



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Coinage metals are well known for their versatile coordination chemistry, yielding a large variety of structural motifs, which are strongly influenced by ligating donor atoms, denticity and geometry of ligands [1, 2, 3, 4]. Importantly, secondary interactions like van der Waals, dipole, dispersive, metallophilic interactions often play decisive roles in supramolecular arrangements and solid state-packing. In particular, gold(I) is prone to form simple linear coordination compounds with coordination number two (C N = 2), where the gold(I) ion can further engage in moderate to strong metallophilic, i.e. aurophilic, interactions [5]. These exhibit strengths comparable to strong hydrogen bonding and allow stabilization of inter- and intramolecular arrangements alike. The former often leads to preferred conformations, while the latter can lead to extended supramolecular structures observable in the solid-state and solution [6, 7]. Observation of these effects in multinuclear clusters can also be interpreted using the premise of cooperativity to further increase their strength, and importance.
Chelating effects are well understood for a variety of ligands possessing energetically favorable binding to a single metal (cation). Nonetheless, multidentate phosphine ligands are also well known to stabilize high nuclearity gold cluster compounds balancing the benefits of aurophilic/metallophilic and chelating effects [8, 9]. However, subtle steric effects of the chelating ligand may have large repercussions on the nuclearity and coordination environment [10, 11], the latter also playing important roles in catalytic applications [12]. Other non-classical interactions such as halogen or hydrogen bonding involving donors and acceptors other than oxygen and nitrogen are of great interest. Halide interactions with acidic hydrogen atoms of “non-coordinating” solvents such as chlorinated alkanes offer a variety of possibilities. In particular, chloroform and DCM have been subject to extensive studies revealing their importance in intermolecular stabilization and packing effects in the solid-state [13]. For example, chloride (and other halide) ions are often found in a hexacoordinated environment stabilized by chloroform X⋯H–C hydrogen bonding interactions [14, 15]. Similar chloroform halide interactions are found in a variety of organic and inorganic molecular systems [16, 17, 18, 19, 20]. Spectroscopic and theoretical analyses of such chloroform and bromoform–chloride interactions clearly showed their affinity towards secondary solid-state interactions [18, 21, 22].
We detail the different aspects of primary and secondary factors that govern the formation of mono- and dinuclear gold clusters based on the rigid bidentate cis-1,2-bis(diphenylphosphino)ethene (= cis-bdppe) ligand. In this combined theoretical and experimental/structural investigation of selected complexes, we elucidate the structural diversity of these deceivingly simple coordination compounds [23].
2. Results
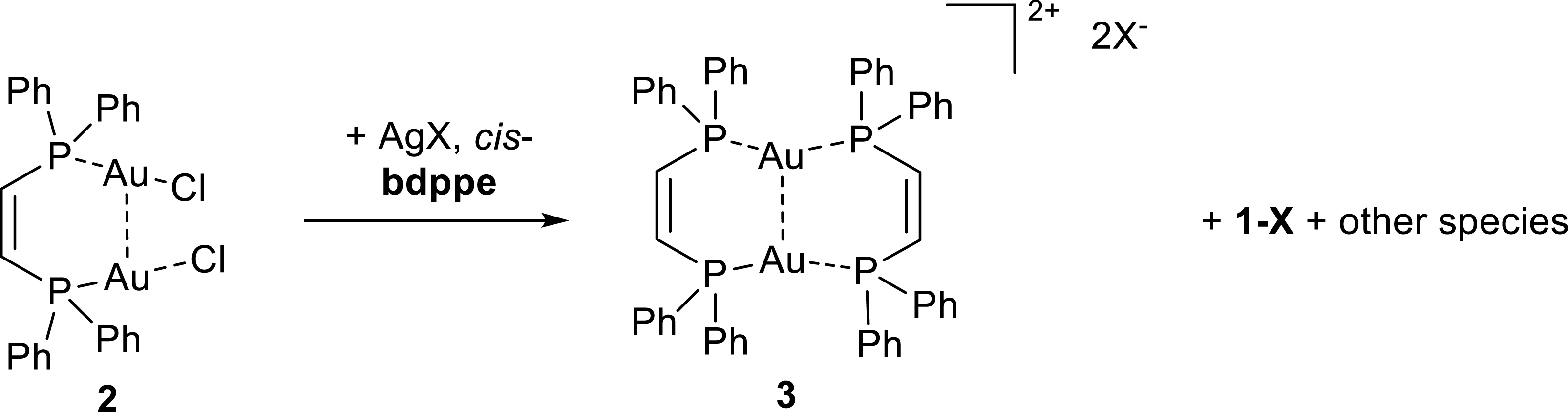
Synthesis of linear phosphine gold halide complexes is typically achieved by the straightforward ligand substitution reaction starting from chloridotetrahydrothiophene gold(I), [AuCl(tht)], or similar thioether precursors with phosphine ligands in a suitable solvent such as DCM, chloroform or THF [24, 25]. Our initial attempts to prepare a dinuclear gold complex based on cis-1,2-bis(diphenylphosphino)ethene (= cis-bdppe) in a 2:1 gold:ligand ratio were unsuccessful. Reactions in concentrated chloroform solutions resulted in the formation of a mononuclear complex along with unreacted gold precursor (Scheme 1).
