



 CC-BY 4.0
CC-BY 4.0
1. Epoxidized natural rubber as a foundation for sustainable material
As industries increasingly seek sustainable alternatives to traditional rubber [1, 2, 3], epoxidized natural rubber (ENR) has gained attention for its unique dual functionality, which enhances its potential as an eco-friendly material. Derived from natural rubber through epoxidation, ENR combines the elasticity and resilience of natural rubber with the added chemical reactivity of epoxides [4].
The introduction of epoxides into the rubber backbone imparts polar functionalities that broaden ENR compatibility with other polymers and improve its resistance to oils, gases, and environmental degradation [5, 6, 7]. These polar groups also enable ENR to interact with crosslinking agents in new ways, facilitating innovative crosslinking strategies that can bypass traditional sulfur vulcanization. This versatility opens the door to creating materials that maintain or surpass the mechanical durability of standard rubber while achieving characteristics essential for modern sustainable design, such as recyclability and extended material lifespan.
Our research from 2009 to 2017 has explored diverse strategies to advance ENR from basic blends to sophisticated multifunctional systems, addressing the complex trade-offs among flexibility, toughness, reprocessability, and environmental sustainability. These efforts have achieved significant progress in developing ENR-based materials that leverage ENR’s unique dual functionality. These advancements include self-vulcanizing ENR blends and novel crosslinking methods that utilize chemical or physical links. Such approaches allow for the formation of robust yet reprocessable networks, setting the foundation for next-generation elastomers that balance mechanical strength with environmental benefits. This manuscript traces the evolution of ENR materials from self-crosslinking blends to crystalline grafts, highlighting how the dual functionality of ENR can transform it into a model for sustainable rubber development.
2. Self-vulcanizing ENR blends with dicarboxylic acids
The research primarily focused on blending ENR with dicarboxylic acids like dodecanedioic acid (DA) for “self-vulcanizable” blends, which cured at high temperatures (around 180 °C) without any of the additives required for sulfur-based crosslinking [8]. This new ENR/DA system was designed to address the fatigue–aging trade-off in rubbers.
Indeed, natural rubber (NR) demonstrates varying resistance to repetitive stress (fatigue) and long-term durability (aging) depending on the crosslinking method used, though these two properties are often incompatible. Two primary crosslinking methods exist for natural rubber: peroxide crosslinking and sulfur crosslinking, the latter known as vulcanization. Vulcanization involves incorporating various ingredients; sulfur-only crosslinking can take several hours, which makes it inefficient for industrial use. Instead, the reaction is optimized to completion in a few minutes at a maximum temperature of 160 °C. To achieve this, accelerators are added, which are classified based on their effectiveness: slow (30–60 min, such as guanidines), moderate (15–30 min, including thiazoles and sulfenamides), and fast (10 min, like thiurams and carbamates). To maximize accelerator performance, activators are added, typically zinc oxide and a fatty acid like stearic acid. Other additives, such as antioxidants and reinforcing fillers, are also included [4].
Adjusting the accelerator-to-sulfur ratio results in distinct crosslinking networks, producing three vulcanization types (Table 1). Conventional vulcanization (CV) uses a high sulfur content, creating a network primarily composed of polysulfide bridges. On the other hand, the efficient vulcanization (EV) system uses a low sulfur content, converting polysulfide groups into di- and monosulfide bridges. Semi-efficient vulcanization (SEV) results from intermediate ratios, incorporating both poly- and monosulfides [4]. The chosen vulcanization method significantly influences the rubber’s final properties, tailoring it to specific applications. For high fatigue resistance—required for parts subjected to frequent mechanical stress—the conventional formulation is ideal, though it has limited aging resistance. Conversely, when thermal and oxidative resistance is prioritized, efficient vulcanization is preferred, sacrificing some fatigue durability (Table 1).
Three types of vulcanization (CV, SEV, and EV) determine the performance characteristics of rubbers (adapted from reference [4]).
| Vulcanization type | Conventional (CV) | Semi-efficient (SEV) | Efficient (EV) |
|---|---|---|---|
| Accelerator/sulfur ratio | 0.1–0.6 | 0.7–2.5 | 2.5–12 |
| Polysulfur bridges % | 95 | 50 | 20 |
| Monosulfur bridges % | 5 | 50 | 80 |
| Aging resistance | Poor | Intermediate | High |
| Fatigue resistance | High | Intermediate | Poor |
Peroxide crosslinking, in contrast, requires no accelerators or activators. The resulting network, formed of carbon–carbon bonds, offers greater aging resistance but lower fatigue durability.
The fatigue–aging balance is influenced by the dissociation energy of the bonds involved [9]. Table 2 provides estimates of these bond energies. Due to the relatively low energy of the S–S bond, disulfide bridges break before the C–C bonds in the polymer backbone but can subsequently re-form at new attachment points, enabling the material to withstand considerable stress. As a result, networks with polysulfide bridges (CV) exhibit higher resistance to repeated mechanical stress compared to networks with monosulfide (EV) or carbon–carbon (peroxide) bonds. In contrast, C–C–C bonds offer greater resistance to thermal and oxidative stress, enhancing the material’s aging durability.
Approximate bond energies in rubbers (data from [9]).
| C–C–C | 93 kcal⋅mol−1 | Peroxide curing: aging > fatigue |
| C–S–C | 50–60 kcal⋅mol−1 | Vulcanization: fatigue > aging |
| C–S–S–C | 35 kcal⋅mol−1 | |
| C–(S)x–C | 27 kcal⋅mol−1 |
With the DA-crosslinked ENR system, we aimed to shift the fatigue–aging compromise by creating a network different from those formed by sulfur or peroxide crosslinking. The crosslinking bridges had to be both flexible and thermally stable, which justified the choice of bridges formed from long aliphatic chains. Dicarboxylic acids were selected as crosslinking agents to exploit the epoxides in ENR, enabling stable network formation.
The crosslinking reaction with dicarboxylic acids exploits the epoxy functionality of ENR to create robust bridges as depicted in Figure 1A. This process results in a chemically crosslinked ENR network, referred to as DA-ENR in the following. To explore the effect of epoxidation on crosslinking, three epoxidation ratios (m) were investigated: 0.1, 0.25, and 0.50, corresponding to ENR10, ENR25, and ENR50, respectively. By modifying crosslinking densities and ratios of ENR to DA, the study identified an optimal ENR/DA ratio of p = 25 for ENR25 (Figure 1B). The ratio epoxide/diacid denoted by p is the number of epoxides for 1 diacid molecule. For ENR25, p = 25 corresponds to 25 epoxide units for 1 diacid, that is, 1 diacid for 100 total monomer units of the polymer backbone. This optimal ratio produced materials with good resilience (60%) and promising stress–strain properties (Figure 1B).
