25 min [39]. Table 2 presents the coded and non-coded values for each factor. The experiments were next carried out with 16 PAHs representative of the log P range of the 24 targeted ones (except for NAPH and ACY, the less hydrophobic compounds because of their higher LOQs) and, it is worthwhile to notice it, with a synthetic serum (Earle’s balanced salt solution containing albumin at 25 g/L). Indeed, to carry out this DOE with sera, 4.5 mL (300 μL × 15 experiments) would have been necessary whereas sera are highly precious samples.
2 × 2 × 3 factorial design with the different factors studied for their potential impact on the PAH recoveries during the protein precipitation (test with a synthetic serum: Earle’s balanced salt solution containing albumin at 25 g/L spiked with 16 PAHs at 4 μg/L each)
| Order | Coded values | Experimental values | |||||
|---|---|---|---|---|---|---|---|
| [CH3CN] | Precipitation conditions | [SDS] | [CH3CN] (%, v/v) | Precipitation conditions | [SDS] (%, w/v) | ||
| 1 | +1 | +1 | 3 | 70 | 25 min at 60 °C | 0.2 | |
| 2 | +1 | +1 | 1 | 70 | 25 min at 60 °C | 0 | |
| 3 | +1 | +1 | 1 | 70 | 25 min at 60 °C | 0 | |
| 4 | −1 | −1 | 2 | 50 | 2 min at 25 °C | 0.1 | |
| 5 | +1 | −1 | 2 | 70 | 2 min at 25 °C | 0.1 | |
| 6 | +1 | −1 | 3 | 70 | 2 min at 25 °C | 0.2 | |
| 7 | +1 | −1 | 3 | 70 | 2 min at 25 °C | 0.2 | |
| 8 | −1 | −1 | 3 | 50 | 2 min at 25 °C | 0.2 | |
| 9 | −1 | +1 | 1 | 50 | 25 min at 60 °C | 0 | |
| 10 | −1 | +1 | 1 | 50 | 25 min at 60 °C | 0 | |
| 11 | +1 | −1 | 1 | 70 | 2 min at 25 °C | 0 | |
| 12 | +1 | +1 | 2 | 70 | 25 min at 60 °C | 0.1 | |
| 13 | −1 | +1 | 2 | 50 | 25 min at 60 °C | 0.1 | |
| 14 | −1 | +1 | 3 | 50 | 25 min at 60 °C | 0.2 | |
| 15 | −1 | −1 | 1 | 50 | 2 min at 25 °C | 0 | |
The considered responses were the recoveries of the 16 PAHs and these data are reported in Table S1. A polynomial model was used to identify the factors impacting the PAH recoveries during the protein precipitation step. Using a classical least-squares multiple linear regression, the model coefficients were estimated to evaluate factor effects. Coefficient significance was evaluated using a t-test comparing the coefficient estimate to its standard-deviation obtained from the repeated experiments and the results are presented Figure S3. A factor or an interaction was considered as significant with a 5% risk of the first kind if the value of P > t was lower than 0.05. First, the change from 50 to 70% of CH3CN had no significant effect for all the studied PAHs except for FLO. It seems then that 50% of CH3CN are sufficient to disrupt the PAH–protein interactions. Similarly, the SDS content had no significant effect on the PAH recoveries. Considering temperature, a negative effect on PAH recoveries was observed for all the PAHs except DahP. Moreover, the interaction between SDS content and temperature had also a significant negative effect for 11 of the 16 studied PAHs. Finally, the interaction between the SDS content and CH3CN percentage had a negative significant effect for seven PAHs.
The Derringer’s desirability function was next used to determine the precipitation conditions giving rise to the highest PAH recoveries. Each response was transformed on a scale between 0 (corresponding to the most undesirable value) and 1 (representing the value that most fulfilled the requirements). The value of each transformed response was called “desirability value”. The global desirability was calculated by multiplying all the individual desirability values. Finally, this global desirability was maximized to determine the predicted optimum (see Figure S4): the coded values of the predicted optimum were (1, −1, 3), which correspond to 70% of CH3CN (v/v), 0.2% of SDS (w/v), and precipitation done at rt for 2 min.