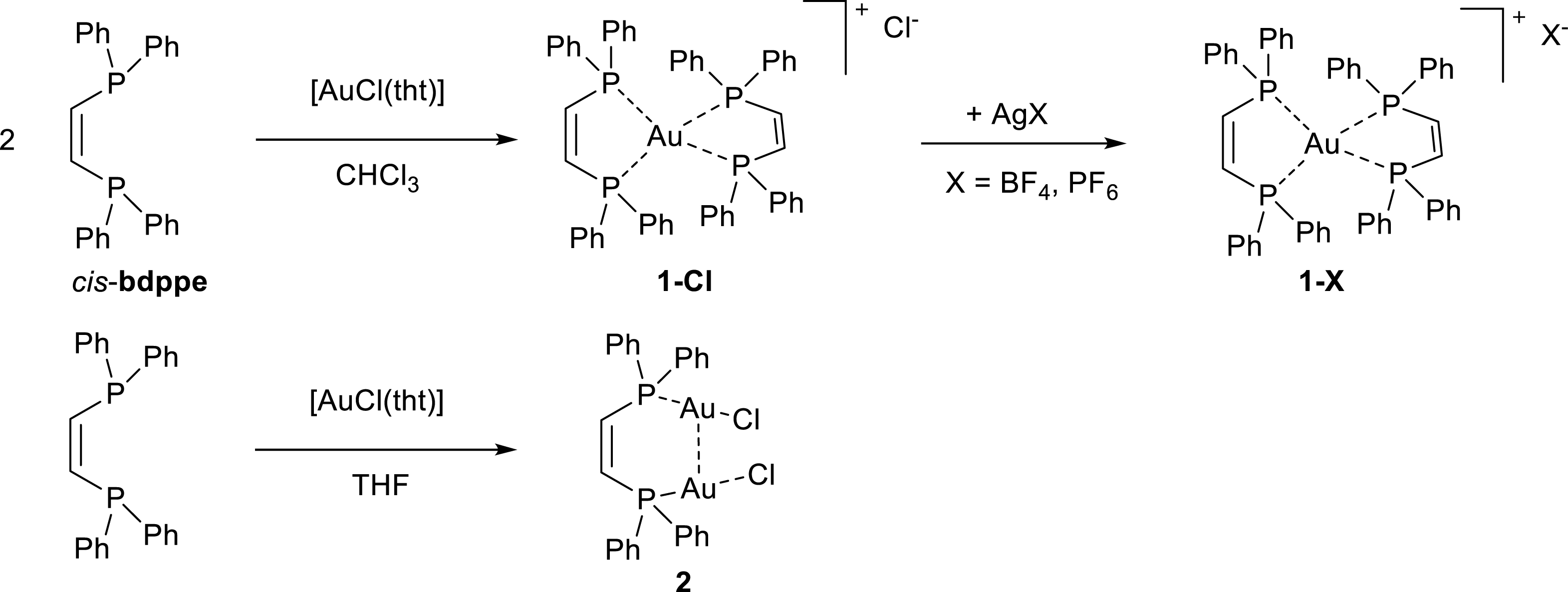
Synthesis of mono- and dinuclear gold complexes of diphosphine ligand cis-bdppe.
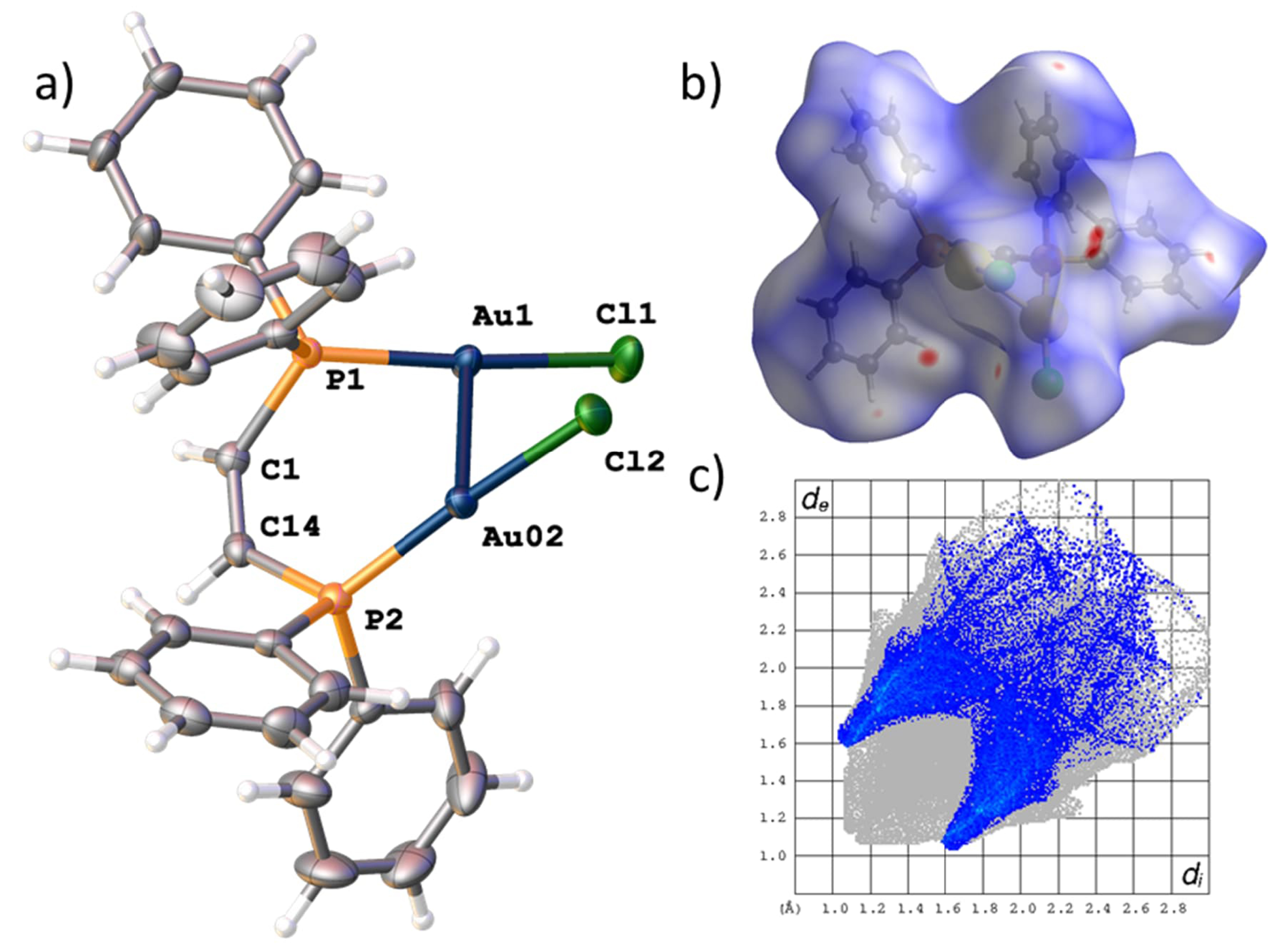
Single crystals of this complex have been obtained by slow evaporation of the solvent, revealing the molecular structure of the complex as a gold(I) center tetrahedrally coordinated by two cis-bdppe ligands. (Figure 1) Complex 1-Cl crystallizes in the monoclinic space group C2/2 with half of the molecule present in the asymmetric unit. The two ligands form a slightly distorted tetrahedral arrangement around the gold center (twist of the least squares planes 104.25(5)°). The gold–phosphorus distances are in the expected range of gold(I) phosphine complexes (P1–Au1 2.375(1) Å and P2–Au1 2.372(1) Å). Notably, the chloride counterion is stabilized by four chloroform molecules illustrating the ability of the acidic solvent proton to engage in hydrogen