(A) Crosslinking of ENR by dodecanedioic acid (DA) in the presence (or not) of 1,2-dimethylimidazole (DMI). The epoxidation ratio m is equal to 0.1 (ENR10), 0.25 (ENR25), or 0.50 (ENR50). The imidazole in parenthesis was not involved in the initial experiments. (B) Strain–stress measurements after curing ENR25 with DA for 3 h at 180 °C in a press, at various epoxy/acid p ratios (after reference [10]). (C) Acceleration of ENR25/DA crosslinking with stoichiometric amount of DMI (red curve). (D) Effect of the DMI/DA ratio—from 0 to 400%—on the stress–strain properties of DA-ENR25 (p = 25) after curing at 180 °C. For each ratio, the indicated curing time was adjusted at the G′ plateau (after reference [11]).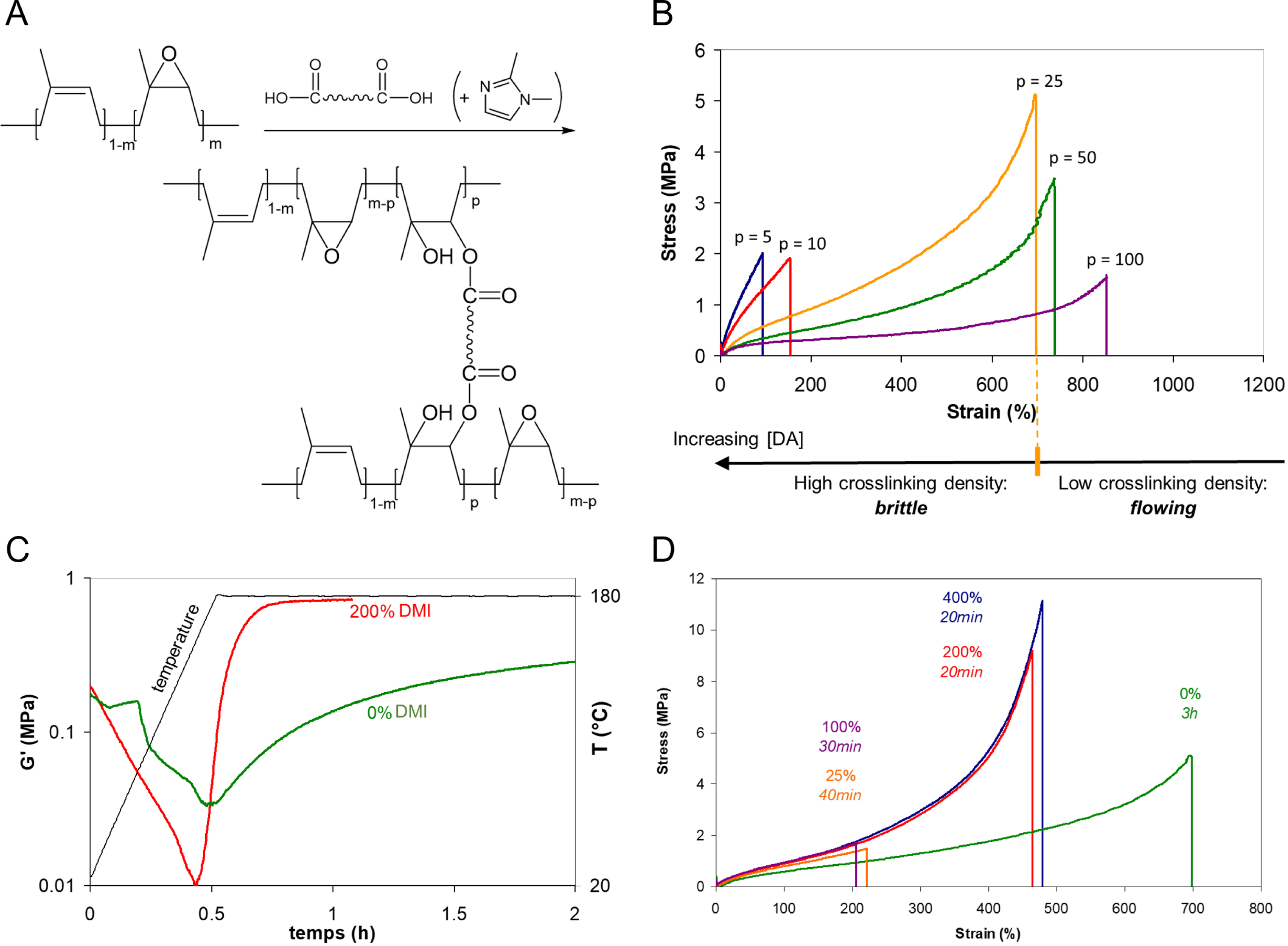
The entire DA-ENR synthesis was conducted in bulk, using an internal mixer for blending and a hot press for crosslinking, the whole process being performed without solvents. This method minimizes environmental impact by eliminating additional reagents and waste, offering a green alternative to traditional sulfur-based vulcanization.
3. Accelerating crosslinking with imidazole compounds
Nevertheless, the initial synthesis required long curing times and high temperatures, deleterious to material properties. A breakthrough came with the introduction of imidazole compounds, particularly 1,2-dimethylimidazole (DMI), as accelerators in the crosslinking process [11]. To ensure homogeneous dispersion of DMI, which is in liquid state at the operating temperature, kaolin—a chemically inert filler—was incorporated during the compounding step in the internal mixer. DMI enabled the curing of ENR/DA blends at significantly lower temperatures and reduced curing times (Figure 1C).
Mechanical testing revealed that DMI-accelerated ENR/DA blends achieved optimal tensile properties at the stoichiometric DMI/CO2H ratio—one DMI molecule per CO2H group, equivalent to 200% DMI per diacid (Figure 1D). For each ratio, the indicated curing time was determined at the point where the storage modulus (G’) stabilized, indicating the completion of the crosslinking process. Since one DMI molecule per CO2H group was required to activate the crosslinking, it was hypothesized that imidazole acted as an accelerator rather than a catalyst. The ionization of carboxylic acid seemed to be crucial to accelerating the esterification reaction. However, a simple acid–base reaction leading to the formation of carboxylate groups did not explain the acceleration of the ring opening of the epoxide. Parallel experiments using other bases like KOH or a tertiary amine did not show any increase in the crosslinking rate. It was hypothesized that the formation of reactive imidazolium carboxylates would facilitate the esterification reaction between the epoxides of ENR and the dicarboxylic acid. This hypothesis was validated through subsequent 13C solid-state NMR analysis, which revealed the ester nature of the crosslinks and confirmed the formation of an intermediate imidazolium dicarboxylate species. These species were found to be well dispersed within the elastomeric matrix, contributing to the uniformity and stability of the crosslinked network [12].
The shortened crosslinking time obtained with DMI not only improved material performance but also significantly reduced the energy and time required for curing, providing a greener alternative to traditional sulfur vulcanization. This innovation significantly enhanced ENR’s appeal for industrial applications by opening up new possibilities for energy-efficient production of elastomers.
An industrial version of the ENR/DA/DMI system was developed, incorporating antioxidants and reinforcing fillers. Its fatigue and aging properties were evaluated at the Hutchinson Research Centre (Montargis, France) and compared to those of the ENR crosslinked with sulfur or peroxide. This study resulted in a material with properties intermediate between sulfur- and peroxide-crosslinked systems (Figure 2) [13, 14].
Comparison of properties achieved through ENR25 crosslinking using dicarboxylic acid (DA), peroxide, or sulfur under conventional (CV), semi-efficient (SEV), or efficient (EV) vulcanization conditions. Inset shows ENR25 test specimens undergoing fatigue testing at the Hutchinson Research Centre.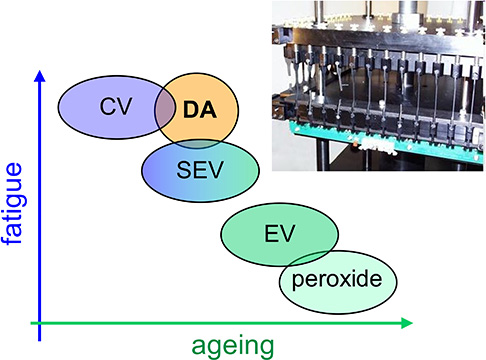
In summary, the successful crosslinking of ENR using carboxylic diacids, accelerated by stoichiometric proportions of DMI, has opened new avenues for the development of advanced systems. This approach enables ENR to be efficiently crosslinked with various functionalized carboxylic diacids or modified with chains bearing reactive or functional groups, such as reversible, supramolecular, or crystallizable grafts. In the following sections, we delve into further functionalizations: first, a diacid incorporating an exchangeable S–S bond, and second, a monoacid bearing a crystallizable tail. Since this research, the ENR/DA derivative/DMI system has attracted significant attention in the literature, particularly for its potential in producing eco-friendly, self-healing ENR materials [15, 16, 17, 18, 19, 20].
4. Thermoreversible chemical ENR networks using exchangeable bonds
With the establishment of DA-crosslinked ENR as a versatile, functionalized elastomer, the next major advancement was to address the recycling challenges associated with rubber. Traditional crosslinked rubber materials are not reprocessable due to their permanent covalent bonds [2]. We introduced reversible crosslinking by using disulfide bonds within the ENR network, which can undergo bond rearrangements when heated, allowing partial reprocessing and extension of the rubber usable life [21].
