



 CC-BY 4.0
CC-BY 4.0
1. Introduction
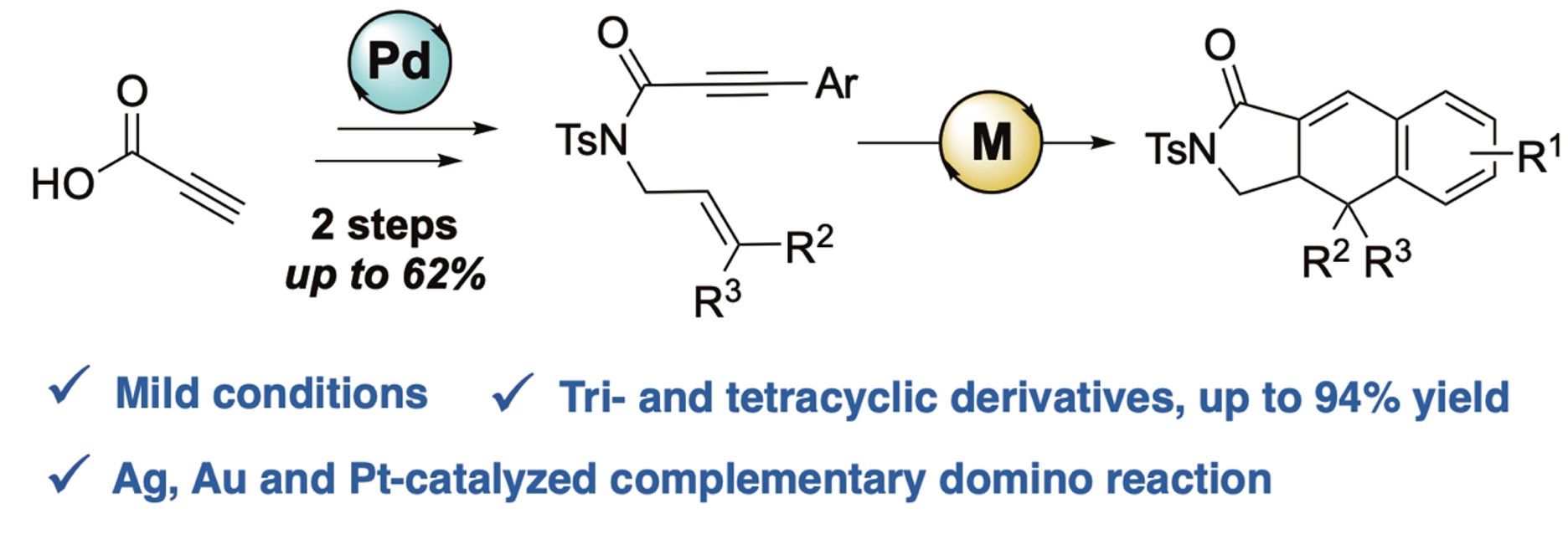
Silver made its first appearance around 5000 BC, recognized as a soft, malleable, ductile, shiny white precious metal. Owing to its outer orbital 5s1 electronic structure, silver is able to form a variety of silver(I) salts/complexes with diverse counterions, such as oxides, sulfides, phosphides, olefin complexes, and others. The typical d10 electronic configuration enables silver to coordinate with many π-donors, including N-, O-, and S-donor groups like alkynes, alkenes, and various unsaturated bonds containing carbon and heteroatoms [1, 2, 3, 4, 5]. Furthermore, because of the vacant f orbitals and relativistic contraction, Ag demonstrates σ- or π-Lewisacidity with a tendency for σ-coordination over π-coordination [6, 7]. The first documented instance of a silver-catalyzed reaction, converting ethylene into ethylene oxide, was noted in the 1930s [8]. Historically, silver was perceived to have lower catalytic activity and efficiency compared to other transition metals. However, in recent decades, the exploration of silver-based catalysts has progressed swiftly under both heterogeneous and homogeneous conditions [9, 10, 11, 12, 13, 14, 15]. Regarding 1,6-enynes, the literature contains a limited number of cases of silver-catalyzed cyclizations. Porcel and Echavarren reported in 2007 the first silver-catalyzed intramolecular carbostannylation of 1,6-enynes utilizing [AgOTf(PPh3)]3 as a catalyst [15] (Scheme 1, Equation (1)). In 2013, Shin’s group disclosed silver-catalyzed cycloisomerization reactions of propiolamide derived 1,6-enynes in the case of N-arylamides [16] (Scheme 1, Equation (2)). For this kind of substrate, the carbonyl group was demonstrated to be a key group for the silver-catalyzed 5-exo-dig selective cyclization. The employment of Au(IPr)NTf2 led to lower selectivity, with 1,4-diene accompanied by 1,3-diene. A 6-endo-dig cyclization of 1,6-enyne systems was proposed in the presence of a gold(I) cationic system [17, 18]. Moreover, theoretical calculations showed the importance of both the aryl group in the terminal position of the alkyne and the carbonyl group (Scheme 1, Equation (3)). Following our endeavor towards organometallic catalysis and more specifically silver- and gold-catalyzed cyclization [19, 20, 21, 22, 23, 24], we became interested in using propiolamide as the linker for enyne derivatives. We and other authors recently described preliminary results on intramolecular [4+2]-cycloaddition of N-(3-methylbut-2-en-1-yl)-amide-1,6-enyne in the presence of Ag catalyst [16, 23, 24]. We report herein our full study including the complementary activity of silver, gold, and platinum complexes (Scheme 1, Equation (4)).