bonded interactions (C–H⋯Cl donor acceptor distance and angles of 3.294(3)/3.354(2) Å) and 149.1(2)°/144.3(1)°, respectively. Moreover, slightly longer hydrogen bonding interactions with the ethene bridge of the ligands are also observed (C21⋯Cl 3.574(2) Å and C1⋯Cl 3.639(2) Å). A Hirshfeld surface analysis of the chloride fragment (Figure 1b) illustrates the hydrogen bonding interactions of the solvent molecules and the ligand backbone. Notably, the yellow single crystals of 1-Cl show discoloration and lose their crystallinity over time, presumably due to solvent loss.
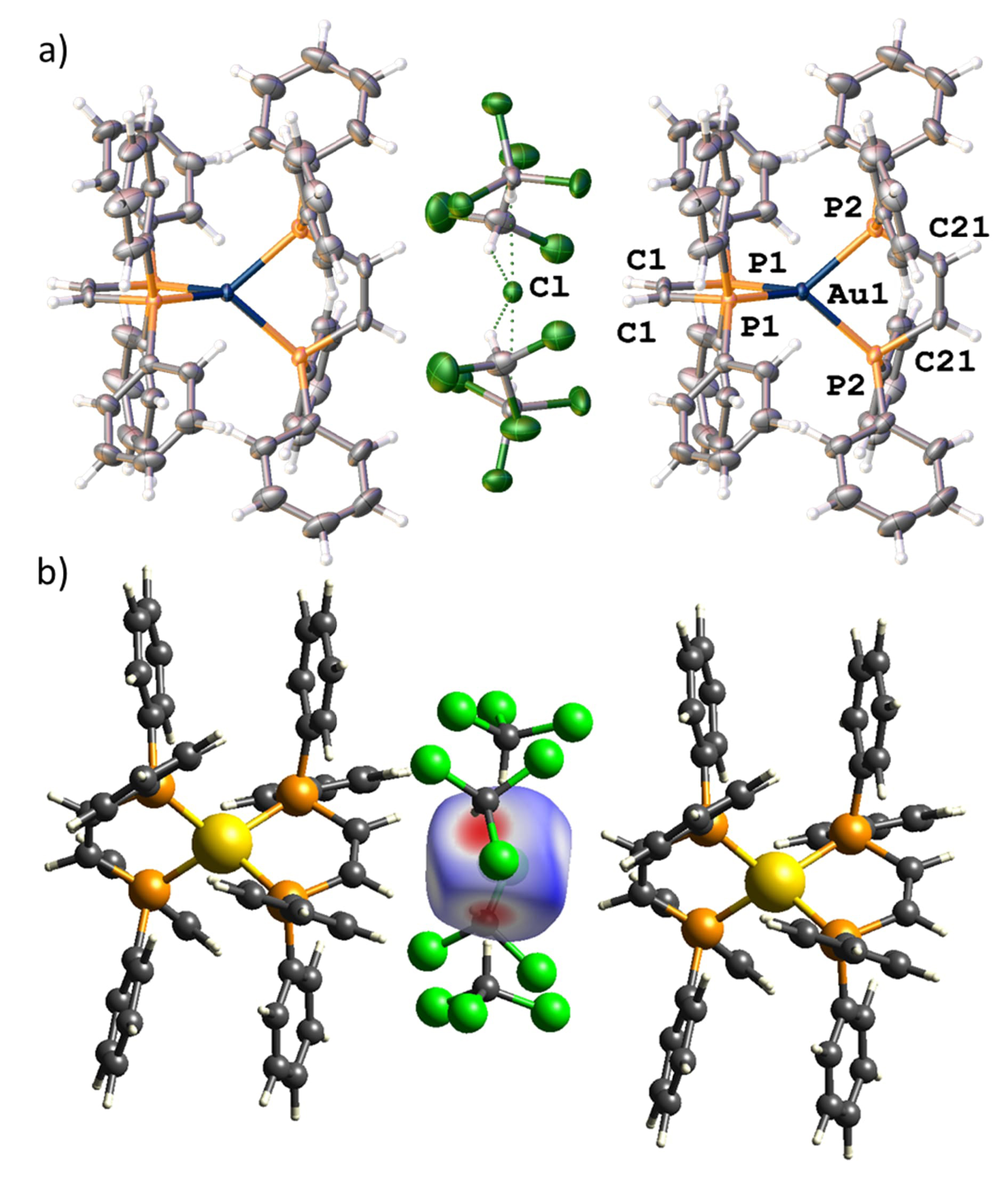
(a) Solid-state structure of mononuclear complex 1. ORTEP style representation with ellipsoids drawn at a 50% probability level. Further crystallographic details are elaborated in the experimental section. (b) Hirshfeld surface analysis highlighting the hydrogen bonded interactions of the chloride fragment.