Disulfide groups were introduced into the rubber using dithiodibutyric acid (DTDB) as a crosslinker (Figure 3A). DTDB-cured ENR presented strong elastomeric properties due to the high molecular weight of the rubber and efficient DA crosslinking. The material behaves like a standard natural rubber until 100 °C and becomes reprocessable above 150 °C owing to disulfide rearrangements. Mechanical tests confirmed that these disulfide-incorporated ENR materials exhibited self-healing capabilities, retaining their mechanical integrity even after multiple cycles of stress relaxation and reprocessing. Rheological analysis further highlighted that these materials could be recycled at 180 °C. After grinding to a fine powder, the reprocessed crosslinked rubber recovers most of its initial mechanical properties (Figure 3B).
(A) Crosslinking of ENR25 with DTDB/DMI and schematic representation of the disulfide-containing network. (B) Recycling experiment. Tensile tests before (line) and after (dashed) the recycling experiment (red: DTDB-cured; blue: DA-cured). The image illustrates dumbbell-shaped cured rubber sample finely grounded and then reshaped by compression molding (after reference [21]).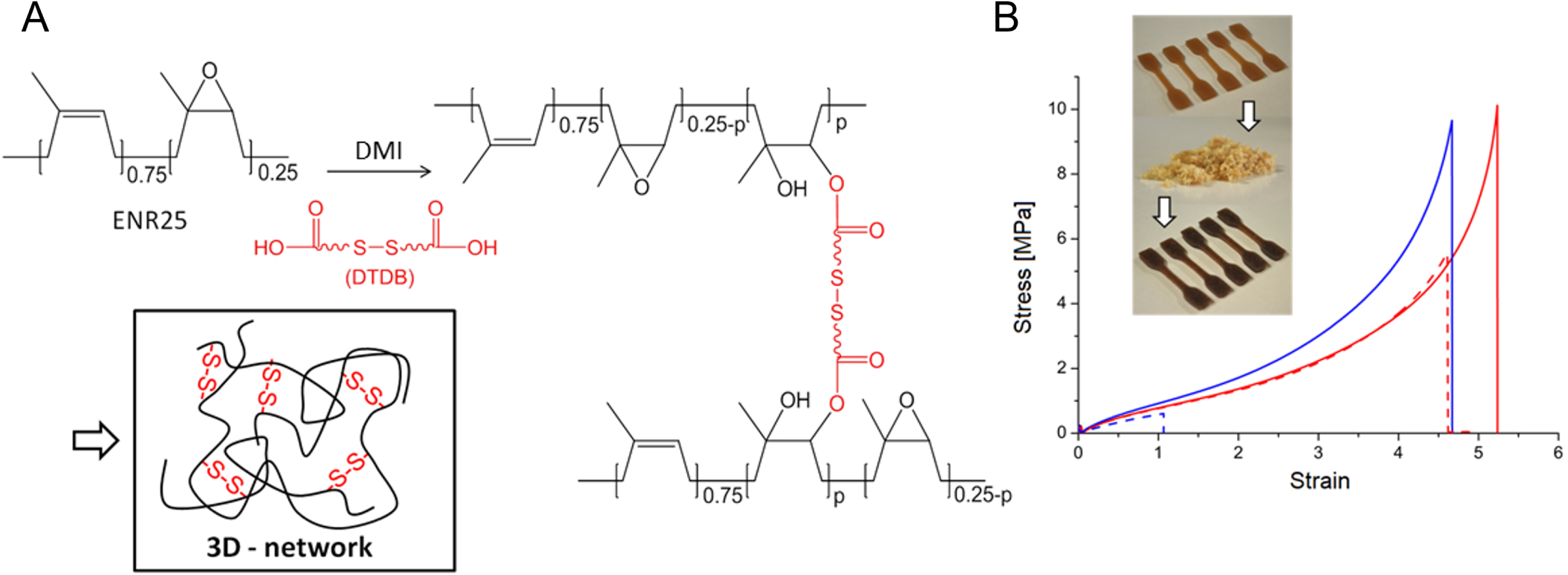
Perspective: comparison with sulfur vulcanization
Since ENR can be crosslinked and recycled through disulfide bond rearrangements, it is important to compare this method with traditional sulfur vulcanization in terms of mechanical performance, aging behavior, and recyclability to evaluate its environmental impact.
Sulfur vulcanization is an established industrial process, yet its chemistry remains incompletely understood. Polysulfide rearrangements are known to enhance rubber fatigue resistance, but this mechanism has seen limited use in rubber recycling. Recycling via hot compression of pulverized vulcanized rubber, without adding new rubber or additives, has been sparsely documented, typically focusing on industrial applications rather than on sulfur network behavior [22, 23, 24]. Historically, sulfur-vulcanized rubber stress relaxation at high temperatures was examined by Tobolsky et al. [25] and later by Berry and Watson [26], who debated the mechanisms behind this phenomenon [27, 28]. They attributed stress relaxation to network degradation, directly correlated with the number of elastically active chains. Later on, Tamura and Murakami highlighted the role of oxygen in this relaxation, noting the impact of polysulfide bonds, which can undergo exchange causing stress relaxation [29].
To compare our DTDB crosslinking process with sulfur vulcanization, we crosslinked ENR under CV conditions, which favor polysulfide bridges due to an excess of sulfur relative to the accelerator (sulfur/accelerator ratio of 7), enhancing fatigue resistance. The ingredients were mixed at 40 °C for 20 min and then cured at 150 °C under 8 tons of pressure for 30 min as determined kinetically in the rheometer. All details of this study are reported in reference [30]. Key findings include the following:
- Crosslink density and stress relaxation: Both ENR/S and ENR/DTDB exhibited similar crosslink densities and stress relaxation patterns, reaching a plateau indicative of secondary reactions in both systems.
- Creep-recovery performance: Creep-recovery tests revealed a slight advantage for DTDB at 150 °C, where ENR/DTDB showed linear deformation unlike ENR/S, which displayed curve flattening due to side reactions occurring at lower temperatures in polysulfide bonds.
- Recyclability: ENR25/S and ENR25/DTDB showed comparable recyclability, retaining about 80% in deformation and 50% in tensile strength post-recycling. However, ENR/S exhibited modulus loss, possibly due to chain scission, not observed in ENR/DTDB.
- Aging resistance: Aging tests indicated that ENR/DTDB degraded less than ENR/S at 150 °C, attributed to the lower reactivity of disulfides against oxidation, unlike the polysulfides in sulfur vulcanization, which produced degradation-promoting acids.
In conclusion, as regards the comparative study of ENR/DTDB and ENR/S, both materials performed similarly across tests with dynamic bonds allowing partial recycling of samples. However, notable advantages favor disulfide bonds. ENR/DTDB withstands temperatures up to 100 °C with minimal residual deformation. The lower reactivity of disulfides compared to polysulfides does not hinder thermal rearrangements but provides better protection against thermo-oxidation. Most importantly, DTDB eliminates the need for toxic additives like N-cyclohexyl-2-benzothiazolesulfenamide (CBS) and zinc oxide (ZnO), essential in sulfur vulcanization, thus supporting the eco-friendly benefits of crosslinking ENR with dicarboxylic acids.
5. Thermoreversible physical ENR networks using crystallizable grafts
ENR development progressed by incorporating crystalline grafts to form a network with physical crosslinks. This approach involved grafting ENR with crystallizable behenic acid (BA), leveraging ENR epoxides for attachment through esterification accelerated using DMI.