[4+2]-cycloaddition reaction of N-alkenylamide-1,6-enynes.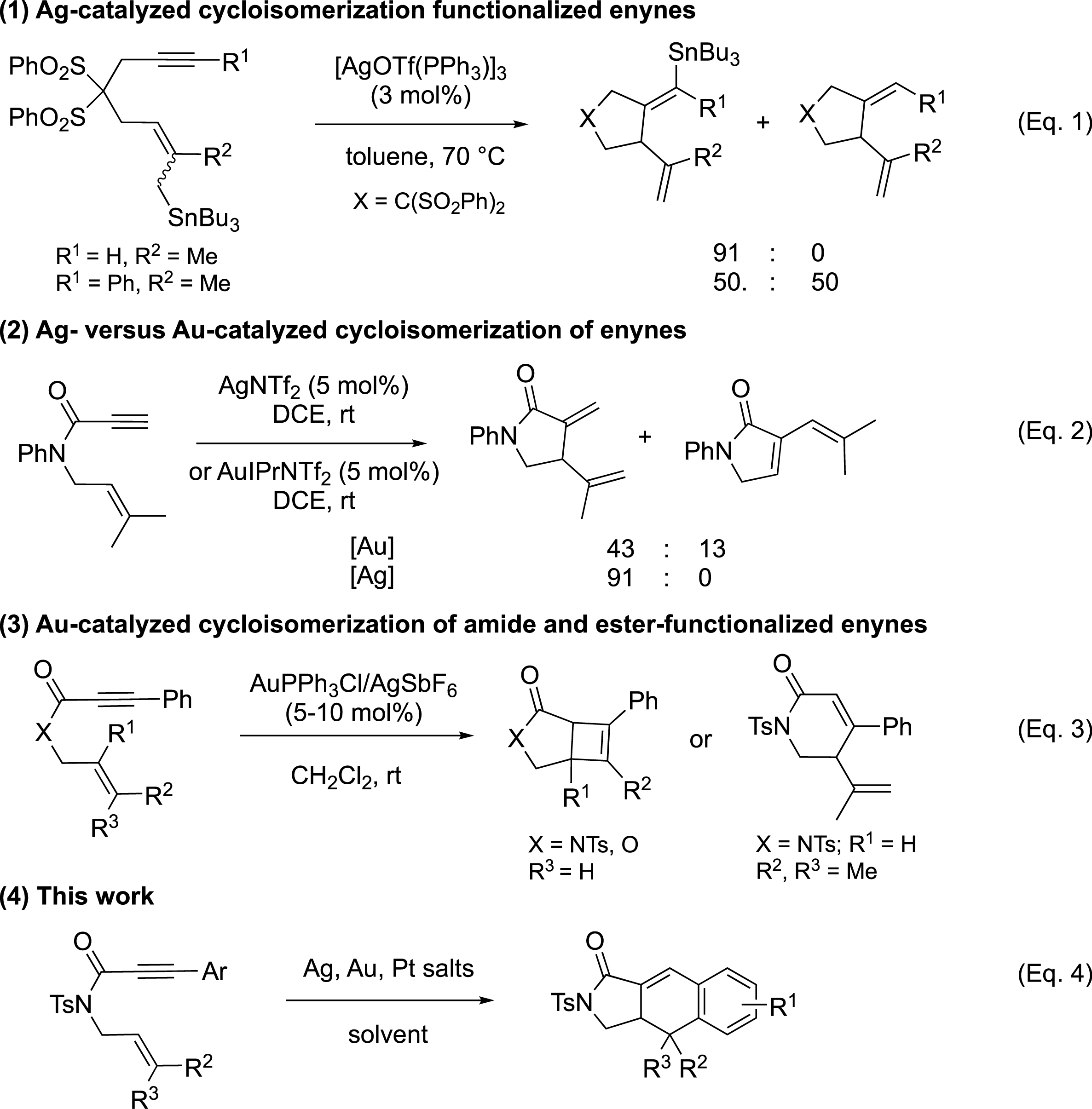
2. Results and discussion
The syntheses of the starting enyne derivatives 5a–5l were accomplished through a three-step reaction sequence (Scheme 2). The strategy to prepare N-tosylpropiolamides was realized starting from the aryl alkynyl carboxylic acids 3a–3e, which were either commercially available (3a) or obtained by Sonogashira cross-coupling from propiolic acids 3b–3e and aryl iodides (Scheme 2). Different aryl alkynyl carboxylic acids were prepared via a copper-free Sonogashira coupling from propiolic acid and aryl halides. This method implied the use of 1,8-diazabicyclo[5.4.0]undec-7-ene/DBU (2.4 equiv) and Pd(PPh3)4 (2.5 mol%) in DMSO. As shown in Scheme 2, the aryl alkynyl carboxylic acids 3b–3e bearing electron-donating groups such as ortho-methyl, para-methyl, and para-methoxy and