We utilized the geometry obtained from X-ray diffraction as the starting point of computational analysis. We preoptimized the geometry using the semiempirical tight-binding GFN2-xTB method, which is known to provide optimal molecular geometries for gold complexes [26], followed by a second geometry optimization at the r2SCAN-3c level. As for the tight-binding approach, this composite method is reported to provide fast and reliable geometries for organo-gold species [27]. The structure was characterized as a true minimum, with no relevant imaginary frequencies.
We studied the orbital contributions governing the intermolecular interactions in the metal complex by applying Weinhold’s Natural Bond Orbital (NBO) analysis. In particular, we studied the second-order energy terms (E(2)) that arise from the interaction of two unperturbed NBOs, which provide a quantitative value for the stereoelectronic stabilization associated with that specific orbital interaction. As expected, one source of intermolecular stabilization comes from the negative hyperconjugation of the lone pairs of the chloride anion with the C–H σ∗ orbitals of chloroform. We found an analogous negative hyperconjugative effect affording
Relevant E(2) values associated with selected hyperconjugative interactions in one of the orbitals depicted in Figure 3
| NBO number (Donor orbital) | Donor orbital | NBO number (Acceptor orbital) | Acceptor orbital | E(2) (kcal⋅mol−1) |
|---|---|---|---|---|