This optimum corresponded to one of the experimental points tested during the DOE, for which it appeared that the obtained PAH recoveries are very close to the ones obtained with another experimental condition using the same content of CH3CN (70%) and no heating, but with only 0.1% of SDS. Therefore, both experimental conditions were tested again, but with the 24 PAHs at a spiking level of 10 μg/L. For the predicted optimum, extraction recoveries were between 28 and 82% with SD values lower than 16%, in agreement with the predicted recoveries, whereas for the other experimental condition, they were between 33% and 81% with SD values lower than 14%. An ANOVA test (𝛼 = 5%) demonstrated that the extraction recoveries were not significantly different for both conditions (data not shown). Therefore, the predicted optimum by the DOE was preferred.
3.4. Extraction of the PAHs from maternal and umbilical cord sera
After the optimization of the different steps of the sample handling with spiked pure media and a synthetic serum, the extraction efficiency of this procedure was evaluated in real biological fluids for the 24 PAHs by spiking pooled maternal and umbilical cord sera at 10 μg/L for each PAH except for CPcdP and IcdP at 20 μg/L. 100 μL of pooled maternal and umbilical cord sera were used for each experiment carried out in triplicate. The protein precipitation was made by adding 233 μL of CH3CN plus 0.2% SDS. 300 μL of the supernatant were taken and water was added to decrease the CH3CN proportion from 70 to 50%. 400 μL of this mixture were percolated through the SPE cartridge and were recovered after elution and evaporation in 100 μL of H2O:CH3CN (1:1, v/v) before analysis. Figure 7 shows the resulting chromatograms and Table 3 the calculated extraction recoveries. It is worthwhile to notice that the pooled sera were also analyzed without spiking to evaluate the potential presence of endogenous PAHs and correct the resulting extraction recoveries, but no PAHs were detected in this pooled sample.
