



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Biomedical applications of X-rays were and still are closely linked to cancer imaging and therapy. Radiotherapy (RT) is an extremely important first-line treatment for cancer therapy which, unlike other treatments such as chemotherapy or immunotherapy which affect the whole body, is generally a local treatment. Despite the spectacular progress made in the field of X-ray sensitization and improvement, radiotherapy as a monotherapy generally fails to eliminate the entire tumor, as cancer cells can undergo DNA repair and regrow [1, 2]. For this reason, current research is gradually shifting from monotherapy to multimodal X-ray therapy, combining two or more therapeutic modalities, producing synchronous/synergistic anticancer effects [3, 4]. Increasing synergistic effect also optimizes several shortcomings of drug-based therapies by improving systemic effect, nonspecific biodistribution, poor bioavailability and limited circulation time, which can lead to severe side effects and low therapeutic index. Chemoradiotherapy has been widely used clinically, offering substantial advantages in terms of treatment and prognosis. Research on the simultaneous administration of drugs and the application of therapeutic beams dates back several decades [5, 6] and is used to treat certain stages of head and neck cancer, lung cancer, and gynecological cancers. Now the field has reached a next-level maturity to experiment with locally activable prodrugs and drug carriers, thus limiting the general chemotoxicity and efficiency of the treatment. Stimuli-responsive biodegradable nanoagents, which can not only deliver and control chemotherapies, but also attenuate hypoxia, or play as antimitotic agent to enhance chemoradiotherapy, have enormous potential. Key to the use of these smart materials is the presence of an interplay that is able to switch from one state to another, triggering the release of the covalently or non-covalently attached compound. Intelligent drug delivery systems can be designed around chemical functions or fragments of molecules that modify their structural or functional properties in engineered materials in response to specific external or internal factors [7, 8]. These dynamic properties can be activated by chemical/biochemical, electrical, magnetic, electromagnetic/optical, ultrasonic, dielectric, redox or piezoelectric signals, some of internal (biological) origin, others of external origin. These interplay units, which are small chemical compounds, may undergo isomerization, rearrangement or cleavage of chemical bonds, such as UV-sensitive azobenzene and o-nitrobenzyl derivatives, or coumarin esters, leading to direct or indirect drug release. Some of these molecules were historically developed for neuroscience where these “caged” compounds revolutionized functional neurophysiology from the late 1970s. They are now commonly used to design prodrugs and light-controlled drug delivery systems, although their application in therapy is limited [9, 10, 11]. Light activation has inherent shortcomings. While the tissue penetration capacity of visible and near-infrared (NIR) light is superior to the more often used UV activation, their penetration in deep tissues does not extend beyond a few hundred micrometers using intensities below cell toxicity. The main obstacles are strong absorption and diffraction in turbid tissues. Fortunately, longer and especially shorter wavelengths than UV/Vis/NIR penetrate tissues better (Figure 1). As longer waves access vibrational and translational excitations, thus have not enough energy to break a chemical bond, shorter waves can provide sufficient energy to induce structural changes at the molecular level. As organic compounds have no electronic transitions in this energy window, they are transparent. Several ingenious strategies have recently been developed to translate short-wave photons into a trigger signal applicable to organic compounds that will be discussed in this review [12, 13, 14, 15, 16, 17, 18, 19]. This strategy also opens up new perspectives for personalized medical care, bringing the dream of tailor-made medical treatments for individuals closer to reality.
Electromagnetic map and modeled soft-tissue penetration capacity (calculated values).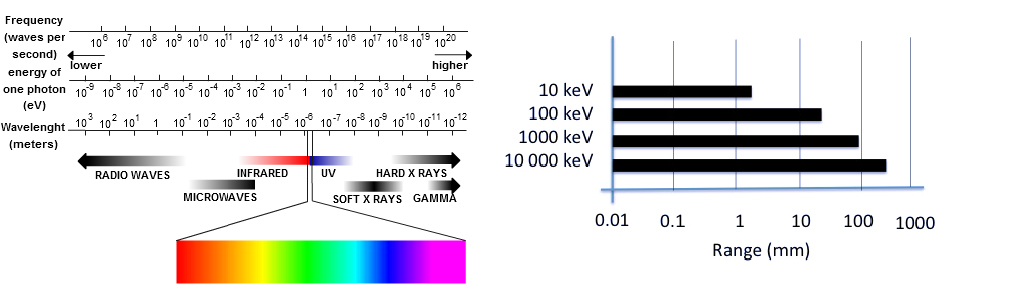
1.1. On the nature of X- and γ-rays
Although X- and γ-rays are both electromagnetic radiation (photons), there is no consensus on how to precisely differentiate between them. They differ mainly in their origin: γ-rays are produced during the nuclear decay of atomic nuclei from the atomic core and particle annihilation, while X-rays are produced when the velocity of electrons (or other charged particles) slows down, and the energy difference between these two kinetic states is dispersed as high-energy photons by Bremsstrahlung and characteristic radiation [20, 21]. X-rays are generally generated in a vacuum tube, where electrons emitted by the cathode, accelerated under high voltage, collide with a tungsten metal anode. X- and γ-rays are ionizing radiations but less so than alpha and beta particles. The energy ranges of X- and γ-rays overlap considerably, with X-rays essentially covering the “lower” part of the spectrum with energies above a few tens of eV, typically between 120 eV and 120 keV (thus the shortest ultraviolet spectrum) and γ-ray energies extending up to a few tens of MeV. In other words, X-rays (generally) have longer wavelengths than γ-rays, mainly ranging from 0.01 to 10 nm. Both γ- and X-rays have a high penetration power in soft tissue, whose extent depends on their respective wavelengths. Penetration is greater at higher energies but also depends on the tissue type. The impact of tissue thickness on X-ray penetration is dramatic. Small increases in tissue thickness led to large attenuations in the intensity of the incident beam. Because of the critical importance of penetration into (biological) media, highly accurate transport equations were developed from the early 1950s and are currently used to estimate the exposure of biological tissues to radiation [22]. Indeed, an utmost important value in biomedical applications is the dose absorbed by the living body. The gray (symbol: Gy) is the unit of ionizing radiation dose that measures the energy deposited/absorbed (in joule per kilogram of matter).
Radiotherapy is used in cancer therapy as ionizing radiation can break down the DNA of cancer cells more effectively than that of healthy cells. This is made possible by the natural repair mechanism, which is more effective for normal cells than for cancer cells, which function in an intrinsically dysfunctional way. The upper limit of a “safe dose” has been defined as a total dose of 55–60 Gy administered to a focal field with fractions of 1.8–2 Gy per day. This means that the total irradiation dose is delivered in small pieces to be gentler on normal tissues and to allow normal cells to repair DNA damage after each treatment. The radiation threshold depends sensitively on the tissue/organ/type of cancer/general condition of the patient and is assessed individually for each patient. An example of a dose-response relationship is illustrated in the treatment of stage III non-small cell lung cancer, where the administration of a total dose of 40 Gy is applied in fractionated treatment (4 Gy/day for five days, followed by a two-week break and a further dose of 4 Gy/day for five days), or the administration of a total dose of 40 Gy, 50 Gy or 60 Gy is applied in continuous treatment (2 Gy/day) [23]. For this reason, drug delivery probes for simultaneous activation must meet the threshold of 2 to 4 Gy, meaning that they must release the maximum payload within this dose for biomedical use.
How can this high-energy radiation be used for prodrug activation and for drug delivery? Three conceptually different approaches have been developed, based either (i) on transformations mediated by energy or by an electron transfer-promoted change in the electronic structure (thus in the stability) of the carrier; (ii) on the incorporation of ROS-sensitive functions into the drug delivery matrix that react chemically and undergo fragmentation; (iii) on the upregulation of enzymes that have recently been explored for selective prodrug activation, but the scope of such activation is beyond the scope of this study [24].
1.2. Drug release triggered by energy- or electron transfer-mediated structural changes
Organic probes are transparent to X-rays, as organic materials do not exhibit absorption transitions in this high-energy spectrum. Consequently, irradiation can be used for sterilization to eliminate microbial growth and prepare pyrogen-free samples without altering the structure of the biomedical sample or equipment [25, 26]. To be able to explore X/γ-rays as a trigger, an intermediate transduction mechanism is needed, which bridges the gap between high-energy electromagnetic radiation and what organic compounds can “understand” to react. Fortunately, there are a few such interactions. Noteworthily not necessarily the “simplest” solutions are the most effective. For example, the light produced by Cerenkov radiation, which is emitted when a charged particle passes through a dielectric medium at a speed greater than the phase velocity of light in that medium, would be useful as a local light trigger signal if the intensity of the UV light generated by this phenomenon could be increased [27]. Similarly, parametric down-conversion (an instantaneous nonlinear optical process that converts a higher-energy photon into a lower-energy photon pair, in accordance with the law of conservation of energy and the law of conservation of momentum) would be an interesting solution, although it cannot yet be used due to the lack of efficient generation of idler photons at optical wavelengths [28]. The conversion of X-rays to UV can be achieved more easily using scintillators (see above) [29, 30]. These materials play an important role in X-ray-induced photodynamic therapy (PDT), enabling low doses of radiation to be applied, as do a variety of well-designed irradiation modes and intelligent strategies for modifying the tumor microenvironment [31]. This activation modality generates 1O2 and ROS similarly to conventional photosensitizers in PDT under ultraviolet (UV) or visible light (scintillator-derived strategies will be discussed in the paragraph on redox/electron transfer reactions).