| 156 | LP Cl102 | 350 | BD∗ C4–H5 | 1.25 |
| 156 | LP Cl102 | 447 | BD∗ C78–H79 | 1.03 |
| 157 | LP Cl102 | 350 | BD∗ C4–H5 | 0.25 |
| 157 | LP Cl102 | 447 | BD∗ C78–H79 | 0.46 |
| 158 | LP Cl102 | 350 | BD∗ C4–H5 | 0.28 |
| 158 | LP Cl102 | 447 | BD∗ C78–H79 | 0.13 |
| 155 | LP Cl102 | 479 | BD∗ C106–H107 | 1.64 |
| 157 | LP Cl102 | 479 | BD∗ C106–H107 | 2.48 |
| 158 | LP Cl102 | 479 | BD∗ C106–H107 | 4.32 |
| 155 | LP Cl102 | 483 | BD∗ C111–H112 | 0.71 |
| 156 | LP Cl102 | 483 | BD∗ C111–H112 | 1.86 |
| 157 | LP Cl102 | 483 | BD∗ C111–H112 | 3.11 |
| 158 | LP Cl102 | 483 | BD∗ C111–H112 | 2.98 |
| 155 | LP Cl102 | 487 | BD∗ C116–H117 | 0.79 |
| 156 | LP Cl102 | 487 | BD∗ C116–H117 | 1.21 |
| 157 | LP Cl102 | 487 | BD∗ C116–H117 | 3.51 |
| 158 | LP Cl102 | 487 | BD∗ C116–H117 | 3.00 |
| 155 | LP Cl102 | 491 | BD∗ C121–H122 | 1.59 |
| 157 | LP Cl102 | 491 | BD∗ C121–H122 | 1.92 |
| 158 | LP Cl102 | 491 | BD∗ C121–H122 | 4.46 |
LP: lone pair; BD∗: antibonding orbital; the index as a subscript for the atoms of each bond corresponds to the atom index in the cartesian coordinate file.
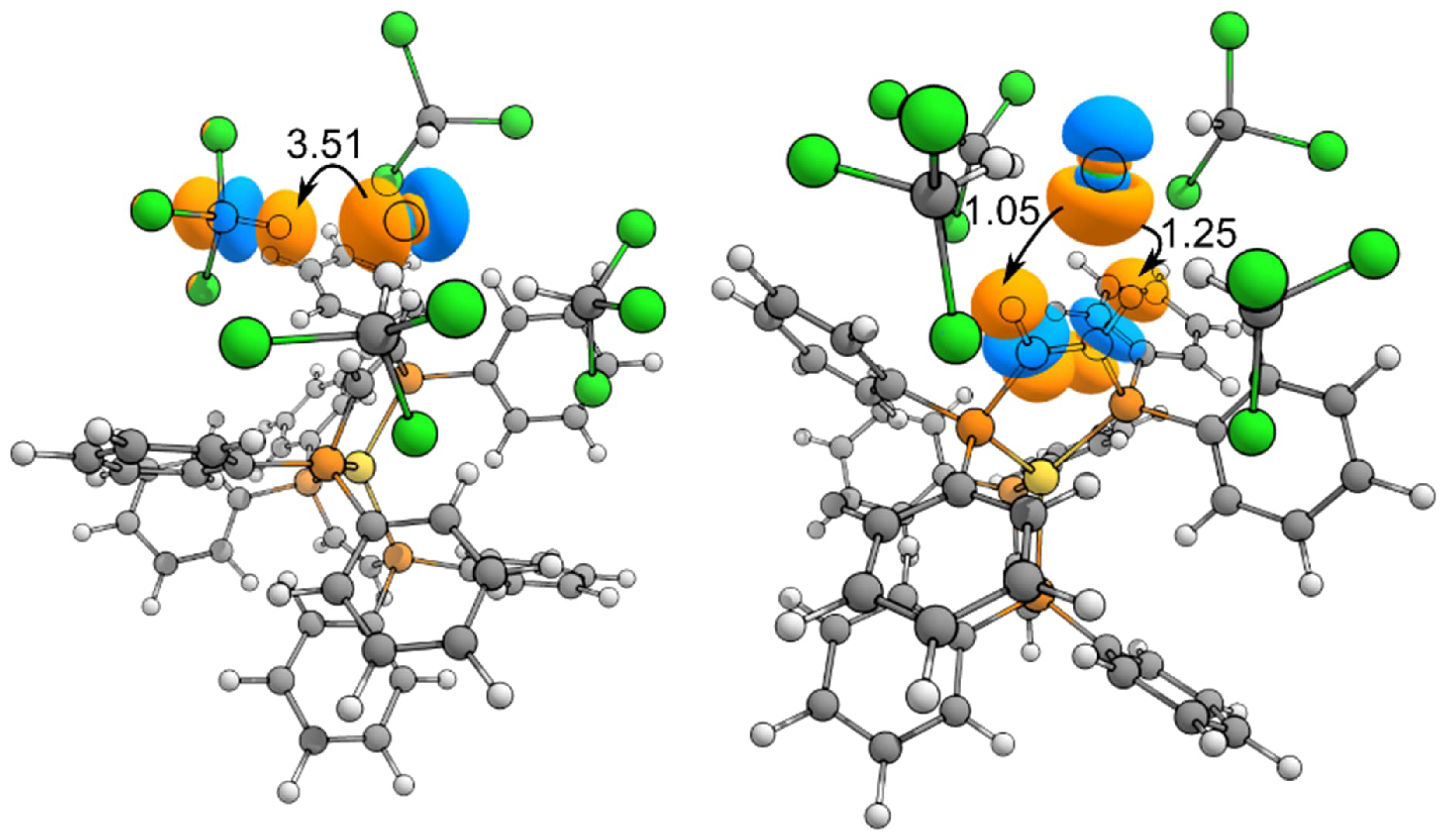
Geometry of the gold complex obtained at r2SCAN-3c level of theory. The main
Geometry of the gold complex obtained at r2SCAN-3c level of theory. The main
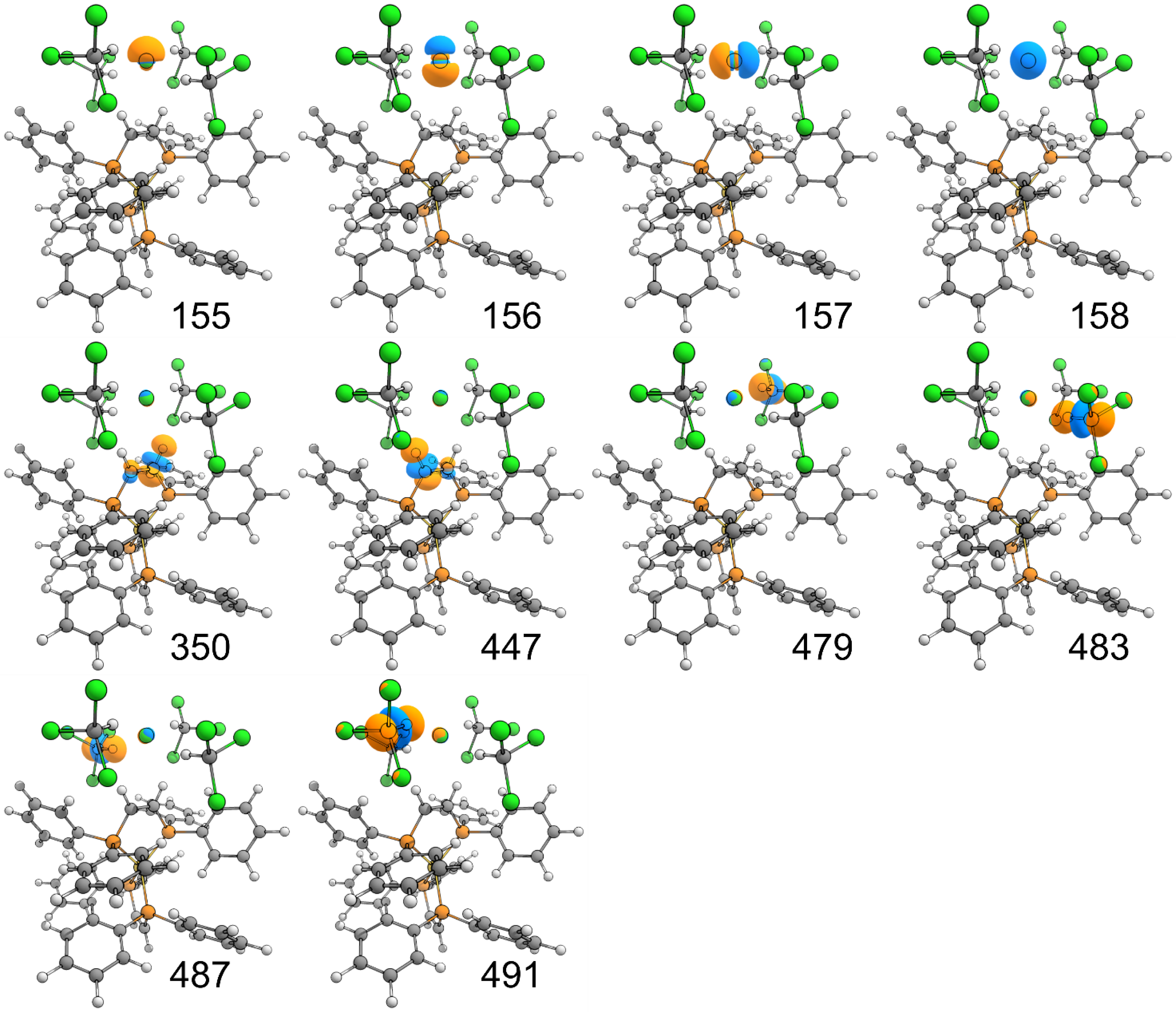
Depiction of the relevant NBOs (isovalue 0.05) listed in Table 1. Each number corresponds to the index value of the NBO.