Several hypotheses guided this work. (i) If BA retained its crystalline structure within the ENR matrix, the grafted BA domains could form a physical network with reversible crosslinking points. These crystalline regions would act similarly to those in thermoplastic elastomers, where crystallites or physical bonds provide elasticity while enabling thermal re-formation. (ii) ENR crosslinked with dicarboxylic acids might also develop strain-induced crystallization (SIC) similar to natural rubber. (iii) ENR might retain such an intrinsic SIC property alongside the BA crystallites, creating a dual crystallization mechanism. This dual crystallization would enable BA-grafted ENR to transition between an elastic, rubbery state at ambient temperatures and a plastic, meltable state above the BA melting point. Such a design would allow the material to benefit from both flexibility at lower temperatures and reprocessability at higher temperatures.
5.1. Examining SIC in DA-crosslinked ENR
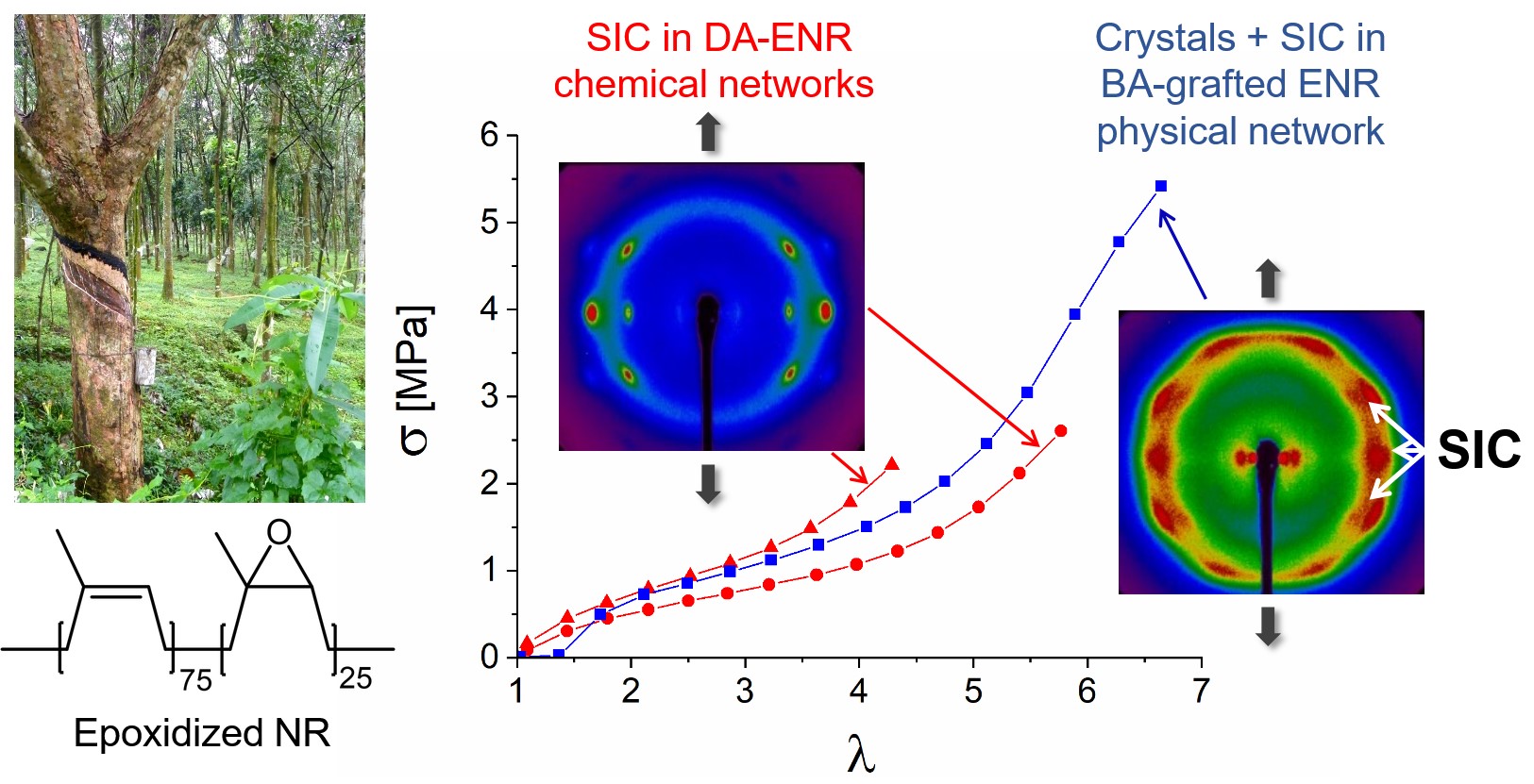
SIC is a unique property of natural rubber, contributing to its high resistance to crack propagation and making it valuable in demanding applications like automotive tires. Our research demonstrated that ENR crosslinked with DA retained the ability to crystallize under strain, using X-ray scattering techniques in situ during cyclic stretching [31].
Figure 4 displays the stress–strain curves for the chemical network DA-ENR25 crosslinked at a p = 50 rate (50 epoxides or 200 ENR25 monomers per diacid molecule). The experiment was carried out at 20 °C over three cycles until rupture at the third cycle. Representative wide-angle X-ray scattering (WAXS) patterns revealed the structural evolution during the second tensile cycle. At the initial state (𝜆 = 1), the pattern exhibited an isotropic amorphous halo. As tensile strain increased (𝜆 = 4.6), the scattering intensity became concentrated along the equator, indicating the alignment of ENR chains in the direction of the applied force (Figure 4A). Upon further stretching beyond 𝜆 = 5, characteristic crystalline peaks appeared, signifying the formation of strain-induced crystals. Detailed peak indexation at 𝜆 ≈ 7 is provided in Figure 4B. These strain-induced crystals subsequently melted as the sample relaxed, returning to its original shape. This second mechanical cycle exhibited a slight shift compared to the first, attributed to the Mullins effect. However, after the second unloading, the sample fully recovered its shape, with the unloading curve eventually aligning with the loading curve, indicating excellent elasticity of the chemical network. The final cycle, extended until break, aligned perfectly with the second loading.