The most frequently used electron/energy transfer-related activation strategies are based on inelastic scattering, which involves a loss of energy from the incident primary photon: the shorter wavelength primary photon is transformed to a longer wavelength secondary photon. Inelastic scattering occurs via a number of mechanisms. The energy is transferred to the sample, generating a series of useful signals that can be exploited in a number of analytical methods and chemical transformations. The interaction of X- and γ-rays with matter is described by the photoelectric effect, Compton scattering, and pair production. The photoelectric effect is a phenomenon whereby electrically charged particles are released from or within a material when it absorbs electromagnetic radiation (Figure 2). The effect is often defined as the ejection of electrons from an atom when light falls on it, and called the Auger–Meitner effect at higher frequencies, such as X- and γ-rays. The effect consists in the occupation of a cavity in the inner shell of an atom, accompanied by the emission of an electron from the same atom. When an electron is removed from the atom, leaving a vacancy, an electron of a higher energy level can fall into the vacancy, resulting in a release of energy (Figure 2). Although this energy is most often released in the form of an emitted photon, it can also be transferred to another electron, which is ejected from the atom; this second ejected electron is called an Auger electron. Since an electron vacancy initially produced by the incident photon leads to two new electron vacancies, which in turn can produce the same number, a complete cascade of electrons can be produced according to tabulated probabilities. The atom is finally left in a state of multiple ionizations. The Auger effect (non-radiative relaxation) is the predominant mode of relaxation in light target atoms such as C, N, O (Z < 20) or for L-layer ionizations.
The photoelectric effect is the consequence of inelastic scattering between the photon and the electron, which generates a non-radiative relaxation (Auger effect) and a radiative relaxation in the target atom.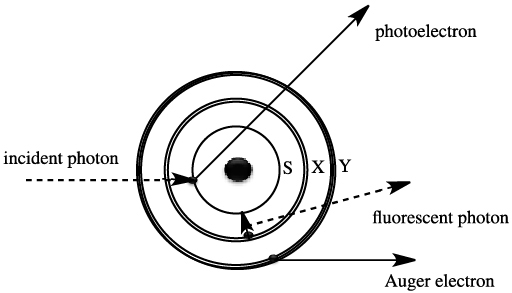
The radiative mode corresponds to the emission of fluorescence radiation, whose spectrum of discontinuous lines is characteristic of the material. In heavy materials (Z > 50) and for K-layer vacancies, fluorescence is the main relaxation mode.
Compton scattering results from the collision of a photon with an electron at rest: this is the combination of an incident X-ray photon and a weakly bound electron in the atom, whose binding energy is negligible compared to the incident energy. During this interaction, the incident X-ray photon disappears: part of its energy is given to the peripheral electron, which is then ejected: this is known as a Compton electron; if the other part of its energy is transmitted in the form of a new X-ray photon, this is known as a scattered or recoil photon. At the energies used in radiology, the Compton effect is favored in low-density, water-equivalent (low Z) media (fat, soft tissue, etc.). The Compton effect also dominates the interaction processes of photons with energies between 0.2 and a few MeV, whatever the atomic number of the medium through which they pass. The probability of occurrence of these scatterings decreases as the energy of the incident photon increases. The photoelectric effect and Compton scattering can be enhanced by the presence of a captive sensor and can also be exploited by Förster-type energy transfer, which can take place between the sensor (often derived from a metal) and the organic effector, resulting in a change in the electronic configuration of the organic probe.
Pair production only occurs at high energies and is statistically rare under normal biomedical conditions. Through this effect, the γ-ray is transformed into matter in the form of a pair of negatively and positively charged particles (electron and positron). Since an electron has a rest mass equivalent to 0.511 MeV of energy, a minimum γ energy of 1.02 MeV is required to produce this pair. Any excess energy of the γ-ray producing the pair is given to the electron–positron pair in the form of kinetic energy. In most cases, the positron will undergo annihilation by reaction with an electron in the detector material, creating two γ photons of 0.511 MeV each.
Photoelectric absorption dominates at low energies, followed by Compton scattering, then pair production as energy increases. Absorption of very high-energy photons leads to nuclear decay. The above processes (with the exception of photodisintegration) all result in the production of electrons (or their antimatter equivalent, positrons) and lower-energy X-rays, which undergo further absorption and scattering. In addition, these heavy nanoparticles can selectively scatter and/or absorb high-energy radiation [32, 33, 34], leading to energy transfer to photosensitizers, often porphyrin derivatives, metal complexes, scintillators or quantum dots that are sufficiently stable under radiative conditions [35].
1.3. Chemical transformation of ROS-sensitive functions
Materials sensitive to reactive oxygen species (ROS) can form covalent bonds with the transiently generated intermediates formed by the ionizing radiation. This can be facilitated by the presence of electron donor shuttles, or simply the presence of large quantities of water, ubiquitous in living organisms, which produce a large variety of reactive intermediates by irradiation [36]. When exposed to light or ionizing radiation, water undergoes a series of transformations involving electronic excitations, ionization of solvated species and the formation of free radicals and solvated electrons [37, 38, 39, 40, 41, 42]. The radiolysis of water generates thus various reactive particles, in which the ∙OH and the e are the dominant products. These processes occur, for example, in (photo)electrochemical cells [43, 44], biological molecules [45], as well as in atmospheric water [46]. Many of these are governed, at least in part, by the nature of electron transfer and binding energies in aqueous solutions. Among the many reactive species formed during this reaction are hydrated electrons and ROS [47] such as peroxides, singlet oxygen and oxyradicals, which are considered the chemically active component of radiation therapy. The HO∙ radical (hydroxyl radical) is the most abundant radical, which can then react with surrounding tissues to create organic radicals (usually alkyl radicals). Historically, iodine and gold-based nanoparticles (AuNPs) were the first radiosensitizers due to their high X-ray absorption, synthetic versatility and unique chemical, electronic and optical properties [48]. The radiosensitization dates back to the pioneering work of Hainfeld, who demonstrated that gold nanoparticles behave as effective radioactive agents, since tumor-bearing animals survive longer when treated with radiotherapy after intravenous injection of ultra-small gold nanoparticles [49]. The energy of the initial photon is thus transferred to electrons, which create ionization that produces in fine ROS in the presence of water and oxygen, leading to significant chemical and biological effects such as DNA degradation. The range of radiosensitizers now extends to other heavy elements with high atomic numbers, such as hafnium, gadolinium or bismuth atoms/nanoparticles [34, 50, 51, 52, 53] which are well-known radiosensitizers, capable of amplifying radiation doses in tumor tissue [54, 55, 56].
As mentioned in the previous paragraph, γ/X-ray activation enables “second-generation” photodynamic therapy (PDT), which relies essentially on ROS generation and is driven by the activation of a photosensitizer (PS), followed by the transfer of excitation energy to molecular oxygen to result in the generation of cytotoxic ROS. Noteworthy, the metal–photosensitizer combination is superior to the use of metal particles alone for ROS generation in terms of biological efficacy, and has a wider reach than the more conventional UV- and IR-triggered ROS generation, as it overcomes the penetration limits of UV or IR into tissue [16, 57, 58].
Fragmentation of ROS-sensitive bonds, such as sulfide/disulfide or selenide/diselenide bonds, or other chemical functions, as well as changes in the mechanical properties (rigidity) of the nanomaterial under the influence of radiation, can lead to the release of the sequestered ligand.
2. Activation of prodrugs or nanocarriers by electron or energy transfer
The prodrug strategy that was developed steadily in the second half of the last century to improve how the drug is absorbed, distributed, metabolized, and excreted in the living body involves masking one of the key parts (usually polar functions) of a biologically active compound, necessary for the activity, and restoring it when the masking group is cleaved [59, 60, 61]. This strategy was also developed in neurosciences, physiology, and molecular biology by using light as trigger signal, making it possible to control drug activation in space and time. By analogy with light, tissue-penetrating X-ray can be used under particular conditions.
2.1. Electroreduction of indolequinones
Historically, the first report on X- and γ-ray molecular fragmentation by electron transfer was reported by the Tanabe group using tethered 5-fluorouracil (5-FU) and 5-fluorodeoxyuridine (5-FdUrd) with 2-oxoalkyl groups covalently linked to radiation-sensitive quinone derivatives enabling the release of the antitumor agents 5-FU and 5-FdUrd, respectively (Scheme 1) [62, 63].