electron-withdrawing groups such as para-bromo, meta-bromo, and para-CF3, on the phenyl ring, were efficiently obtained in 58% to 85% yields. The next step was the condensation of commercially available 4-methylbenzenesulfonyl isocyanate and various bromo-alkenes. This one-pot process was performed on the prepared carboxylic acids 3b–3e by treatment with allylic bromides, and several functionalized enynes were isolated (5a–5l) in modest to good yields (29–79%).
A three-step synthesis of 5a–l.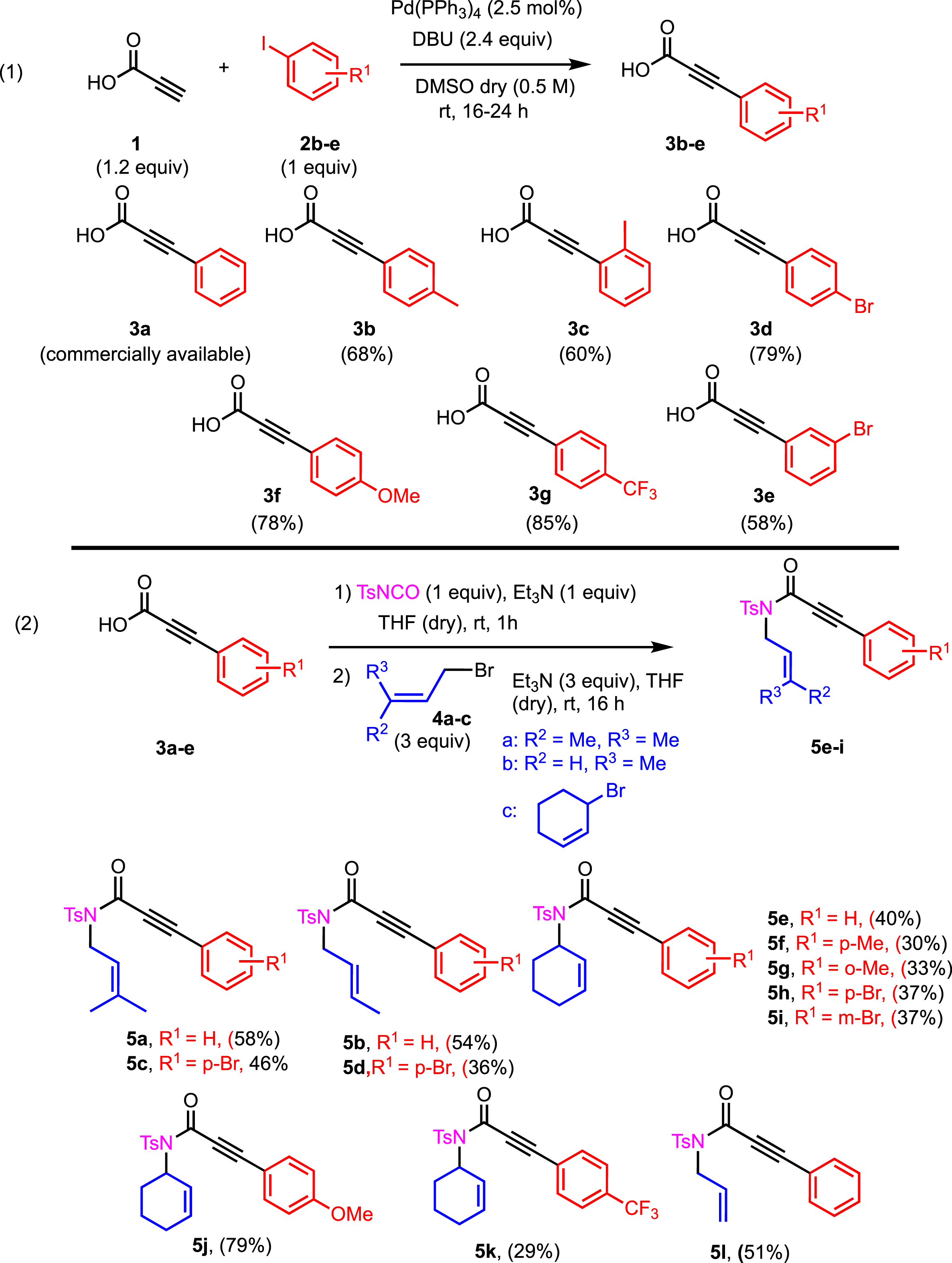
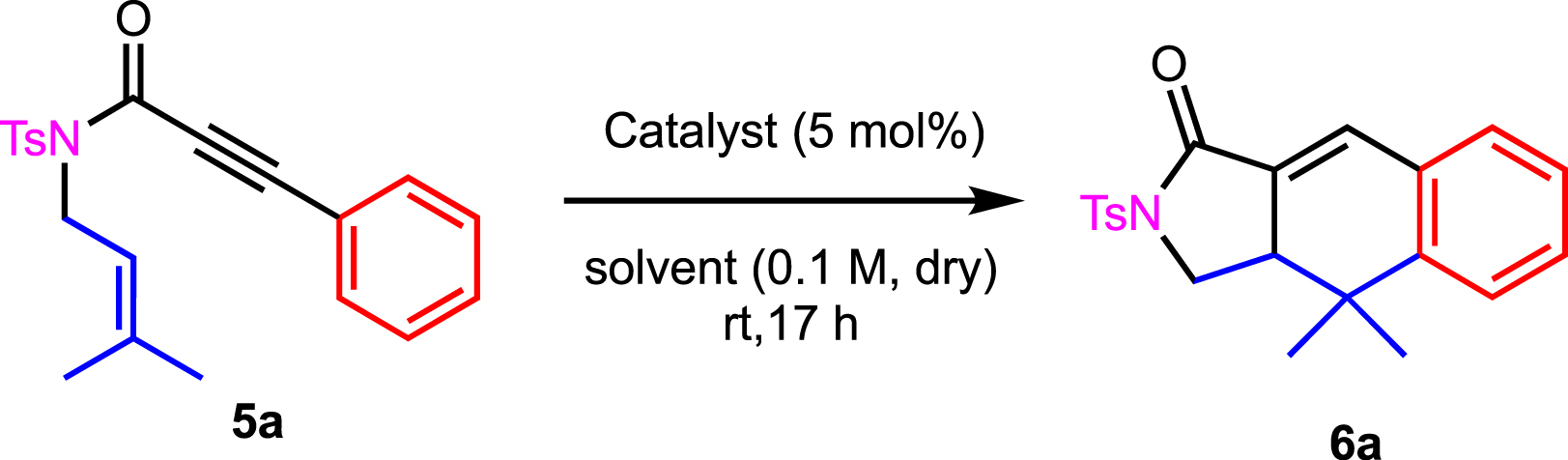
We started to investigate the reaction of enyne 5a in the presence of a catalytic amount of silver salt (Table 1). Previous studies have shown that dichloromethane and nitromethane were the best solvents for such transformation [23, 24]. We therefore made a large screening of silver salts (Table 1, entries 1–16) in both solvents. The best results were obtained in the case of AgSbF6 and AgNTf2 in dichloromethane (Table 1, entries 1 and 3, respectively). As anticipated, AgBF4 and AgOTf gave similar results (Table 1, entries 5 and 6; 7 and 8, respectively). Some salts like AgF and AgClO4 gave no conversion, most probably because of low solubility in the solvents (Table 1, entries 9 and 10; 13, respectively). We tried to correlate the activity of silver salts with the pKa of their respective counteranion [25], but no trends were obtained. We also wanted to compare the reactivity of 5a in the presence of silver and gold complexes. We therefore engaged enyne 5a in the presence of the cationic PPh3AuNTf2 complex, which prevented any presence of silver salt. A modest activity of gold was observed, allowing the formation of 6a in 50% and 36% yield, respectively, in dichloromethane and nitromethane (Table 1, entries 17 and 18, respectively).