Exclusive formation of this complex can be improved by maintaining a strict 1:1 ratio of metal precursor to ligand and performing the reactions in chloroform (Scheme 1). The crude product was purified by washing with cold ethanol yielding 96% of analytically pure compound 1-Cl. We hypothesized that the poor solubility of the gold precursor and the chloride-stabilizing nature of chloroform are the main reasons for the formation of this product. Hence further reactions were conducted in different coordinating solvents. Reactions in THF at higher concentrations (>0.1 M) gave a product mixture, while reactions in THF at lower concentrations (1–2 mM) exclusively resulted in formation of dinuclear complex 2 in good yields. Upon removal of all volatiles and washing with ethanol, the THF insoluble portion contained mainly 1-Cl, and the soluble fraction left for crystallization provided analytically pure material of complex 2 (yield 78%). The complex crystallized in the monoclinic space group Cc with one molecule in the asymmetric unit (Figure 4). The dinuclear complex 2 showed a slight decrease of the gold phosphine distance to 2.231(1) Å and 2.237(1) Å for P1–Au1 and P2–Au2, respectively. The spatial arrangement of the diphosphine and moderate aurophilic interactions led to an intramolecular Au1–Au2 distance of 3.0313(3) Å—below the sum of their van der Walls radii (3.32 Å)—which indicates moderate aurophilic interactions. While the ligand backbone is essentially co-planar, the diphenylphosphine-C single bond rotation creates a twisted arrangement of the nearly linear phosphine gold(I) chloride fragments (Cl–Au⋯Cu–Cl torsion angle of 51.96(6)°).
Solid-state structure of mononuclear complex 2 and Hirshfeld surface analysis. (a) ORTEP style representation with ellipsoids drawn at a 50% probability level. Further crystallographic details are elaborated in the experimental section. Red areas indicate dominating intermolecular CH⋯π interactions. (b) Hirshfeld surface plot. (c) Short intermolecular contacts; highlighted in blue H⋯C interactions.
Removing the coordinating chloride ions from the dinuclear complex 2 by non- or weakly coordinating anions using suitable silver(I) salts proved to be difficult and led to complex mixtures of products, including a tetrahedrally coordinated gold ion, determined by 31P-NMR spectroscopy. Unambiguous identification of this species was obtained by single crystal X-ray diffraction of crystal directly obtained by slow evaporation of the crude reaction mixture. Targeted synthesis of these tetrahedral coordination compounds 1-X by treating 1-Cl with suitable silver(I) salts gave essentially quantitative conversions. Compound 1-BF4 and 1-PF6 crystallized in the triclinic space group P-1 and monoclinic space group P21 with one and two molecules in the asymmetric unit, respectively. The solid-state structure of 1-PF6 also contained disordered solvent molecules which were treated as diffuse scattering using solvent masking. Substituting the chloride of complex 1-Cl with a tetrafluoroborate (1-BF4) or hexafluorophosphate (1-PF6) barely affected the direct gold(I) coordination (P–Au distances ranging from 2.3859(4) Å to 2.3996(3) Å) and 2.356(6) Å to 2.394(6) Å, respectively. However the folding angle of the P2Au planes varied significantly: 98.1(1)° (1-BF4) and 110.2(2)° (1-PF6) compared to 104.7(4)° (1-Cl). The tetrafluoroborate anion is positioned in between the diphenylphoshine groups from the two ligands. Weak hydrogen bonding interactions of the tetrafluoroborate and intermolecular π-stacking (centroid distances 3.373(1) Å) contribute to the solid-state packing of 1-BF4. Similarly, π-interactions 1-PF6 (for details, see Supporting Information Figures S1–S2) dominate the solid-state packing. In contrast to 1-BF4, 1-PF6 and 1-Cl have the counterions and solvent molecules (THF and CHCl3, respectively) in close proximity to the ethylene bridge. The differences of the counterion position and additional solvation effects provide a rationale for the large differences observed in the discussed folding angles. The gold (coinage metal) coordination environment is in good agreement with previously reported [M(I)(cis-bdppe)2]PF6 complexes (M = Au and Cu) [28]. In a slight variation of this ligand, the diphosphine 2,3-bis(diphenylphosphino)maleic acid (dpmaa) could also be targeted to form either a mononuclear tetrahedral or a dinuclear gold complex. In view of the high solubility in water, cytotoxicity tests revealed potential applications as anti-tumor agents [29]. The related 2,3-bis(diphenylphosphino)maleic-N-phenylimide ligand showed similar coordination behavior forming both mono- and dinuclear complexes [30, 31].
Despite the easy of formation of the mononuclear complex, treatment of 2 with silver triflate and excess of ligand partially suppressed its formation and a dinuclear complex 3 (Scheme 2 containing two ligands was formed in an inseparable mixture with 1-OTf. Similarly, treatment of 2 with silver tetrafluoroborate led to the identification of 1-BF4. The mononuclear cations 1+ have been identified by their characteristic 31P NMR resonance, as well as their crystal structure (see Supporting Information). Unambiguous identification of the molecular structure of compound 3 was provided by single crystal X-ray diffraction of colorless blocks that crystallized directly from the reaction mixture (Figure 5). Dinuclear complex 3 crystallized in the centrosymmetric space group P-1 with half of a molecule in the asymmetric unit. The gold atoms are modeled with a positional disorder having site occupancy factors of 89.5% and 10.5%. Two gold atoms bridge the ligands with an Au⋯Au distance of 2.9908(4) Å and 3.051(4) for the major and minor component respectively, which is comparable to that observed in 2, despite the shorter intra-ligand P1⋯P2 distance (3.525(2) Å). In addition, π–π-interactions (centroid distances 3.435(8) Å) result in a partially rigidified diphenylphosphine backbone. The inter-ligand P⋯Au⋯P arrangement is slightly contorted (162.3(1)°), forming a near ideal plane plane1 (P1Au1P1′P2′Au1′P2) with an average deviation of 0.1 Å, while the planes spanned by the ligand backbone plane2 (P1C1C21P2) form an overall chair like arrangement and a fold angle 48.8(3)°between plane1 and plane2. In a similar approach, 2 was treated with silver trifluoroacetic acid (2.5 equiv) and subsequently added to a solution of 4,4′-bipyridine; however predominant formation of tetrahedrally coordinated mono-nuclear gold complex was observed.