Activation of indolequinone-tethered 5-fluorodeoxyuridine by electron transfer and subsequent drug release.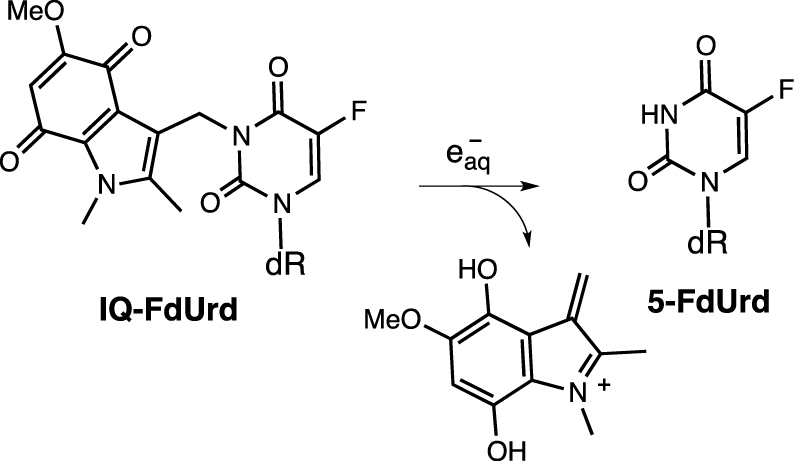
Mechanistic studies have shown that these prodrugs undergo reduction by capturing hydrated electrons (e) generated by the radiolysis of water. Optimization of the anchoring group structure has yielded indolequinones that can release the covalently attached ligand by bioreduction or under radiolytic conditions [64, 65, 66, 67]. This transformation was presented as the first prototype transformation for the release of complex structures from encapsulated compounds sensitive to irradiation [68].
2.2. Intramolecular electron or energy transfer from heavy elements
The Gd-sensitized aminoquinolone probe was found to undergo radiolytic fragmentation with a covalently attached model dihydrocinnamate as substrate [4, 69]. Although the precise mechanism of the fragmentation has not been established, electron transfer or electronic coupling between the irradiated Gd complex and the quinolinium probe has been postulated. Radiolysis of the covalently attached probe by 17.5 keV and 1.17 MeV X-rays resulted in the liberation of the dihydrocinnamate model (Scheme 2).
Radiolysis of gadolinium-bound quinoline cage with release of dihydrocinnamate ligand.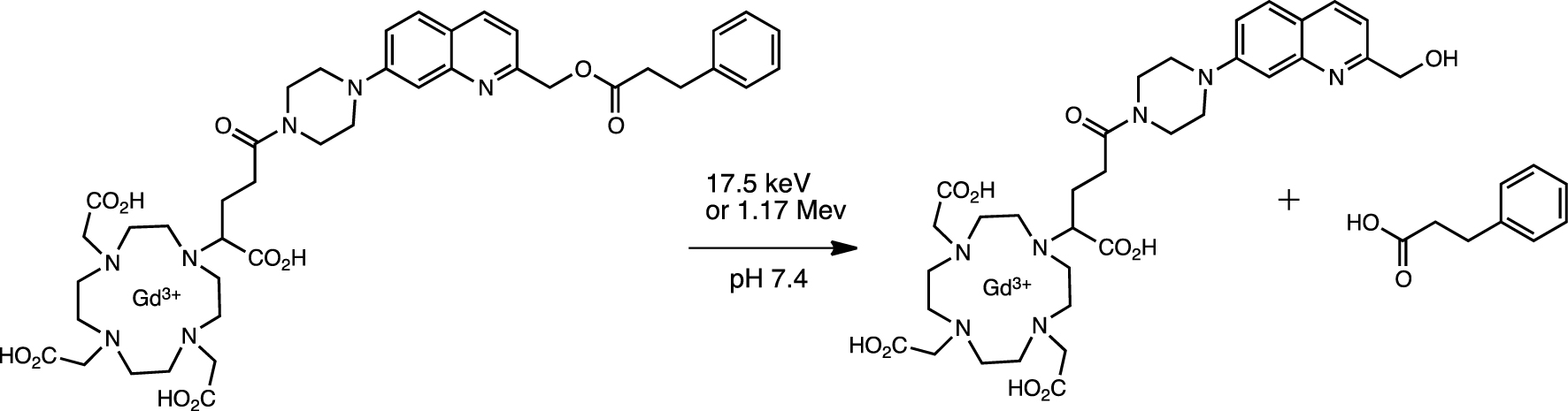
In a somewhat analog entry, a Gd chelate and azobenzene were combined in a radio-switch (Scheme 3) [70]. Azobenzene was isomerized by low-dose X-rays, offering the possibility of implementing trigger systems based on radiation penetrating the isomerization.
Activation of cis-GdAzo to trans-GdAzo using a 1 keV source.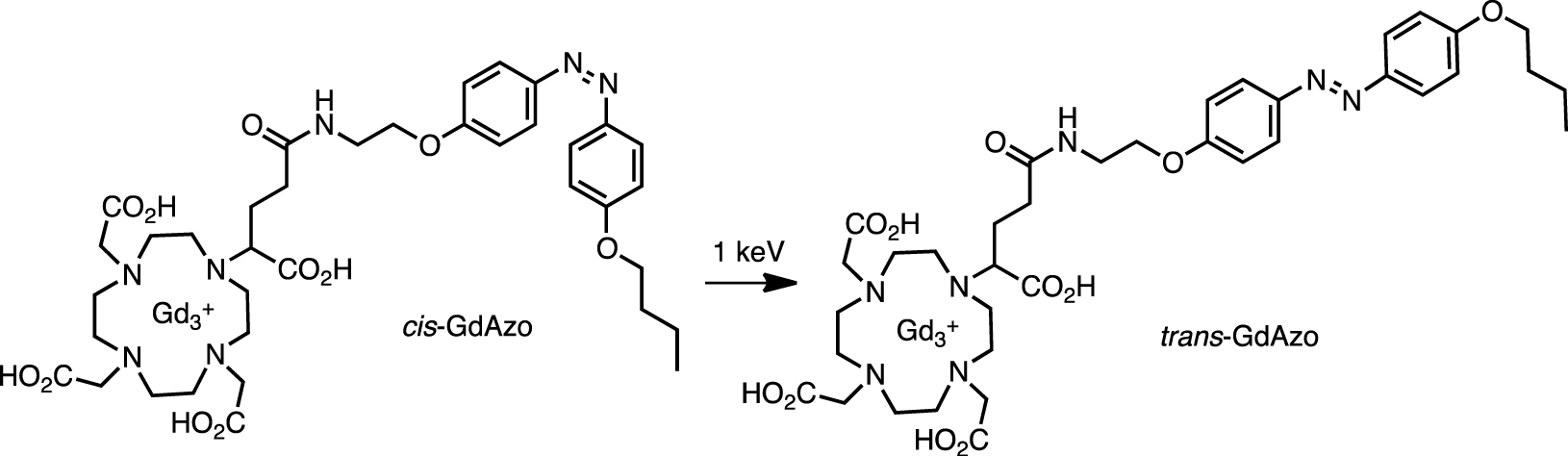
The activation efficiency (i.e., part of the isomerized (trans) compound compared to the starting (cis) material) was 33% and 69% at irradiation doses of 2 and 20 Gy, respectively, by using 1 keV source (∼1.24 nm). No compounds other than trans-GdAzo were observed spectrophotometrically. This molecular activation based on a radio-switch seems promising and opens the way to new types of applications such as actionable tools in radiotherapy.
Solvated electrons and transiently formed radical species generated by X/γ-rays have been exploited in the fragmentation of sensitive redox probes derived from picolinium and immobilized on the surface of ultra-small superparamagnetic iron oxide nanoparticles (USPIO) [71]. The iron-derived nanoparticle enabled Auger/Compton electron generation upon X-ray or γ irradiation, as well as monitoring of probe biodistribution by T2-weighted MRI and by the appearance of fluorescence from the released probe (Scheme 4). The functionalized particles showed high colloidal stability at physiological pH. The picolinium group effectively quenched the fluorescence of the covalently attached pyrene reporter in the starting material, probably via Förster-type quenching and dark complex formation. Probe activation was validated under pulsed radiation and also by using a conventional Cs-137 source. Although fragmentation was observed under both conditions, pulsed sources proved considerably more effective: 82% of the fluorescence of the pyrene reporter was recovered upon 30 Gy irradiation, compared with 27% of the fluorescence under conventional conditions.