Optimization of reaction conditions
| Entry | Catalyst | Solvent (M) | Yielda (%) |
|---|---|---|---|
| 1 | AgSbF6 | CH2Cl2 | 97 |
| 2 | AgSbF6 | CH3NO2 | 54 |
| 3 | AgNTf2 | CH2Cl2 | 77 |
| 4 | AgNTf2 | CH3NO2 | 69 |
| 5 | AgOTf | CH2Cl2 | 65 |
| 6 | AgOTf | CH3NO2 | 28 |
| 7 | AgBF4 | CH2Cl2 | 61 |
| 8 | AgBF4 | CH3NO2 | 55 |
| 9 | AgF | CH2Cl2 | Traces |
| 10 | AgF | CH3NO2 | nr |
| 11 | AgOOCCF3 | CH2Cl2 | 34 |
| 12 | AgOOCCF3 | CH3NO2 | 9 |
| 13 | AgClO4 | CH2Cl2 | nr |
| 14 | AgClO4 | CH3NO2 | 48 |
| 15 | AgPF6 | CH2Cl2 | 6 |
| 16 | AgPF6 | CH3NO2 | 49 |
| 17 | PPh3AuNTf2 | CH2Cl2 | 50 |
| 18 | PPh3AuNTf2 | CH3NO2 | 36 |
a 1H NMR yields with 3,4,5 trichloropyridine as internal standard.
With the optimized conditions in hand (Table 1, entry 1), the scope and limitations of this domino transformation were evaluated (Scheme 3). The dimethyl derivative 6a was isolated in 89% yield. It was obvious to note that the cyclohexenyl side chain was compatible with the whole process and led to the tetracyclic derivatives 6e–k in moderate to good yields. The presence of a bromide was