Attempted synthesis of 3, under the formation of various side products including 1 and unidentified species.
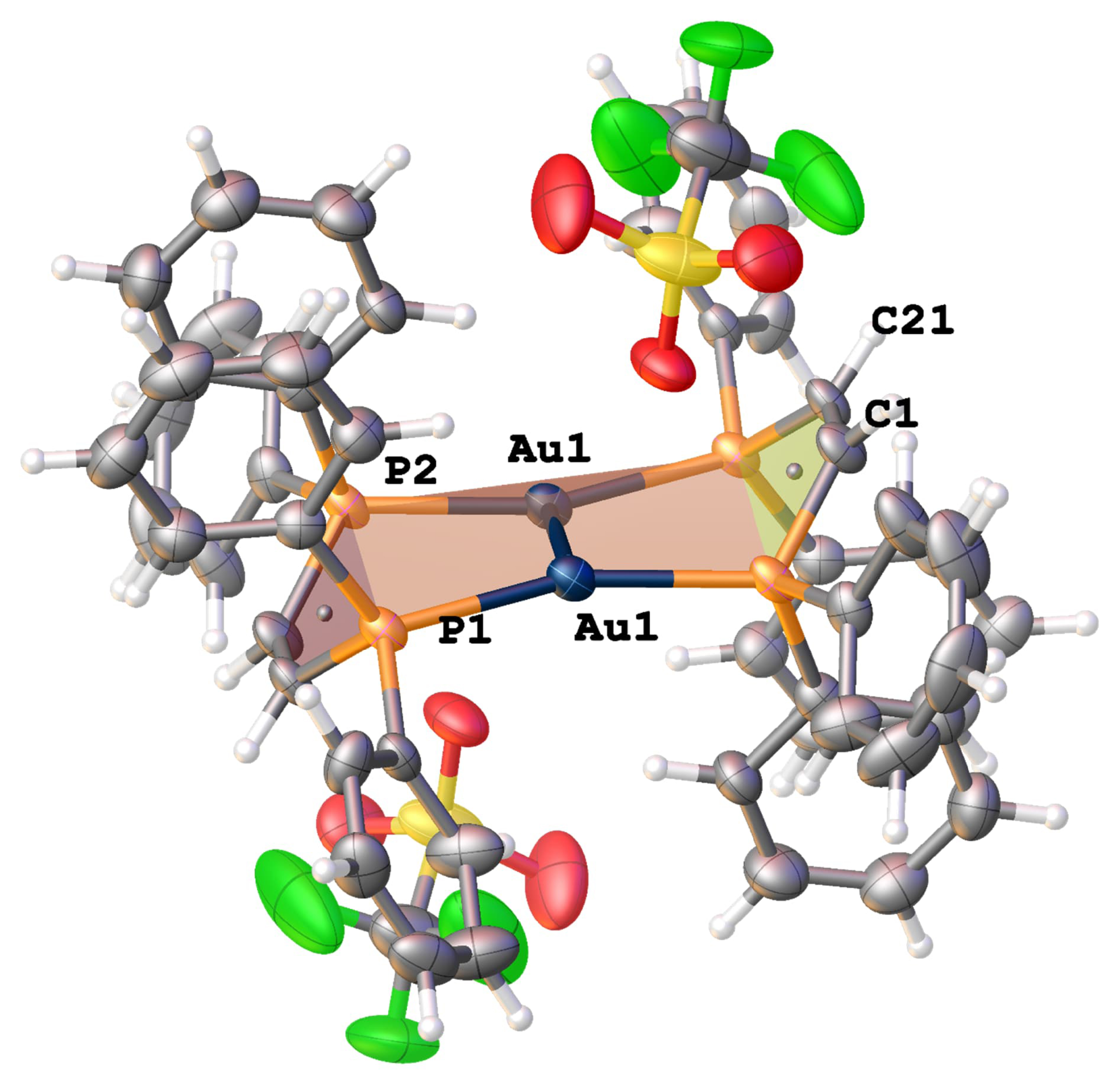
Solid-state structure of dinuclear complex 3. ORTEP style representation with ellipsoids drawn at a 50% probability level. Further crystallographic details in the experimental section.
Neither the mononuclear nor the dinuclear complexes show any appreciable emission with UV irradiation in solution and solid state. This is, in particular, surprising given the short intramolecular Au⋯Au distances of the dinuclear complexes, which is indicative of aurophilic interactions and contrasts the observed emission of higher nuclearity clusters using this and similar ligand systems [32].
3. Conclusions
The chemistry of these rigid diphosphine ligands having an unsaturated spacer is dominated by the balance of chelating and aurophilic effects that lead to mono and dinuclear complexes. Primarily, reaction conditions such as ligand:gold ratio, solvent and concentration of reagents also lead to different products. Further, non-covalent interactions play decisive roles in the solid-state packing and may impact their solution behavior. Notably, the prepared dinuclear gold(I) complexes are non-emissive, which hints at dominating non-radiative decay pathways. With this study we have elucidated few of the factors that direct the coordination behavior, the simple bidentate phosphine ligand towards Au(I) centers, and provide more insights into the rational synthesis of related derivatives.