Activation of pyrenebutyric acid immobilized by USPIO by X- or γ-ray irradiation.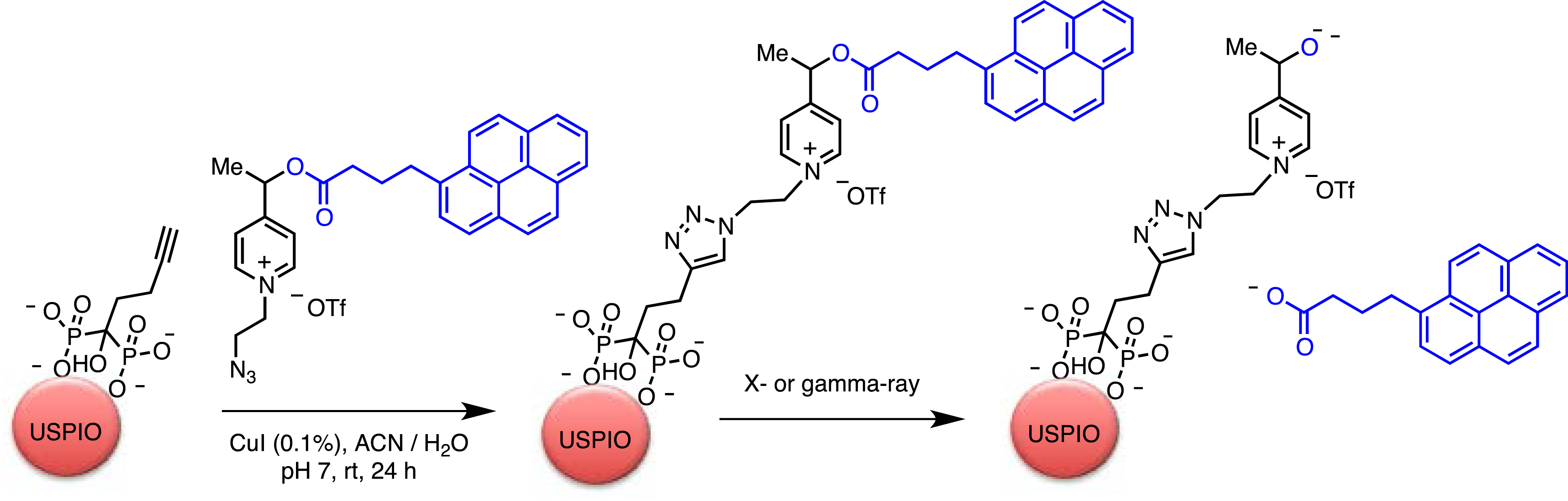
Several redox-sensitive compounds derived from picoline, quinolinine and picolinium were examined for X-ray activation [72]. 4-Hydroxymethylene pyridinium was selected as the most efficient probes for releasing client molecules in a dose gradient ranging from 0 to 60 Gy (Scheme 5). The cytotoxic payload (monomethyl auristatin E (MMAE)) was attached via a carbamate linker to the probe. A humanized antibody (sibrotuzumab) was attached to the release complex for better targeting (NAPC-ADC) (Scheme 5). The therapeutic efficacy of the radiation-induced activation was then assessed in vivo. Mice were irradiated with 3 × 4 Gy of X-rays on days 2, 4 and 6 post-injection. It should be noted that the overall 12 Gy of X-ray dose was supposed to trigger the complete release of MMAE. Tumor growth inhibition in the NAPC-ADC + X-ray group was better: mean tumor size was around 85 ± 55 mm3 at 21 days, whereas tumor size in the control (PBS only) and radiation-only groups was up to 1387 ± 463 and 433 ± 120 mm3, respectively (n = 5). Administration of NAPC-ADC (5 mg⋅kg−1) alone had no significant impact on tumor growth, suggesting that radiation-induced payload release contributed to tumor treatment.
The NAPC-ADC conjugate undergo fragmentation by X-ray induced electron-transfer reaction in vivo to release a highly toxic MMAE.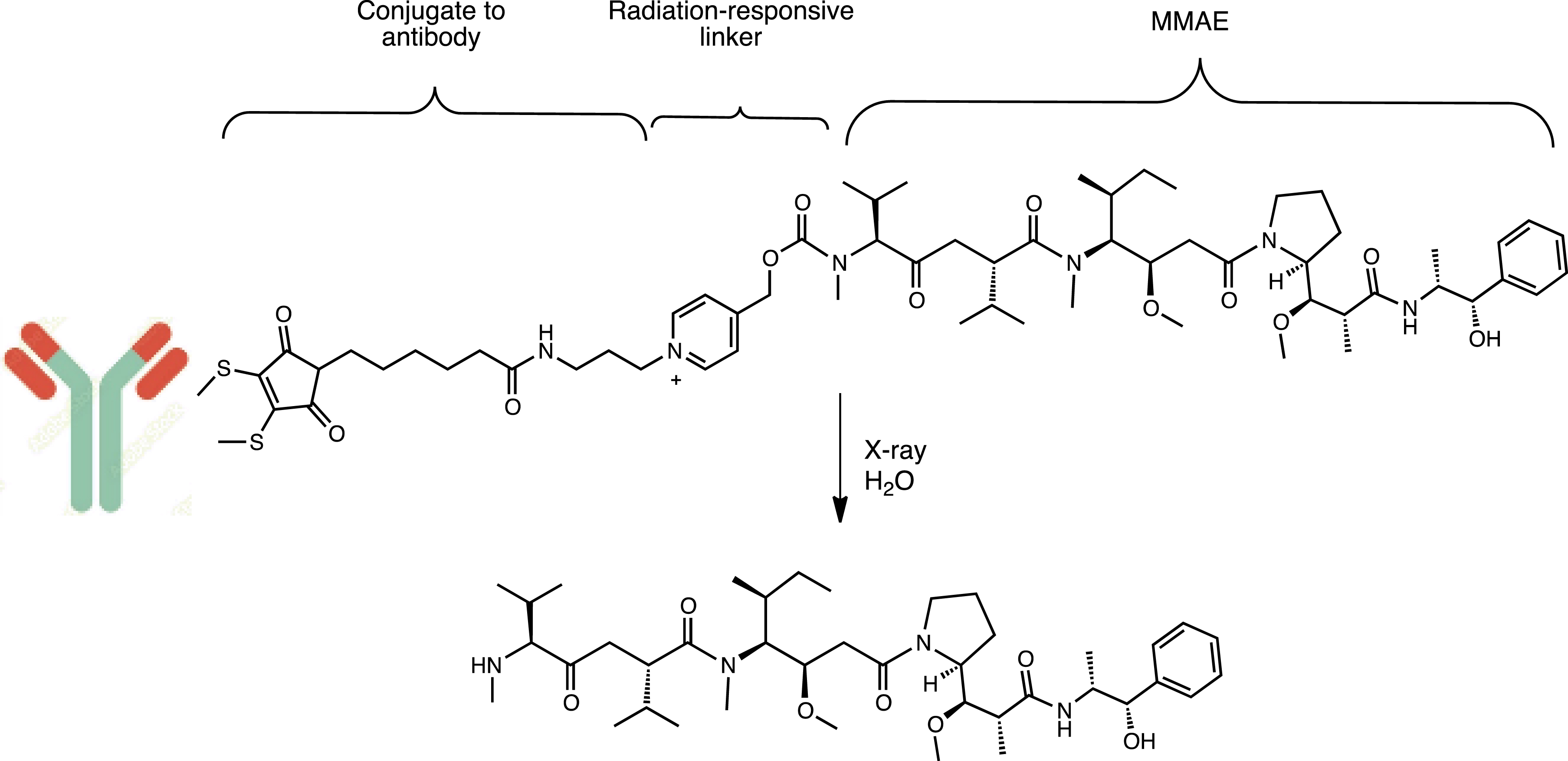
2.3. Fragmentation of the quaternary ammonium group
Similar to the C–N bond cleavage of quaternary organoammonium salts under photochemical conditions, quaternary ammoniums can be converted to tertiary amines under X-ray radiative conditions [73]. Fragmentation alters the polarization of the central benzyl group (i.e., through the transformation of an electron-withdrawing group (EWG) into an electron-donating substituent), triggering the 1,6-fragmentation of the benzyl-bound carbamate, thereby releasing the ligand (Scheme 6) [74].