WAXS experiments in DA-ENR25 (p = 50) over three traction cycles (red: first cycle; green: second cycle; blue: third cycle extended to rupture). (A) Patterns observed during the second loading and unloading phases, with the traction direction oriented vertically. Strain rate: 1.25 min−1. (B) Close-up of the pattern at 𝜆 ≈ 7, highlighting the anisotropic amorphous halo of extended ENR chains and peak assignment of the SIC crystals within an orthorhombic lattice.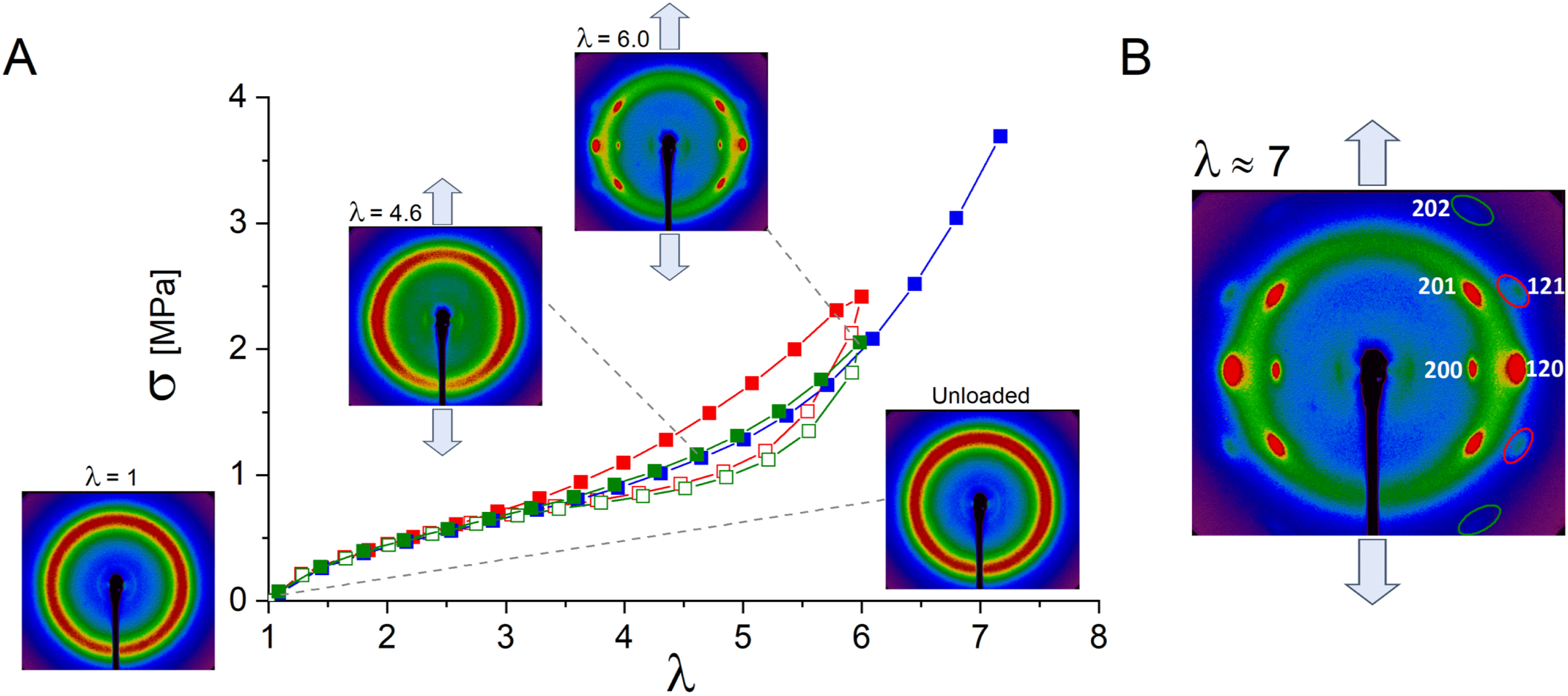
The results showed that the DA-ENR networks exhibited SIC behavior comparable to sulfur-vulcanized natural rubbers. The lattice parameters, calculated in the orthorhombic system (Table 3), are very close to those reported in the case of sulfur-crosslinked ENR25 [32]. They are slightly higher than those reported for S-cured natural rubber [33], resulting in a larger cell volume, which is likely due to the incorporation of the oxiranes of ENR within the structure of the strain-induced crystals [31]. The high fatigue resistance typical of sulfur-vulcanized rubber is traditionally associated with its SIC capability. Experiments demonstrated that ENR crosslinked with DA retained the ability to crystallize under strain in accordance with the good fatigue resistance behavior observed in this material [14]. Moreover, the DA-based crosslinking did not interfere with the lattice structure or crystallization onset, allowing for structural integrity of the material under repetitive stress. These findings confirmed the potential for DA-crosslinked ENR to serve in applications demanding longevity and mechanical robustness, establishing it as a sustainable alternative for conventional sulfur-crosslinked natural rubbers.
Comparison of SIC lattice parameters in the orthorhombic system for DA-ENR25 and BA-g-ENR25. The crosslinking rate in DA-ENR25 corresponds to an epoxy/diacid ratio of p = 25.
| T | a (nm) | b (nm) | c (nm) | Volume (nm3) | |
|---|---|---|---|---|---|
| DA-crosslinked ENR25 | 20 °C | 1.25 | 0.95 | 0.82 | 0.97 |
| BA-grafted ENR25 | 10 °C | 1.22 | 0.93 | 0.84 | 0.95 |
5.2. Integrating crystalline grafts for physical crosslinking in ENR
Detailed results of this section are available in reference [30].
5.2.1. Synthesis of BA-grafted ENR25
The reaction between ENR and BA followed the same mechanism as the DA crosslinking of ENR: an acid group reacts with an oxirane to form an ester bond (Figure 5A). DMI was added in stoichiometric proportions relative to acid functionalities to accelerate esterification, with the BA content set at one acid molecule per two epoxides. This high concentration of grafting molecules was needed to make crystallization observable. The resulting grafted ENR is referred to as BA-g-ENR in the following.
(A) Reaction scheme of ENR25 with BA at an acid-to-epoxy ratio of 1:2. (B) FTIR spectra of the ENR25/BA/DMI blend before (green) and after (red) grafting, zoomed in the carbonyl area. (C) DSC traces of BA-g-ENR25 at 10 °C⋅min−1. (D) Dynamic mechanical analysis of a BA-g-ENR25 sample at 3 °C⋅min−1.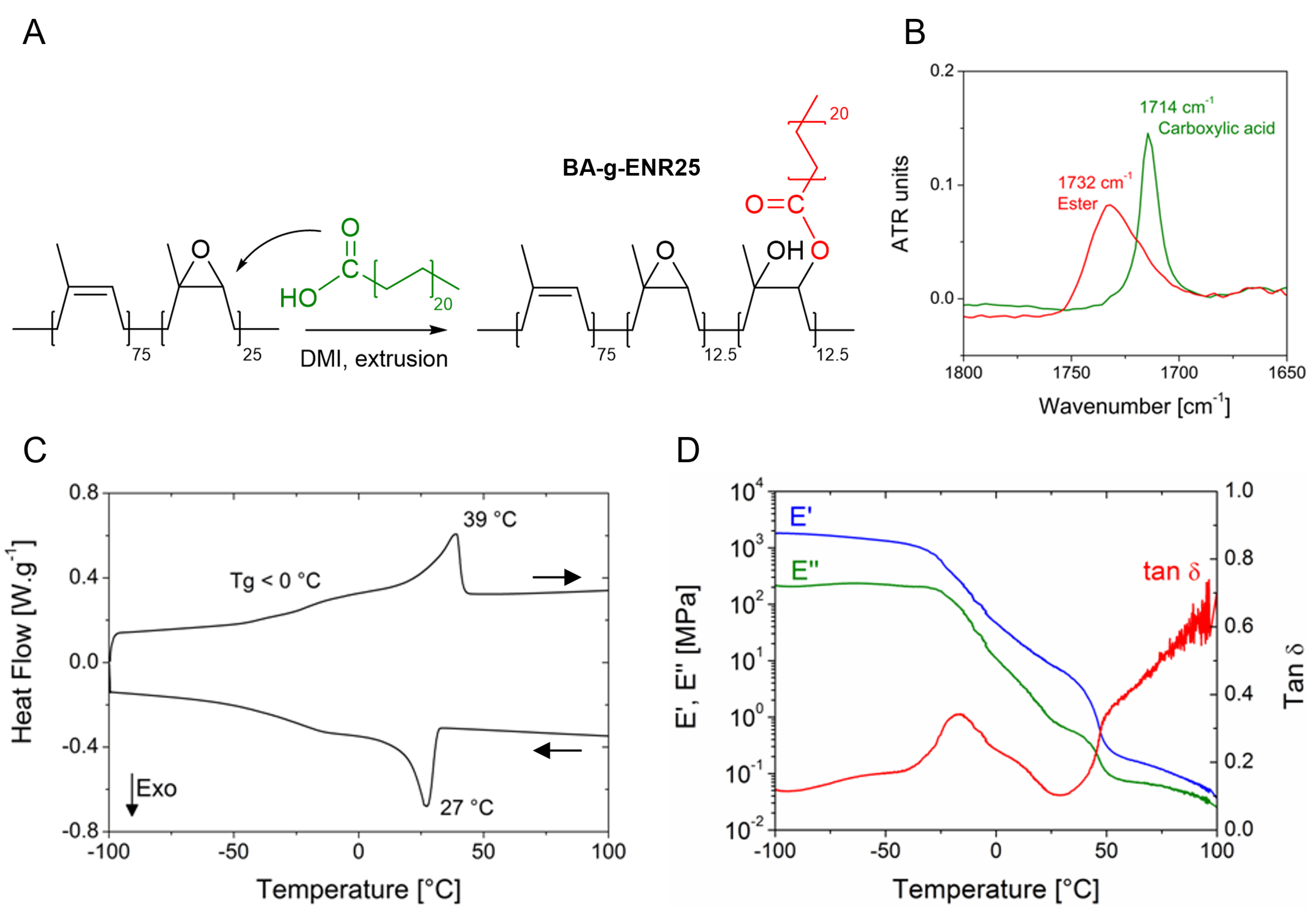
BA was grafted onto ENR25 through a reactive extrusion process at 180 °C for 25 min. The reaction was deemed complete once the torque measurement stabilized. This grafting method is entirely solvent-free, offering substantial industrial and environmental benefits. Although ENR grafting is achievable within the extruder, ENR crosslinking was not feasible due to the adhesion of crosslinked samples in the extruder. For crosslinking, the mixing and curing steps were separated, with curing conducted under a hot press. This two-step approach also remained solvent-free and maintained scalability for industrial applications.