well tolerated, as the cyclic adduct 6c was isolated in an excellent 94% yield. The success of the cyclization appeared to be moderately dependent on the electron-donating or electron-withdrawing character of the substituent R1 on the aryl group. The presence of an electron-withdrawing group in the para position of the alkyne led to a lower isolated yield in the case of the cyclohexyl substituent. Compounds 6h and 6k were obtained in 38% and 34% yields, respectively. The presence of an electron-donating group or an electron-withdrawing group in the meta position allowed the formation of the desired derivatives in 50% (for 6j) and 76% (for 6i) yields.
Scope and limitations for the synthesis of 6a–l.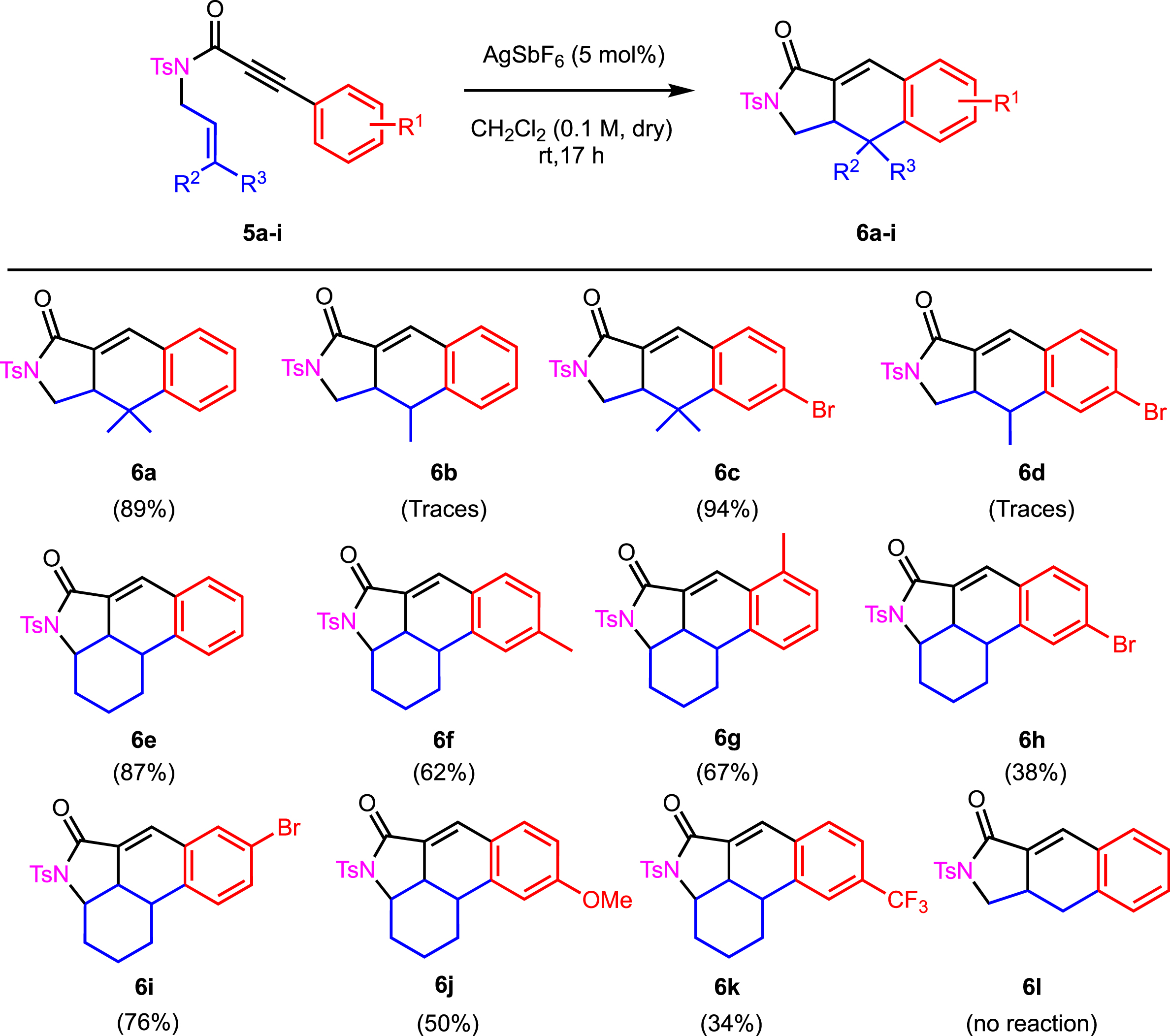
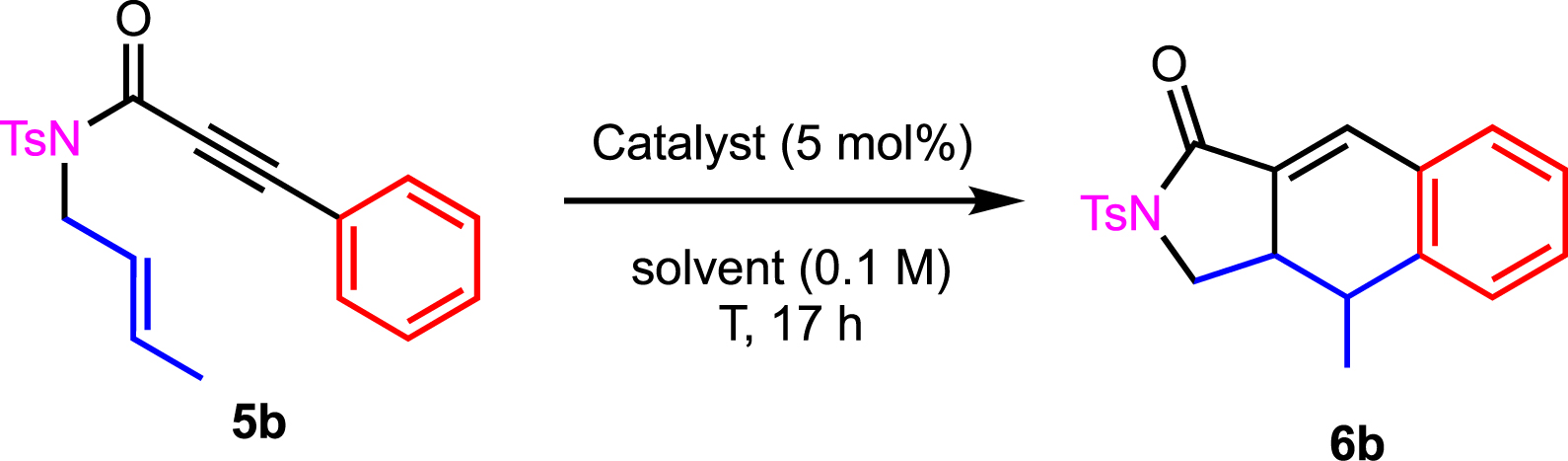
Interestingly, the disubstitution of the alkene moiety was crucial to the cyclization. Traces of 6b and 6d were detected whereas no reaction was observed in the case of 6l. We therefore turned our attention to substrates 5b, 5d, and 5l to further revisit their potential reactivity (Table 2, Scheme 4).