Radiolytic fragmentation of the quaternary ammonium group.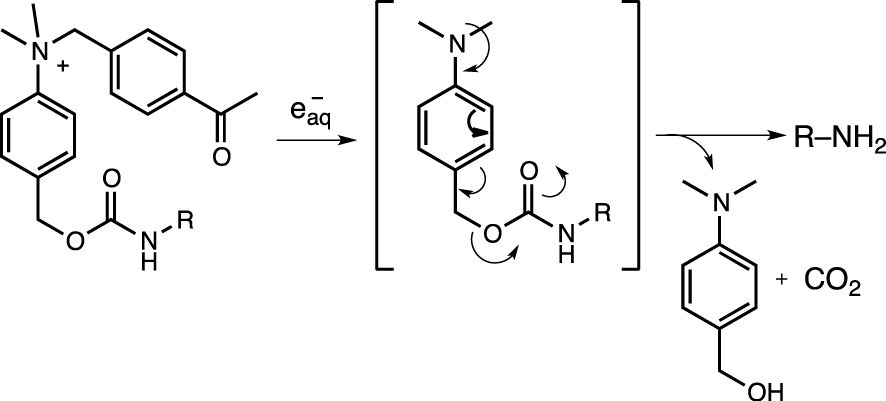
Carfilzomib, an anticancer drug with a morpholine motif, was released as a model substrate to demonstrate the feasibility of the in vitro strategy of radiation-induced prodrug release. Radiation-induced cleavage was demonstrated in living cells and in tumor-bearing mice, but high doses (60 Gy) were required to achieve satisfactory conversions.
3. Reduction of azides to amines
Prodrugs of pazopanib and doxorubicin encapsulated in sulfonyl azide and phenyl azide were released at irradiation doses of up to 60 Gy (Scheme 7) [75]. The driving force behind the transformation is the high reducing potential of e− generated by the radiolysis of water, with a standard electrode potential of −2.77 V. Although the exact reduction mechanism is unclear, it appears to follow a similar pathway to the photolysis of sulfonyl azides in alcohols and water to generate sulfonamides via the formation of sulfonylamido radicals or nitrenes [76].
Radiolytic activation of doxorubicin.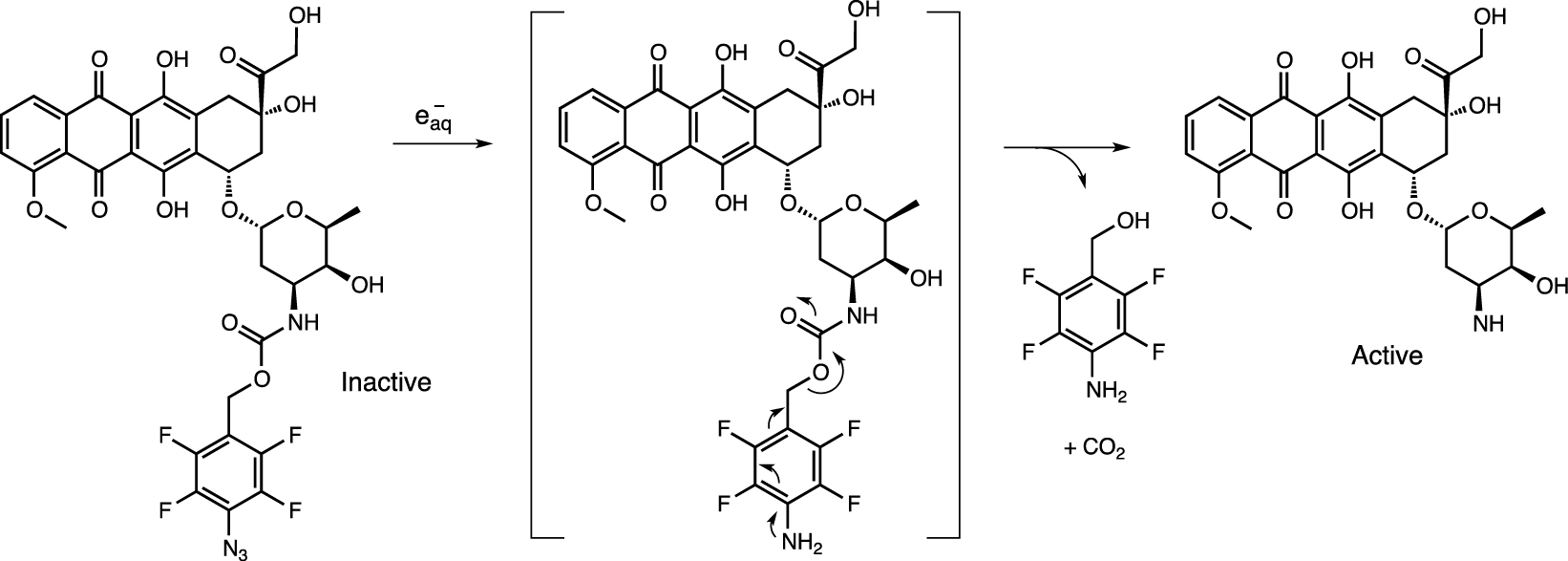
In a similar strategy, a tetrafluorobenzyl azide cage was used to mask the polar functions of the VHL E3 ligase via a carbonate linker. Continuous irradiation up to 60 Gy resulted in conversion of the azide group to an amine and triggered ligand release (Scheme 8) [77].
Activation of VHL E3 ligase by removing the tetrafluoroazidobenzyl carbonate protecting group by X-ray.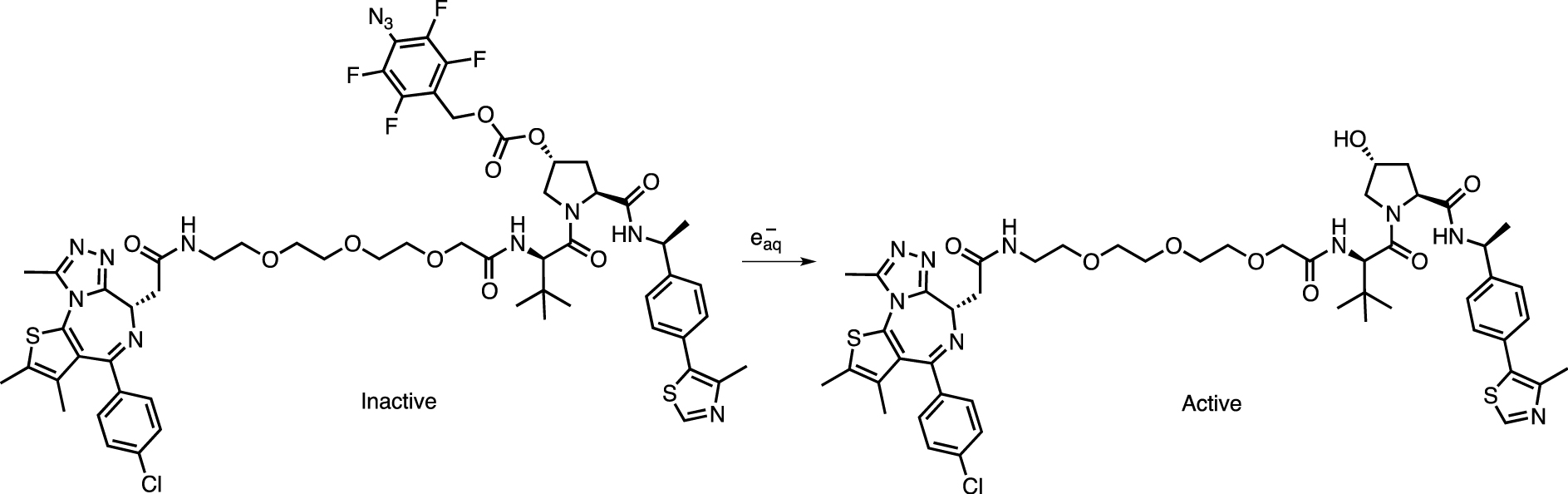
4. Activation of N-oxide prodrugs
The high electrode potential of electrons generated by local radiolysis of water can also effectively reduce nitrogen oxides [78]. The caging strategy involves oxidation of the target drug to N-oxide prodrugs, generating drugs whose biological activity is silenced. Radiolytic activation has been demonstrated from NO-imiquimod, NO-ampiroxicam, NO-pranoprofen, NO-loratadine, and NO-camptothecin by X-ray-induced electron transfer using radiation up to 60 Gy (Figure 3).