5.2.2. Spectroscopic analysis (FTIR and 1NMR)
FTIR spectroscopy provided evidence of successful BA grafting through ester linkages, as new peaks associated with ester groups appeared after reactive extrusion (in particular, an IR broad band at 1732 cm−1 characteristic of the ester carbonyl signal, Figure 5B) alongside a decrease in the intensity of peaks corresponding to carboxylic acids in BA. 1H NMR spectroscopy further confirmed successful grafting by showing diminished signals for epoxy protons, indicative of ring opening of oxiranes (not shown). After 25 min of grafting at 180 °C, approximately half of the epoxy functions were consumed. Since BA was introduced at a 1:2 ratio into the epoxides, this indicates that the reaction reaches completion within about 25 min. To ensure complete grafting of BA onto ENR, the grafting time was fixed at 30 min.
5.2.3. Thermal properties and differential scanning calorimetry (DSC)
DSC analysis provided insight into the melting and crystallization behavior of BA-g-ENR for an acid:epoxy ratio of 1:2. Free BA exhibits a melting point around 78 °C; however, when grafted onto ENR, the BA crystal’s melting signal shifted to approximately 39 °C and the corresponding crystallization peak to 27 °C (Figure 5C). This decrease in melting temperature suggested that the grafted BA crystalline domains were structurally constrained by their attachment to the rubber matrix. Even though modified, these melting and crystallization transitions provided a temperature range over which the material maintained elastic properties as determined using dynamic mechanical analysis (DMA).
5.2.4. Dynamic mechanical analysis
The temperature evolution of the storage modulus (E′) and loss modulus (E′′) was measured in a film tension configuration. Samples with a thickness of 1.5 mm were subjected to a temperature ramp of 3 °C⋅min−1 from −100 °C to +100 °C at 1 Hz under 0.1% strain. The DMA revealed a broad peak in the tan 𝛿 curve corresponding to the first drop in E′, identifying the glass transition temperature (Tg) of ENR25 around −15 °C (Figure 5D). Below this Tg, the high E′ value indicated a robust network structure reinforced by crystalline crosslinks. A second sharper drop in E′ was observed around 50 °C corresponding to the melting of BA crystals. Between these transitions, E′ decreased in the range of values typical of elastomers, showing significant temperature dependence near room temperature. This analysis highlights the dual nature of BA-grafted ENR: it behaves as an elastomer at ambient temperature and exhibits thermoplastic properties above 50 °C.
The functional range of this thermoplastic elastomer extends from its glass transition temperature near −15 °C to the BA crystal’s melting temperature of approximately 40–50 °C. Within this range, the modulus exhibits strong temperature dependence, likely aligning with the degree of BA crystallinity. Mechanical and structural analyses of the grafted material were conducted at 10 °C to leverage the enhanced crystallinity of the material at this temperature.
5.2.5. Microstructure analysis by transmission electron microscopy (TEM)
Blend morphologies were observed using a transmission electron microscope operated under a voltage acceleration of 80 kV. Thin sections (60–100 nm) were cut at −80 °C using a cryo-ultramicrotome and stained by exposing the thin sections to ruthenium tetroxide (RuO4) vapors for 2 min at room temperature. TEM images confirmed the formation of crystalline lamellae within the BA-g-ENR (Figure 6A). These lamellae, visible as white rows in the stained samples, formed a periodic array throughout the ENR matrix, with an interlamellar spacing estimated to be approximately 12 nm. The regularity of these lamellae underscored the uniformity of the grafting process, with BA long aliphatic chains crystallizing consistently across the ENR matrix. These organized lamellar structures contributed to the material’s enhanced mechanical properties at lower temperatures, where the crystalline domains restricted polymer chain mobility as crosslinks would do in a physical network.
Microscopic characterization of BA-grafted ENR25, with BA constituting 37 wt% of the total formulation. (A) TEM image of the physical network stained with RuO4. Crystalline lamellae appear in white. (B) SAXS measurements comparing DA-crosslinked ENR25 (chemical network) and BA-grafted ENR25 (physical network). As the DA-ENR sample (black curve) includes kaolin used during chemical network synthesis, the BA-g-ENR sample (purple curve) was similarly loaded with kaolin for comparison.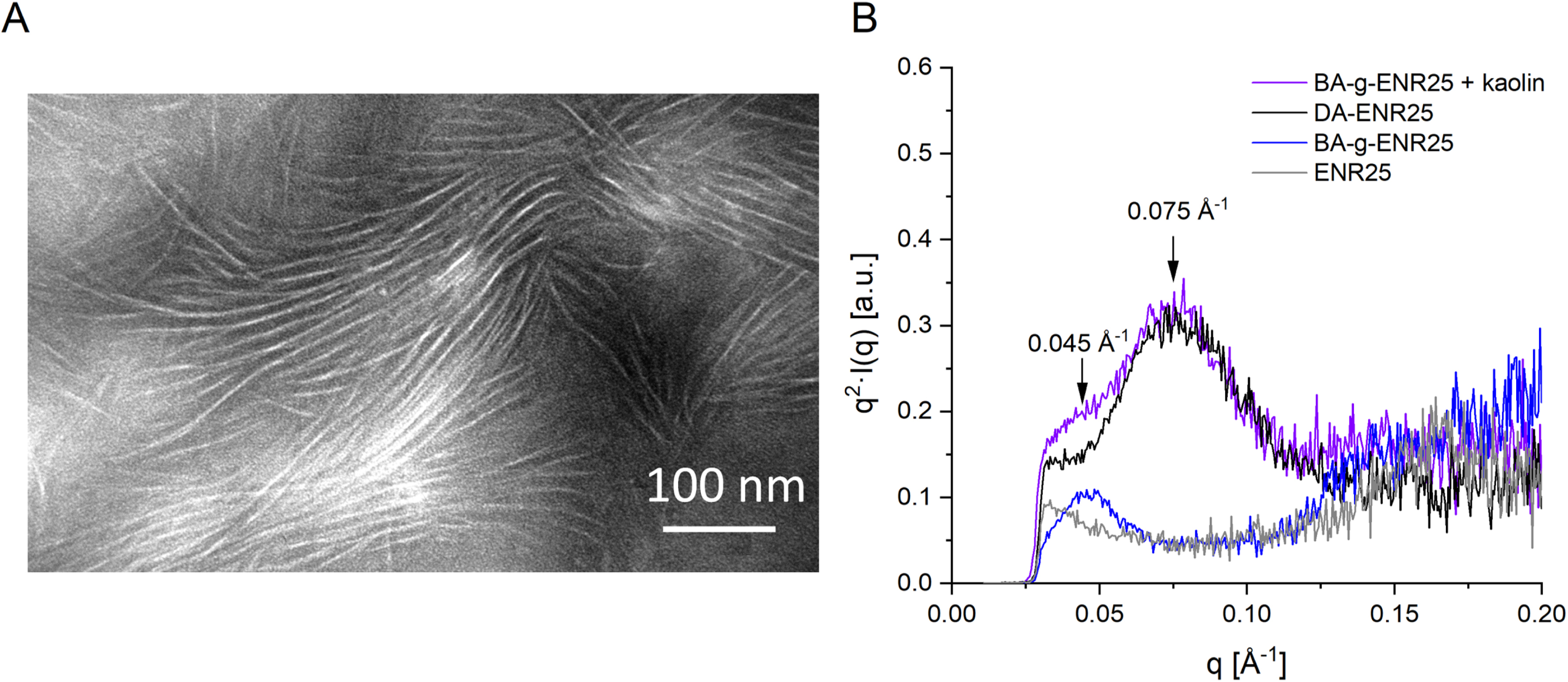
5.2.6. Small-angle X-ray diffraction (SAXS)
SAXS experiments were conducted at room temperature in transmission mode using the Cu Kα radiation from an X-ray generator operating at 40 kV and 25 mA. The SAXS profiles of BA-grafted ENR25 were compared to those of DA-crosslinked ENR25 and unmodified ENR25 as shown in Figure 6B. The spectra are displayed in the Kratky representation, effective for assessing lamellar structures. DA-ENR25 contains kaolin as used in the synthesis of the chemical network. For comparison, a BA-g-ENR25 sample was also loaded with kaolin.