Optimization of reaction conditions for 5b
| Entry | Catalyst | Solvent (M) | T (°C) | Yielda (%) |
|---|---|---|---|---|
| 1 | PPh3AuNTf2 | CH2Cl2 | rt | 26 |
| 2 | IPrAuNTf2 | CH2Cl2 | rt | 10 |
| 3 | PtCl2 | CH2Cl2 | rt | - |
| 4 | PPh3AuNTf2 | CH3NO2 | rt | 9 |
| 5 | PPh3AuNTf2 | DCE | rt | 21 |
| 6 | PPh3AuNTf2 | Toluene | rt | 33 |
| 7 | PPh3AuNTf2 | DCE | 80 | 81 (66)b |
| 8 | PPh3AuNTf2 | Toluene | 80 | 77 |
| 9 | IPrAuNTf2 | DCE | 80 | 75 |
| 10 | PtCl2 | DCE | 80 | 63b |
| 11 | PtCl2 | Toluene | 100 | 67 |
a 1H NMR yields with 3,4,5 trichloropyridine as internal standard; b isolated yield.
Reactivity of 5d and 5l.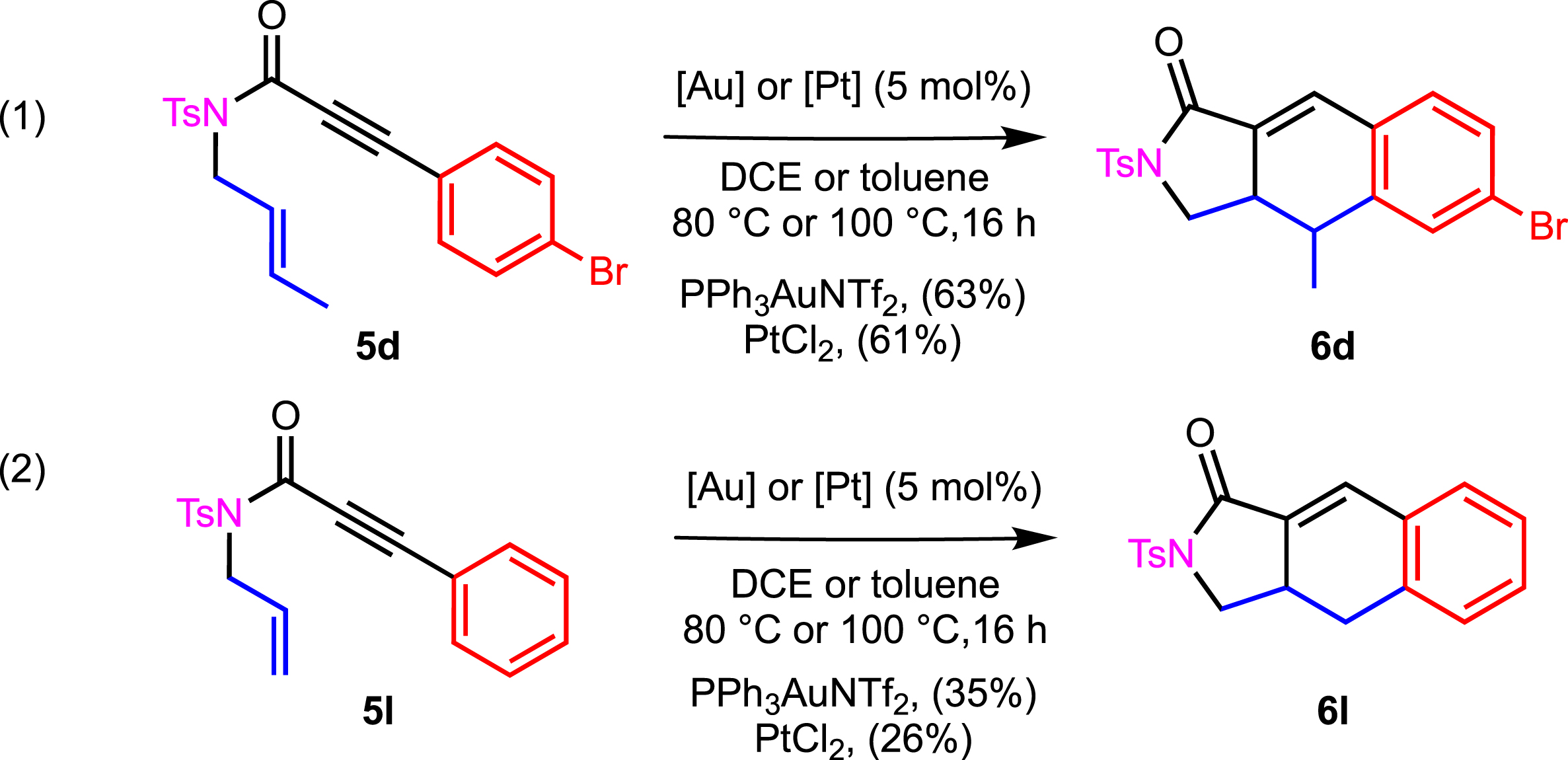
In order to obtain 6b, we initiated this study by investigating the reaction in the presence of gold or platinum complex in dichloromethane at room temperature (Table 2, entries 1–3). Low to no conversions were observed. Switching to other solvents such as nitromethane, dichloroethane, or toluene did not give better results (Table 2, entries 4–6).