N-Oxide prodrugs for radiolytic activation.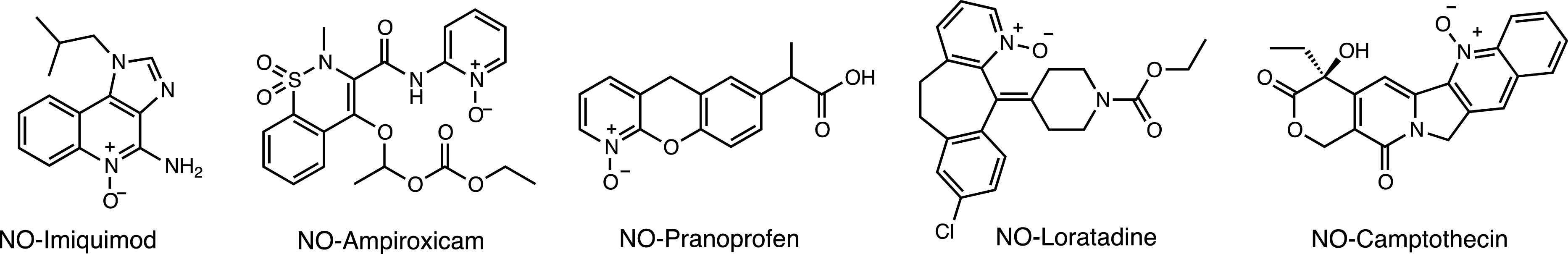
5. Chemical transformation of ROS-sensitive functions
5.1. Oxidation of thioethers
Nanovesicles were assembled from ROS-sensitive poly(propylene sulfide)-poly(ethylene glycol) (PPS-PEG) amphiphilic polymers and hydrophobic Au nanoparticles (Au NPs) linked to X-ray-labile linoleic acid hydroperoxide (LAHP) (Figure 4) [79]. The nanomaterial exhibited burst release of encapsulated doxorubicin (DOX) by irradiation at 8 Gy, up to 46.7%. Remarkably, drug release from Au–LAHP–vDOX incubated in H2O2 progressively increased to 76.7% 24 h after irradiation, while late drug release in PBS was minimal. In contrast, drug release from Au–LA–vDOX showed little or no response to X-ray irradiation. Irradiation (8 Gy) in a mouse subcutaneous tumor model significantly retarded tumor growth in the presence of Au–LA–vDOX and inhibited tumor growth even after irradiation, which could be attributed to the slow release of DOX from vesicles.
Nanoparticles, made by the co-assembly of the hydrophobic Au–LAHP NPs, amphiphilic oxidation-sensitive PPS–PEG polymers and hydrophilic DOX were activated by in situ-formed hydroxyl radicals (∙OH). The oxidation of the thioether chain to sulfoxide and sulphone transformed the hydrophobic chain to hydrophilic, leading to vesicle degradation and the release of the drug.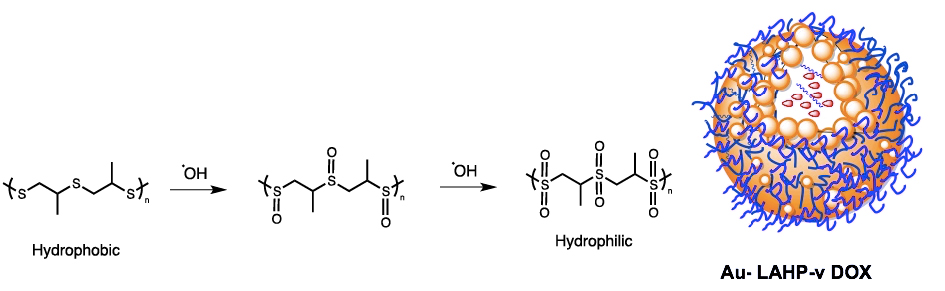
In an analogous study, radiation-generated ROS were the trigger signal for the activation of doxorubicin (DOX)-loaded nanomicelles derived from poly(propylene sulfide) (PPS) and hyaluronic acid (HA) [57]. HA–PPS@DOX nanoparticles exhibited prolonged circulation times of up to 24 h and showed decent accumulation in tumors. When oxidation by X-ray irradiation (8 Gy) converted the lipophilic thioether chain to polar sulfoxides, the ROS-sensitive HA–PPS@DOX NMs disintegrated and released the cytotoxic cargo.
5.2. Fragmentation of disulfide and diselenide bonds
Disulfide and diselenide are not particularly sensitive to direct X-rays without the synergistic effect of a high concentration of ROS. The Tanabe group devised a DNA amphiphile (DAM) composed of hydrophilic oligodeoxynucleotides linked to hydrophobic alkyl chains by disulfide bonds [80]. When DAM molecules are exposed to X-ray irradiation, selective fragmentation of the disulfide bonds leads to dissociation of the aggregates and release of the drug.
Radiolytic oxidation (5 Gy) resulted in the cleavage of the cross-linked polysulfide shell of the cysteine-decorated poly-G4.5 PAMAM dendrimer (a “4th generation” branched polyamidoamine polymer containing 128 surface carboxylate groups), allowing release of the anticancer drug (Figure 5) [81].
Cross-linked polysulfide shell of cysteine-decorated poly-G4.5 PAMAM dendrimer.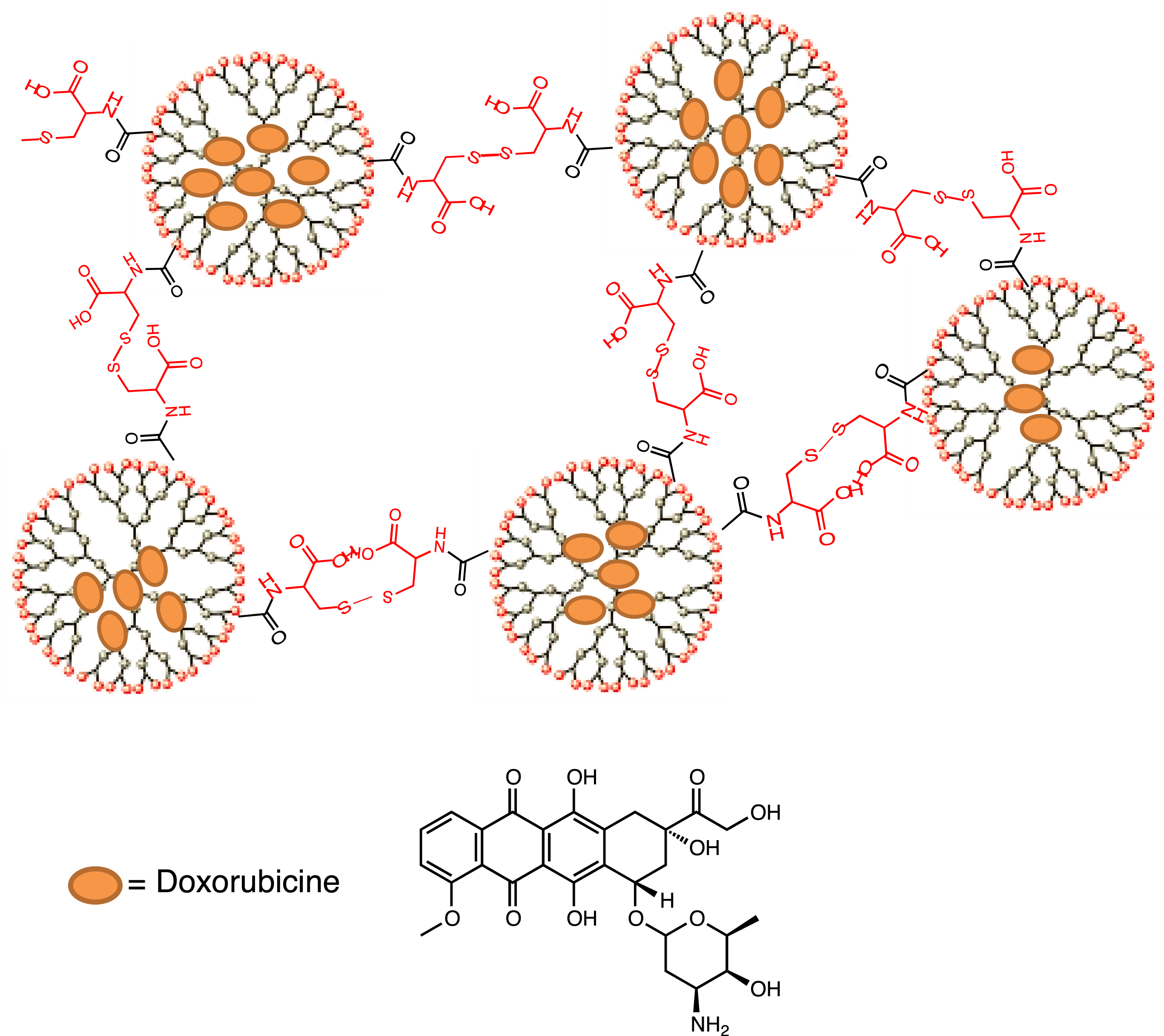
Ma et al. used PEG–PUSeSe–PEG, a radiation-sensitive diblock copolymer, to construct a nanoscale drug delivery system (Figure 6) [82]. On exposure to γ-rays (5 Gy), the generated ROS oxidized the diselenide groups by forming selenic acid and triggering the fragmentation of the PEG–PUSeSe–PEG micellar aggregates/release of the encapsulated drug (PUSeSe stands for polyurethane diselenide hydrophobic block). Unfortunately the PEG–PUSeSe–PEG polymer showed cytotoxicity to HepG2 cells.