LC/UV/FD chromatograms of the extracted pooled maternal (A) and umbilical cord (B) sera spiked with 24 PAHs at 10 μg/L except CPcdP and IcdP at 20 μg/L. Other conditions: see Section 2.2. 1, NAPH; 2, ACE; 3, FLO; 4, PHE; 5, ANT; 6, FLT; 7, PYR; 8, BcF; 9, BaA; 10, CHR; 11, 5MCHR; 12, BbF; 13, BkF; 14, BaP; 15, DahA; 16, DalP; 17, BghiP; 18, DaeP; 19, DaiP; 20, DahP.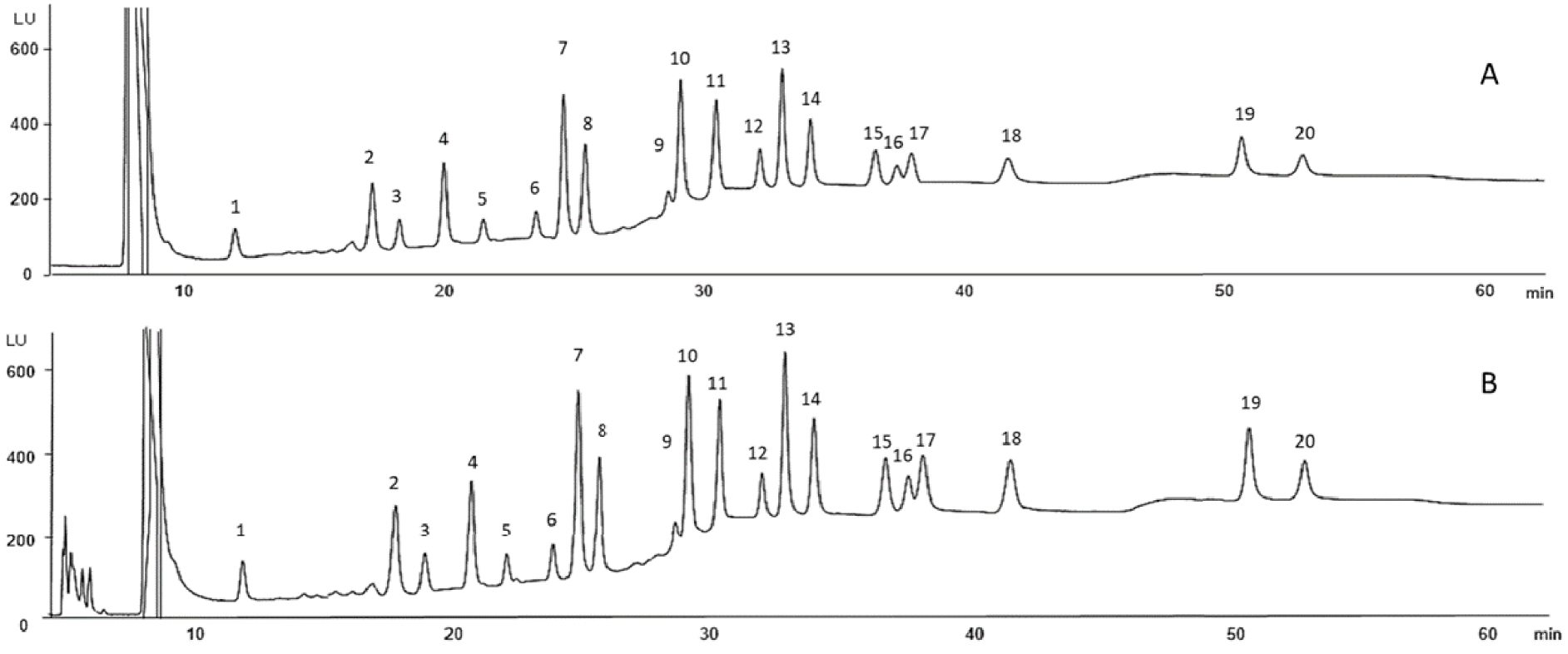
Extraction recoveries (R%) and SD (n = 3) of 24 PAHs in spiked CH3CN:H2O mixture, pooled maternal and umbilical cord sera
| PAHs | Spiked CH3CN:H2O (1:1, v/v) | Spiked maternal serum | Spiked umbilical cord serum | |||||
|---|---|---|---|---|---|---|---|---|
| R (%) | SD (%) | R (%) | SD (%) | R (%) | SD (%) | |||
| NAPH | 27 | 13 | 46 | 11 | 47 | 11 | ||
| ACY | 24 | 5 | 31 | 2 | 34 | 2 | ||
| ACE | 35 | 10 | 40 | 7 | 48 | 4 | ||
| FLO | 38 | 7 | 40 | 5 | 48 | 4 | ||
| PHE | 47 | 5 | 45 | 10 | 51 | 2 | ||
| ANT | 51 | 4 | 39 | 5 | 53 | 2 | ||
| FLT | 64 | 5 | 52 | 8 | 62 | 5 | ||
| PYR | 66 | 9 | 57 | 6 | 68 | 9 | ||
| BcF | 62 | 7 | 53 | 5 | 64 | 4 | ||
| CPcdP | 64 | 6 | NQ | - | NQ | - | ||
| BaA | 55 | 5 | 51 | 4 | 68 | 2 | ||
| CHR | 67 | 7 | 55 | 3 | 69 | 3 | ||
| 5MCHR | 71 | 8 | 51 | 6 | 64 | 4 | ||
| BjF | 71 | 2 | 41 | 6 | 60 | 2 | ||
| BbF | 68 | 7 | 51 | 3 | 69 | 5 | ||
| BkF | 69 | 8 | 51 | 4 | 67 | 4 | ||
| BaP | 68 | 8 | 48 | 3 | 65 | 4 | ||
| DahA | 74 | 9 | 46 | 2 | 68 | 7 | ||
| DalP | 70 | 6 | 33 | 1 | 66 | 10 | ||
| BghiP | 82 | 8 | 40 | 1 | 59 | 3 | ||
| IcdP | 75 | 6 | NQ | - | NQ | - | ||
| DaeP | 77 | 9 | 37 | 2 | 61 | 4 | ||
| DaiP | 72 | 8 | 28 | 1 | 54 | 5 | ||
| DahP | 67 | 8 | 27 | 2 | 47 | 3 | ||
Precipitation step: 0.2% of SDS and 70% of CH3CN. SPE (Hypersep cartridge): percolation of 400 μL of spiked sample with 10 μg/L of each PAH except for CPcdP, BjF and IcdP at 20 μg/L, washing with 100 μL of CH3CN:H2O (1:1, v/v), elution with 300 μL of THF and partial evaporation (addition of 40 μL of toluene in the elution fraction). The extract was further recovered with 100 μL of CH3CN:H2O (1:1, v/v). NQ: Non-quantifiable.