Increasing the temperature was the key parameter, and in DCE as well as in toluene, the desired product 6b was isolated in 66% and 63% yields (Table 2, entries 7–11) with PPh3AuNTf2 and PtCl2 complexes, respectively. We therefore found three sets of optimized conditions (Table 2, entries 7, 9, and 11) for which the yield of 6b did not change significantly.
Under these conditions, the cyclization of 5d and 5l was attempted (Scheme 4). The bromo derivative 6d was isolated in similar yields of 63% and 61% by using gold and platinum, respectively. The case of 5l was still tricky, as non-substituted adduct 6l was obtained only in the case of PtCl2, with a very modest yield of 26%. The yield was slightly increased to 35% in the presence of PPh3AuNTf2, which is a preliminary promising result.
These results appeared to indicate that the reactivity of the propiolamide derived 1,6-enynes derivatives could be realized by a choice of the metal depending on the substrate (Scheme 5). Based on our studies and literature precedents [15, 16, 17, 18, 23, 24], the first step of the mechanism would be a π-activation of alkyne 5 by coordination of the metallic complex to the triple bond and potentially the carbonyl in the case of silver salts. Subsequent 5-exo-dig addition of the alkenyl moiety would lead to intermediate B. This intermediate would be stabilized in the case of R2 and R3 different from an hydrogen. The Lewis acid properties of the metallic catalyst therefore play a crucial role. In the case of disubstituted alkenes, the silver metal may induce further cyclization. In the case of the mono-substituted alkenyl side chain, or the non-substituted chain, the transformation needs a higher electrophilic metal and therefore gold or platinum complexes are efficient. The third elemental step would be the addition of the aromatic ring, according to a Friedel–Crafts type addition, which would therefore be favored in the case of electron-donating groups. At this point, the presence of a substituent on the alkenyl side chain may also stabilize the cationic aryl moiety. Intermediate C would be then transformed to the cyclic adduct 6, and the catalyst regenerated by a protodeauration step.
Mechanistic proposal.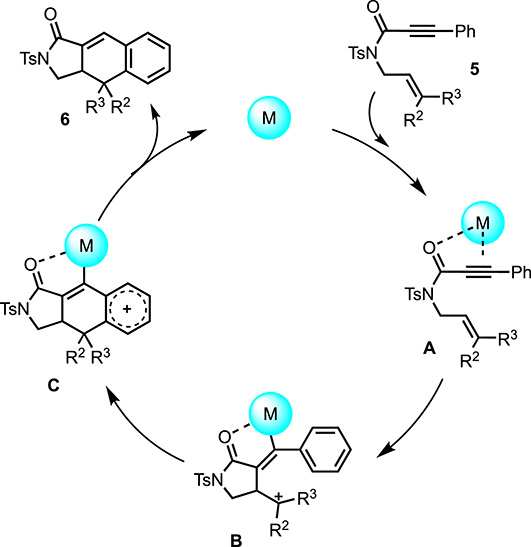
3. Conclusion
In summary, we have extended the method of an intramolecular domino metal-catalyzed sequence of N-(alkenyl)-amide-1,6-enynes leading to 2,3,3a,4-tetrahydro-1H-benzo[f]isoindol-1-one derivatives. The optimization of this process in the presence of silver complexes revealed the importance of Lewis acid properties in view of the alkenyl side chain. For mono- and non-substituted enynes, gold and platinum complexes were found to be efficient. The 5-exo-dig addition followed by a Friedel–Crafts arylation led to tri- and tetracyclic derivatives in good to excellent yields. These results give quite a positive prospect for the improvement of catalytic domino processes. Further studies will be dedicated to the optimization of an asymmetric version and the access to biologically active building blocks.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Acknowledgments
This work was supported by the Centre National de la Recherche Scientifique and the Université Côte d’Azur. We gratefully acknowledge UniCA for a Master grant to LJPP and China Scholarship Council for PhD grants to KM and XC. We are grateful to the MS3U-FR2769 platform (Sorbonne Université) for HRMS analyses.