In the purple SAXS spectrum of BA-g-ENR25 with kaolin, a broad peak appears around 0.075 Å−1 corresponding to distances of approximately 80 Å. This peak is also present in the DA-crosslinked ENR25 network (black curve) but is absent in the spectra of pure ENR25 (gray curve) and kaolin-free physical network (blue curve). Therefore, the peak at 0.075 Å−1 was attributed to the filler. Notably, the kaolin-free physical network displays a distinct peak around 0.045 Å−1 corresponding to distances of approximately 140 Å. This peak is also visible as a shoulder in the spectrum of the kaolin-containing physical network. This distance aligns with the 12 nm interlamellar spacing observed in TEM images, confirming that this peak is due to BA crystalline lamellae.
5.2.7. In situ X-ray scattering experiments during cyclic stretching
WAXS experiments were conducted at 10 °C during loading and unloading cycles of BA-g-ENR25 samples, with three successive cycles analyzed. The final cycle was extended to rupture. All experiments were performed at a constant speed of 25 mm⋅min−1 corresponding to a strain rate of 1.25 min−1. Figure 7 displays the mechanical curves for each cycle along with the X-ray patterns recorded during the initial loading and unloading phases.
Stress–strain curves recorded at 10 °C for three successive stretching cycles of a BA-g-ENR25 sample (red: first cycle; green: second cycle; blue: third cycle extended to rupture). Filled and empty squares represent loading and unloading, respectively, with a strain rate of 1.25 min−1. The in situ 2D X-ray patterns captured along the stress–strain curves during the first cycle correspond to successive elongations 𝜆.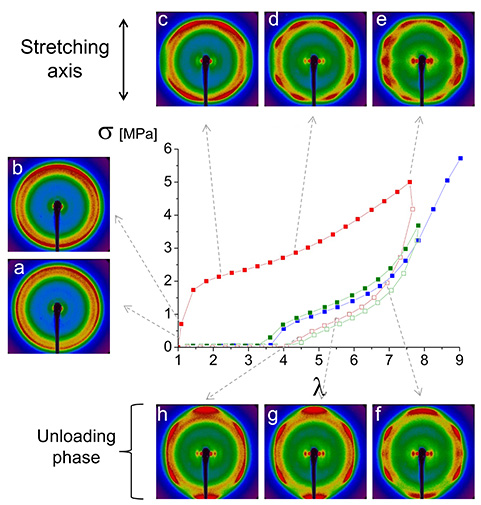
Initial loading cycle
The first loading cycle (red curve in Figure 7) deviates from typical elastomeric behavior, displaying characteristics of a semi-crystalline material. The sample shows poor elasticity, with a substantial residual strain of approximately 300%, indicating limited recovery. Young’s modulus, calculated from the initial slope of the stress–strain plot, is notably high at around 5.5 MPa. In comparison, DA-ENR chemical networks present lower Young’s moduli, typically around 1.5 MPa or less. The increase of modulus in case of BA-g-ENR25 is likely attributed to the presence of graft crystals at 10 °C.
Before stretching (𝜆 = 1), the 2D X-ray pattern (Figure 7a) reveals some crystallization alongside the amorphous halo previously observed in the DA-ENR chemical network (𝜆 = 1 in Figure 4). A secondary ring, attributed to the BA crystals, appears near the halo but at a higher Bragg angle. The crystalline ratio of BA-g-ENR25 calculated from this (𝜆 = 1) ring at 10 °C is 12%.
Upon stretching, this outer ring aligns along the meridian (stretching axis), indicating the orientation of BA crystals, interpreted as clusters of aligned C21 chains. A minimum intensity is observed along the equator, perpendicular to the stretching axis (Figure 7b). Beyond 𝜆 = 2, the orientation along the meridian varies with increasing stretch (Figure 7c, d) as highlighted by dashed oval lines in Figure 8. At the onset of stretching, the short graft BA chains orient perpendicularly to the stretching direction (Figure 8A), resulting in concentrated intensity along the meridian. With increased stretching, these short chains begin to tilt (Figure 8B).
Schematic model illustrating BA crystal orientation during the stretching phase as observed in the 2D patterns of: (A) Figure 7b and (B) Figure 7d. Dashed oval lines highlight the evolution of signal intensity upon stretching. The dashed line denoted as Eq represents the equator, perpendicular to the stretching axis. In (C), the (120) and (201) characteristic peaks from SIC superimpose onto the existing signal from the BA crystals as highlighted by white arrows.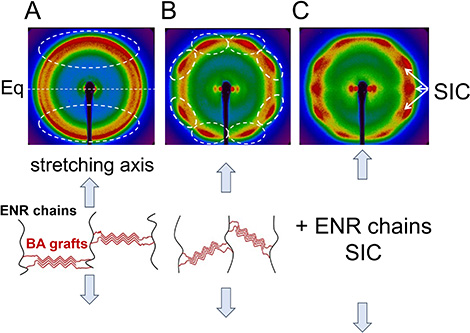
For elongation ratios beyond 𝜆 = 5, peaks associated with SIC of ENR superimpose (Figure 7e). The characteristic (120) and (201) SIC peaks are marked by arrows in Figure 8C. Upon unloading, these strain-induced crystals dissipate while the primary signal from BA crystals gradually reorients along the meridian (Figure 7f–h).
At the end of the first loading cycle, the BA crystals remain oriented although their tilt has been lost.
Subsequent cycles
The 2D X-ray patterns for the second cycle and final stretching are shown in Figure 9. After the second unloading, the material exhibits low hysteresis with a weak residual strain, which dissipates within 5 min. The subsequent cycles align closely with the second cycle, showing very similar 2D patterns. During stretching, the signal intensity, initially concentrated along the meridian, disorients towards the equator, and SIC appears as the loading curve upturns. Since the third loading cycle is extended to rupture, the SIC signal intensifies here. Upon unloading, the SIC signal dissipates while the BA crystal orientation returns to the meridian.