Diselenide-linked micellar aggregates were cleaved in the presence of ROS, generated by γ-rays (5 Gy).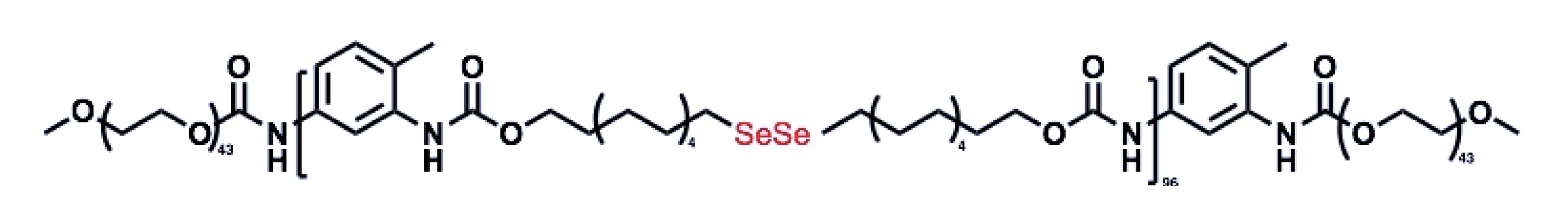
Immunotherapy can be combined with radiotherapy and chemotherapy, providing a new strategy for cancer treatment. A combination of diselenide and pemetrexed has been developed to combine NK cell-based cancer immunotherapy with radiotherapy and chemotherapy in a single system [83]. The assemblies were prepared by co-assembly between pemetrexed and cytosine-containing diselenide via hydrogen bonds (Scheme 9). ROS generated by γ-radiation (5 Gy) not only cleaved the diselenide bonds (converted to seleninic acid), thereby suppressing human leukocyte antigen E (HLA-E) expression in cancer cells and activating the immune response of NK cells, but also released pemetrexed.
Assembly of the diselenide–pemetrexed block.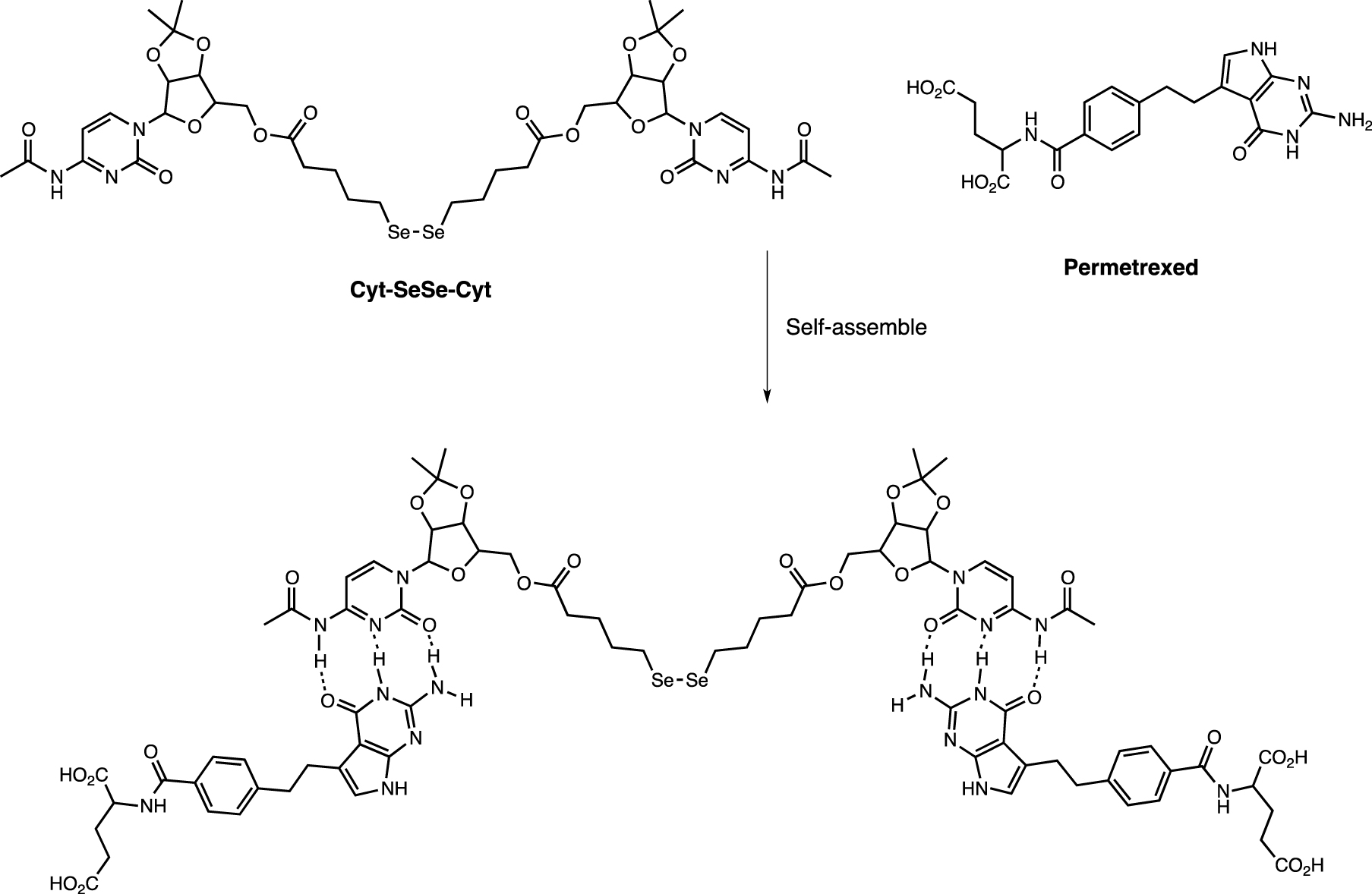
The Chen group has developed a strategy for the targeted delivery of doxorubicin (DOX) to tumor sites using mesoporous organosilica (MON) nanoparticles coated with a cancer cell membrane, which can be activated by X-rays (Figure 7) [84]. This approach involved loading DOX and a PD-L1 inhibitor into the pores of MONs bridged by diselenic linkers. MONs were then coated with cancer cell-derived membrane fragments to produce anti-PD-L1 CM@MON@DOX +. Low-dose X-ray irradiation (less than 1 Gy) resulted in the fragmentation of the diselenic bond, disrupting the structure of the MONs, and releasing DOX and the PD-L1 inhibitor, resulting in DOX-mediated immunogenic cell death at the tumor site [85].
ROS-sensitized mesoporous organosilica nanoparticles (MONs).
Diselenide-derived block copolymers have been used to load doxorubicin for precision chemotherapy [86]. ROS generated by X-ray irradiation (2 Gy) promoted release of the encapsulated drug under in vivo conditions in tumor-bearing mice, which produced a more pronounced overall antitumor effect than the other test groups.
5.3. Dihydroxybenzyl functions sensitive to oxidation
The Co-60 source has been tested in the radiative fragmentation of several carbamate-bound photosensitive compounds [87]. Best fragmentation kinetics were observed with resorcinol derivatives. The hypothesis of radiation-induced ortho- or para-hydroxylation of the aryl ring was put forward as the operative fragmentation mechanism, followed by a 1,4 or 1,6 elimination reaction releasing the covalently attached ligands (Scheme 10). Radiation activation has been studied in cell experiments as well as in tumor-bearing mice, using the ligand MMAE as an anticancer agent with a single-dose activation of 4 Gy. Details of oncological relevance were not provided, however.
Fragmentation of rhodamine DHBO–Me to Me-Rhodamine (green) by γ-ray irradiation.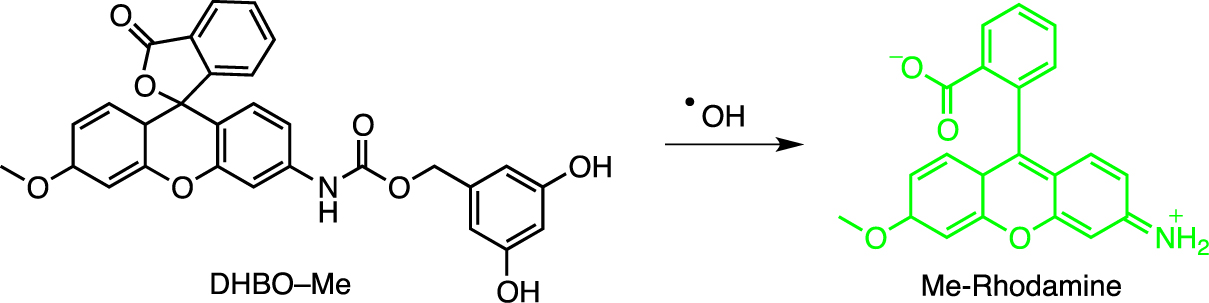
5.4. Release of small molecules
5.4.1. NO synthesis triggered by ROS
The mixing of hafnium tetrachloride (HfCl4) and 2-nitroimidazol in a microemulsion containing hexamethylene, hexyl alcohol, and a surfactant (triton) resulted in the formation of a unique type of nanoscale coordination polymer, HNP NPs [88]. NPs were coated with DOPA (sodium salt of 1,2-dioleoyl-sn-glycero-3-phosphate) and then mixed with 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly(ethylene glycol))-5000] (DSPE–PEG5000). The nanomaterial was shown to act both as a radiosensitizer and as an immune adjuvant for NO donors (2-nIm), which released NO in a controlled manner under X-ray irradiation.