Extraction recoveries were between 27 and 57% for maternal sera and between 34 and 69% for umbilical cord sera, with SD values lower than 11% thus highlighting the reliability of the developed method applied to complex samples. These recoveries are quite inferior to the ones obtained with spiked pure media (between 24 and 82%) (Table 3), but for which no precipitation was involved. This may also result from a matrix effect. Moreover, the extraction recoveries for maternal sera are lower than those obtained with umbilical cord sera, especially for BaA to DahP. Therefore, the matrix effect seems to be different for both kinds of sera. Unfortunately, it was not possible to quantitate CPcdP and IcdP, because some impurities co-eluted with them. This is why it is possible to conclude that the developed whole sample pretreatment led to good extraction recoveries for 22 of the 24 listed PAHs. These extraction recoveries are inferior to the values given in literature [6, 19, 20, 24], ranging from 77 to 122%, but it is worthwhile to notice that here 22 compounds with a wide range of log P were targeted against only between five and 16 US-EPA PAHs in literature.
The whole sample pretreatment led to ratio between the sample volume and the final extract volume of 0.95. Considering the extraction recoveries, enrichment factors ranged from 0.3 to 0.5 and from 0.3 to 0.7 for maternal and umbilical cord sera, respectively. This led to LOQs in sera between 0.2 and 3.1 μg/L for the 20 fluorescent PAHs and between 7.0 and 14.5 μg/L for the two others PAHs (calculation for a signal to noise ratio equal to 10), which are close to the expected concentrations in real samples [6, 14, 21, 27, 28, 29, 30, 31]. In literature, LOQ values between 0.01 and 2.5 μg/L were reached, but requiring a serum volume at least 10 times larger, i.e. 1 or 2 mL of serum [6, 20, 21, 24].
The optimized sample handling, separation, and detection steps require a total analysis time of 1 h 40 and the SPE and analytical steps can be automated. This constitutes also a significant improvement over the literature where serum sample handling always required at least three LLE steps followed most often by an additional SPE step [6, 19, 20, 21, 24, 25]. After validation, it should allow the analysis of a large number of real samples.
4. Conclusions
The analysis of the 24 listed PAHs by LC-DAD/FD was developed. Then, different parameters governing the SPE procedure were studied to optimize the extraction recoveries on C18 silica of the 24 PAHs belonging to a large range polarity (lop P values between 3.3 and 7.7). The use of an aromatic solvent was proposed to minimize the loss of the most volatile PAHs during the evaporation step. A DOE was used to optimize the protein precipitation conditions. The performances of the whole sample pretreatment were determined for 22 PAHs and for a reduced volume of 100 μL of maternal and umbilical cord sera. The LOQs obtained for both matrices were between 0.2 and 3.1 μg/L for the fluorescent PAHs and between 7.0 and 14.5 μg/L for ACY and BjF (UV detection). The optimized sample handling, separation, and detection steps require a total analysis time of only 1 h 40 and the SPE and analytical steps can be automated. The next step will consist in validating this method, which will allow its use for the analysis of numerous real samples. The consistency of the resulting data will enable to carry out epidemiologic studies.
Abbreviations
ACE, acenaphthene; ACY, acenaphthylene; ANT, anthracene; BaA, benzo[a]anthracene; BaP, benzo[a]pyrene; BbF, benzo[b]fluoranthene; BcF, benzo[c]fluorene; BghiP, benzo[g,h,i]pyrene; BjF, benzo[j]fluorene; BkF, benzo[k]fluoranthene; CHR, chrysene; CPcdP, cyclopenta[cd]pyrene; DaeP, dibenzo[a,e]pyrene; DahA, dibenzo[a,h]anthracene; DahP, dibenzo[a,h]pyrene; DaiP, dibenzo[a,i]pyrene; DalP, dibenzo[a,l]pyrene; FLO, fluorene; FLT, fluoranthene; IcdP, indeno[1,2,3-cd]pyrene; 5MCHR, 5-methylchrysene; NAPH, naphthalene; PHE, phenanthrene; PYR, pyrene; SDS, sodium dodecyl sulfate.
Declaration of interests
The authors do not work for advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.