4. Experimental Section
4.1. General
All chemicals were obtained from commercial suppliers and used as received. All reactions and products were protected from prolonged exposure to sunlight using aluminum foil or amberized glass.
4.2. X-ray crystallography
Crystallographic data sets were collected from single crystal samples mounted on a loop fiber and coated with high viscosity Fomblin PFPE (Hampton Research). Collection was performed using a Bruker SMART APEX diffractometer equipped with an APEXII CCD detector, a 3-circles goniometer, and a MoKα-microfus sealed tube. The crystal-to-detector distance was 5.0 cm, and the data collection was carried out in 512 × 512-pixel mode. Cell refinement and data reduction were performed with SAINT (Bruker AXS), absorption correction was done by multi-scan methods using SADABS [33, 34]. The structure was solved by direct methods and refined using SHELX suite of programs [35, 36] using OLEX2 [37]. Full details concerning the data sets and crystal resolutions can be found in the respective CIF files stored at the Cambridge Crystallographic Data Centre under the allocated deposition numbers CCDC: 2291875-2291879.
4.3. Computational methods
Preliminary optimizations of the geometries were obtained using xTB 6.6.0 [38] at GFN2-xTB level002E. Optimization and frequency analysis at the DFT level were performed using ORCA 5.0.3 [39] with r2SCAN-3c method [40]. Finally, NBO analysis was performed using NBO 7.0.9 software package [41]. The cartesian coordinates of the optimized geometry, the Molden file with the NBOs and the ORCA output of the NBO analysis can be found in a figshare repository [42].
4.4. Synthesis
4.4.1. Synthesis of [AuCl(tht)]
Tetrahydrothiophene gold(I) chloride was prepared in a slightly modified way than reported in literature [43]. Tetrahydrothiophene (C4H8S, 45 μL, 0.51 mmol, 1.4 eq) was added to a suspension of tetrachloroauric acid (H[AuCl4], 123 mg, 0.36 mmol, 1 eq) in water (1 mL)/EtOH (5 mL). The initial yellow precipitate changed to a white precipitate within 15 min. The precipitate was filtered and washed twice with cold EtOH (10 mL) to yield 94% of analytically pure product. The solution was filtered and subjected to slow evaporation of the solvent protected from light, giving the corresponding complexes in good yields (95% and 92%, respectively) as microcrystalline powders. Single crystals suitable for X-ray diffraction were obtained by slowing down the evaporation of THF.
4.4.2. Synthesis of 1-Cl
Tetrahydrothiophene gold(I) chloride (44 mg, 0.14 mmol, 1 eq) and Ph2P–C=C–PPh2 (cis-bdppe, 108 mg, 0.27 mmol, 2 eq) were dissolved in chloroform (5 mL) and stirred with protection from light for 24 h. Filtration, followed by slow evaporation of all solvents gave 1-Cl in 96% yield as large yellow crystalline material suitable for single crystal diffraction. Upon further drying of the material under reduced pressure, removal of residual tht also results in complete discoloration of the material which is ascribed to the loss of chloroform solvate molecules. NMR spectroscopic data is in agreement with previously reported data of the cationic complex 1-OTf and 1-PF6:32 1H NMR (400 MHz, CD2Cl2) δ 7.53–7.40 (m, 4H), 7.33 (dd, J = 10.1, 7.9 Hz, 8H), 7.15 (td, J = 8.0, 6.6 Hz, 25H) ppm. 31P NMR (161.8 MHz, CD2Cl2) δ 23.1 ppm.
4.4.3. Synthesis of 1-BF4 and 1-PF6
A solution of 1-Cl (40 mg) in THF (10 ml) is treated with 1 equivalent of [Ag(MeCN)4)]BF4 or [Ag(MeCN)4)]PF6 for 24 h. The solution is filtered and slow evaporation of the solvent protected from light gives the corresponding complexes in good yield (95% and 92%) containing single crystals suitable for X-ray diffraction.
4.4.4. Synthesis of 2
Importantly, [AuCl(tht)] (70 mg, 0.22 mmol, 2 eq) must be fully dissolved in THF (ca. 80–100 ml) before cis-bdppe (43 mg, 0.11 mmol, 1 eq in 5 mL THF) is added. The reaction is stirred overnight, followed by slow evaporation of the solvent (at all time protect from light), upon which small colourless crystals suitable for x-ray diffraction are obtained. Residual tht is removed under reduced pressure, while impurities arising from formation of 1-Cl can be easily removed by extracting 2 using a minimal amount of THF, which upon solvent removal gives 78% of 2, while the THF insoluble fraction consists mainly of 1-Cl. 1H NMR (400 MHz, CD2Cl2) δ 7.59 (m, 16H) 7.52 (d, J = 7.3 Hz, 8H), 7.47–7.38 (m, 20H) ppm. 31P NMR (161.8 MHz, CD2Cl2) δ 13.6 ppm.
4.4.5. Attempted synthesis of 3
In an attempt to remove the coordinating chloride complex 2 (50 mg, 0.06 mmol) was stirred for 24 h with a silver salt, [Ag(OTf)], in THF (36 mg, 2.5 equiv.) in the presence of excess ligand (24 mg, 1.1 equiv.). The reaction mixture was filtered, and after removal of all volatiles the crude was crystallized from THF and chloroform, giving a product mixture containing 3 (crystalline) and 1-X (where X is either OTf or Cl). Satisfactory spectroscopic data could not be obtained. 31P NMR (THF): −22.6 ppm.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
The authors would like to thank the Swedish Research Council (Vetenskapsrådet project grant 2017-03727 to AO, and 2021-05414 to SC) for financial support. OYOF would like to thank the ERASMUS program for funding their exchange. The computations were made possible by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at the National Supercomputer Centre (Linköping University, project NAISS 2023/22-567) partially funded by the Swedish Research Council through grant agreement no. 2022-06725.