A template-free method was devised for the synthesis of Bi–SH nanoparticles (Bi–SH NPs), which were then coated with SiO2 to improve aqueous solubility, and finally functionalized with the NO precursor S-nitrosothiol (Bi–SNO NPs) [89]. The particular advantage of this system is the local release of NO: the short half-life of NO and its high sensitivity to biological substances considerably limit the development of NO-based therapeutic platforms for clinical application. The release of NO by X-rays (5 Gy) has been validated in rodents.
5.4.2. Carbon monoxide release triggered by X-rays
A multifunctional X-ray-activatable theranostic nanoplatform has been developed based on Ce-doped LiLuF4 scintillant and UV-sensitive Mn2(CO)10 for the controlled generation of CO and manganese dioxide (MnO2) in tumors [90]. Under X-ray irradiation, radioluminescent UV light from SCNPs triggered the photolysis of Mn2(CO)10 by luminescent resonance energy transfer (LRET). In this process, the Mn–Mn and Mn–CO bonds of Mn2(CO)10 were cleaved photochemically to release CO forming MnO2 as a by-product. In vitro and in vivo experiments demonstrated the potent antitumor activity and low systemic toxicity of this nanomedicine.
5.4.3. Peroxynitrite generation
Peroxynitrite (ONOO∙−), a reaction product derived from nitric oxide (NO) and superoxide (), is a powerful oxidizing and nitrating agent that modulates complex biological processes and promotes cell death. A multifunctional X-ray-controlled ONOO∙− generation platform based on scintillating nanoparticles (SCNPs) and UV-sensitive NO donors, Roussin black salt, has been reported [91]. In this system, Ce-doped LiLuF4 acts as a radiosensitizer by promoting ROS, including , under X-ray irradiation, and it also converts X-rays to UV light to activate Roussin’s black salt to release NO. The simultaneous release of NO and ensures efficient X-ray-controlled production of ONOO∙− in tumors.
6. Miscellaneous
Doxorubicin (DOX), gold nanoparticles (AuNPs), DNA-coated AuNPs (DNA–AuNPs), and DOX conjugated to DNA–AuNPs (DOX–DNA–AuNPs) were synthesized and incubated with MCF-7 breast cancer cells [92]. Strong internalization was observed with only residual leaching of immobilized doxorubicin. The extinction efficiency of DNA strand-attached DOX on AuNPs was estimated at 99.7%. Under the influence of X-rays, the DOX–DNA–AuNP DNA strands were cleaved, probably by the presence of ∙OH radicals, and the DOX molecules were removed from the AuNP surface. The drug-carrying nanoparticles became more toxic as the X-ray dose increased, from −1% ± 15% at 2 Gy to +19% ± 13% at 5 Gy and +33% ± 21% at 10 Gy. Noteworthily the observed toxicity was greater than the sum of the radiation toxicity and the toxicity of the drug-carrying nanoparticles. Given the amount of AuNPs in the cell (≈30 wt%), the small increase in toxicity observed seems to suggest that these nanoparticles remained in the cytoplasm, as this amount of AuNPs in the cell would cause significant damage only if the increase in X-ray absorption was taken into account. It should be noted that this report is the first demonstration of increased cytotoxicity due to X-ray-triggered release of chemotherapeutic drugs from nanoparticles in cells.
ROS can oxidize unsaturated lipids and destabilize liposomal membranes, releasing the encapsulated cargo. The Deng laboratory devised a drug delivery system using sensitized liposomes containing 3–5 nm gold nanoparticles (GNPs) and the photosensitizer verteporfin (VP), and X-rays to trigger liposome rupture [93]. This system takes advantage of the intensity of the radiation and the ability of liposomes to adapt to the environment. This system takes advantage of the radiation intensification effect of GNPs to amplify the production of singlet oxygen (1O2) and other ROS from verteporfin under X-ray irradiation, triggering thus the rupture of liposomes. The results indicate that liposomes loaded with GNPs and VP exhibit higher rates of 1O2 production than those loaded with GNPs or VP individually. The antitumor efficacy of this drug delivery system was validated in vitro and in vivo, demonstrating that LipoDOX, a drug delivery system based on DOX-loaded liposomes, displayed superior tumor inhibition efficacy following X-ray irradiation.
The liposome was prepared from dioleoyl-glycerophosphocholine (DOPC) and dioctadecenoyl-trimethylammonium-propane (DOTAP) [93]. Cellular uptake of the nanomaterial was facilitated by its positive charge at neutral pH. In vitro gene inhibition by X-ray activation of the liposome was demonstrated using an antisense oligonucleotide for a pituitary receptor specific for adenylate cyclase-activating polypeptide (PACAP) in rats. The system was also tested in X-ray-triggered chemotherapy, using doxorubicin in liposomes on human HCT 116 colorectal cancer cells.
It has been shown that low-dose X-rays (1–4 Gy at 6 MeV) can trigger the release of doxorubicin (DOX) from liposomes co-loaded with gold nanoparticles and photosensitizers [94]. ROS produced by gold nanoparticles and photosensitizers under X-ray irradiation destabilized the lipid membrane and led to the release of encapsulated DOX.
In a similar system, the anticancer drug paclitaxel (PTX) and radioluminescent CaWO4 (CWO) nanoparticles were coencapsulated with poly(ethylene glycol)-poly(lactic acid) (PEG–PLA) block copolymers (“PEG–PLA/CWO/PTX NPs”) [94]. Upon exposure to X-rays, the PEG–PLA coating layer degraded and PTX molecules were released. It should be noted that radioluminescent CWO nanoparticles emit UV-A/blue light (350–525 nm) under X-ray excitation, which also produced radio-enhancement/radio-sensitization effects in tumor tissues.
GFP reporters were attached to silica-coated gold nanoparticles (AuNP@ SiO2) by DNA strands as linkers [95]. The DNA strand is broken by reaction with ROS, releasing the GFP reporters. Although proteins also react with ROS at high rates, the low concentrations of ROS rendered this damage (less than one reaction per protein) undetectable in this work. The authors hypothesized that the efficiency of AuNP@SiO2 release could potentially be improved if the thickness of the SiO2 layer was reduced and DNA strand breaks could be chemically enhanced. This work has shown that radiation activation can be compatible with the delivery/activation of therapeutic proteins in biological systems.
Alkyne-modified dextran hydrogel (500 kDa) was irradiated with γ-rays (14.4 kGy), resulting in densification of cross-linking by the formation of reactive terminal alkyne radicals, generated directly by irradiation or indirectly by reaction products from radiolysis of water [96]. Polymerization controlled hydrogel rigidity, contraction, release, and fusion. Hydrogel stiffness increased twofold for hydrogels swollen with H2O and threefold for hydrogels swollen with phosphate buffer after γ irradiation, due to the increase in the cross-linking density.
7. Conclusion and outlooks
Tissue-penetrating X- and γ-rays have the potential to selectively reach specific tissues. Simultaneous release of organic compounds with the therapeutic beam presents a conceptually different approach compared to standard protocols that combine radiotherapy and chemotherapy. This approach may allow further reducing radiation doses and systemic cytotoxicity compared to current protocols. Since organic compounds are transparent at this wavelength, and direct irradiation is inefficient, the search for different transduction mechanisms has led to many ingenious drug delivery strategies. To overcome the difficulties associated with the biocompatibility, degradability and cellular toxicity of several nanocarriers, strategic solutions are still needed. One of the main challenges is to minimize the X-ray doses required to trigger cargo release close to unity. Even though the release efficiency is one of the most important parameters, in most reports the quantitative analysis of the drug release is neglected claiming technical difficulties. This should be certainly improved in the near future.
The X-ray activation method is likely to be applied clinically in the near future, provided that issues of toxicity (of nanomaterials) and efficacy (enabling the necessary activation doses to be reduced) are optimized. Also, the nanoparticle formulation can serve as a simultaneous multifunctional platform for chemo-, thermo-and immuno-radiotherapy; it can not only enhance tumor irradiation responses during RT but also leverage combination therapy to achieve a synergistic effect. We believe that this mode of activation is still in its infancy, while opening up the potential of nuclear medicine to the control of smart materials. Future developments can be extended to proton and carbon ion beams, although they have a very narrow Bragg peak, meaning that they do not scatter along the radiation path and reach the biological region without energy loss [97]. This young field is developing rapidly and, with access to increasingly powerful and safe experimental methods and equipment, promises a breakthrough in research and clinical applications.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.