2D X-ray patterns captured at 10 °C along the stress–strain curves during (A) the second stretching cycle and (B) the third cycle, which is extended to rupture. Filled and empty squares represent loading and unloading, respectively, with a strain rate of 1.25 min−1.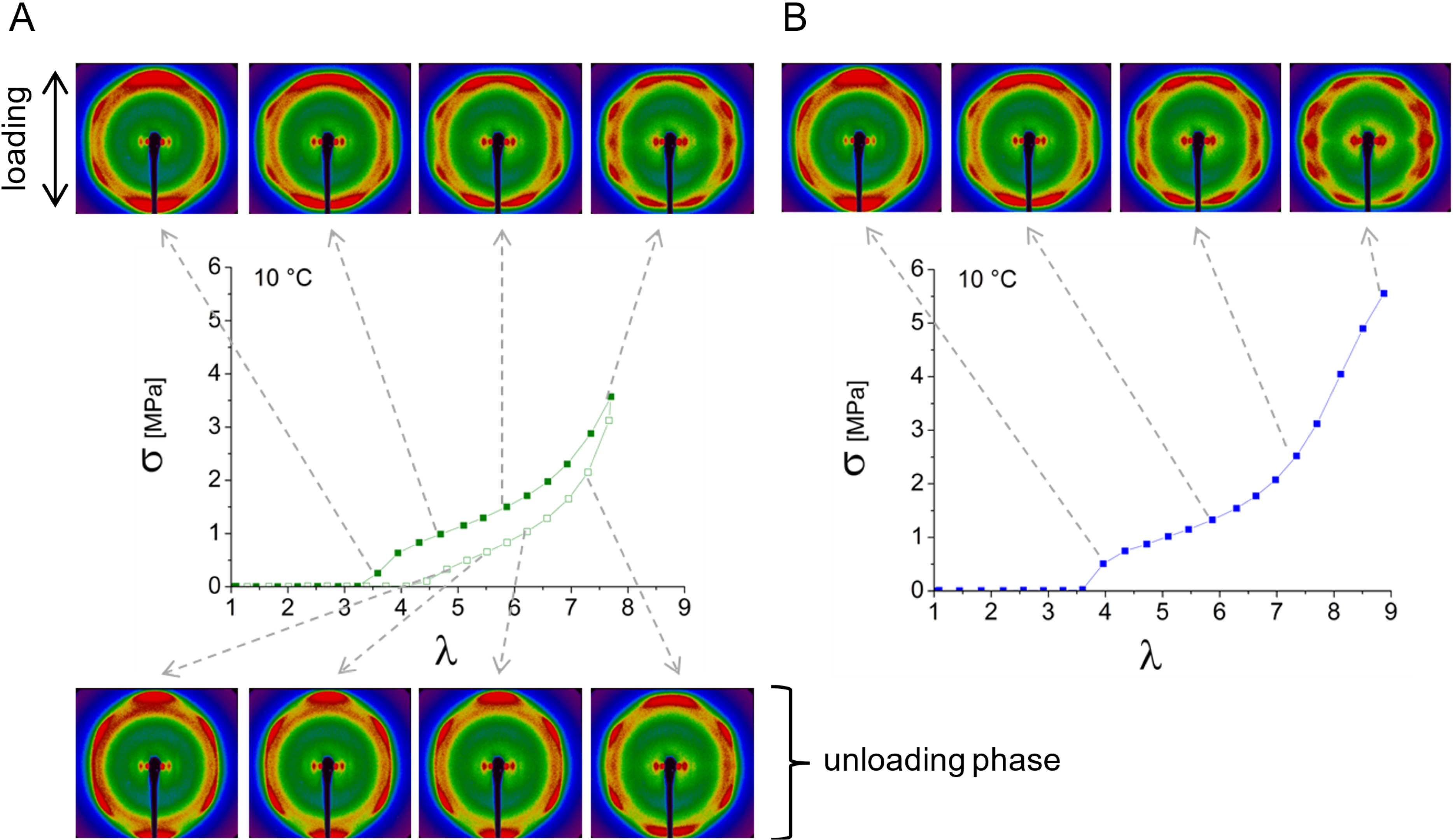
Overall, this combined mechanical and structural analysis demonstrates that a pre-stretching step breaks and orients BA crystals, aligning grafts perpendicularly to the ENR chains. During subsequent stretching, these crystals reversibly tilt, returning to their original orientation upon unloading, but do not dissipate. Consequently, BA crystals act as physical crosslinks providing elastomeric properties to the ENR.
Analysis of the SIC signal
The lattice parameters for SIC crystals calculated within the orthorhombic system are presented in Table 3 alongside those for ENR25 crosslinked with DA (Section 5.1). The parameters for DA-crosslinked ENR25 were derived from measurements at 20 °C while those for BA-grafted ENR were obtained at 10 °C due to insufficient sample crystallinity at 20 °C (9%, compared to 12% at 10 °C), which would not provide reliable data. Nevertheless, the calculated lattice parameters closely align with those observed for the chemical network, indicating that the crystallized grafts do not interfere with SIC behavior of the rubber.
6. Comparison between chemical and physical networks
Stretching cycles for the DA-ENR25 chemical network were recorded at 10 °C and plotted along with the BA-g-ENR25 third cycle until rupture as shown in Figure 10. For comparison, the BA-g-ENR curve was reset to 𝜆 = 1 after recalculating the strain at the end of the first cycle of pre-stretching, which retained a 300% strain following the breaking and orientation of the BA crystals. Two chemical networks with different crosslinking densities were considered corresponding to epoxy/diacid ratios of p = 25 and p = 50. The latter exhibited a lower crosslinking density. The second cycle for DA-ENR25 demonstrated excellent reversibility (green curves), attesting to the elastomeric character of both chemical networks at 10 °C.
Comparison of mechanical properties for chemically crosslinked DA-ENR25 and physically crosslinked BA-g-ENR25. Two crosslinking densities were considered corresponding to epoxy/diacid ratios p = 25 and p = 50. Green curves: second loading cycle. Blue curves: third loading cycle until rupture. All experiments were conducted at 10 °C with a strain rate of 1.25 min−1.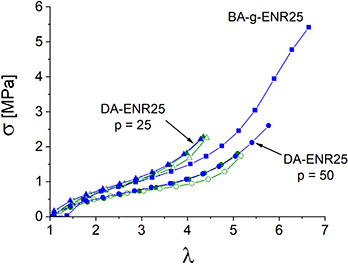
The physical network cycle closely aligns with that of the chemical network, displaying stress–strain behavior at deformations below 𝜆 = 5, which falls between the two crosslinking densities. However, the physically crosslinked rubber exhibits superior ultimate mechanical properties. WAXS patterns indicate that both chemical and physical networks undergo SIC. In the BA-grafted ENR, however, two distinct reversible physical networks coexist: one formed by SIC within the rubber matrix and another arising from physically connected crystallizable grafts. This dual-network structure not only enhances the mechanical properties compared to the chemical network alone but also imparts reprocessability to the physical network, offering a significant functional advantage.
7. Concluding remarks
ENR25 was efficiently grafted with BA through a solvent-free reactive extrusion process, resulting in self-organized crystallite lamellae observable via electron microscopy and SAXS. Mechanical tests revealed that after a single pre-stretching cycle, the resulting material exhibited elastomeric behavior, with crosslinking solely through BA crystals. During pre-stretching, grafts oriented into a stable configuration, enabling further cycles and yielding a thermoplastic elastomer exhibiting SIC properties. This combination provided enhanced tensile strength and flexibility, outperforming chemically crosslinked versions. Our physically crosslinked ENR, using crystallizable BA grafts, exhibits the outstanding mechanical properties of natural rubber, such as tear and crack propagation resistance due to SIC, alongside the reprocessability of thermoplastic elastomers. With enhanced toughness at low temperatures, BA-g-ENR is well suited for applications demanding flexibility and strength. The remeltable BA crystalline domains make this modified ENR a sustainable choice for recyclable rubber products that maintain structural integrity upon remolding.
Although this approach marks a significant step in sustainable elastomer development, a limitation remains: the melting temperature of BA-g-ENR around 40 °C is slightly too low for consistent room-temperature applications. A potential solution involves creating a doubly crosslinked ENR by introducing chemical reprocessability alongside physical crosslinking. For instance, crosslinking BA-g-ENR with DTDB, which contains exchangeable disulfide groups, could produce a network stable at temperatures above 40 °C while retaining elasticity. This would allow the material to maintain its unique properties over a wider temperature range while enhancing recyclability.
To conclude, ENR exemplifies sustainable rubber development. Our research from 2009 to 2017 transformed ENR from simple self-vulcanizing blends to advanced multifunctional materials. Each stage deepened our understanding of how ENR can combine traditional rubber properties with modern demands for sustainability, durability, and reprocessability. Through the integration of chemical or physical crosslinking, ENR has evolved into a versatile material well-suited for applications ranging from tires to flexible electronics, where both resilience and reduced environmental impact are essential. This progress has paved the way for ENR to lead innovations in eco-friendly rubber solutions.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Acknowledgments
The authors warmly thank Pierre-Antoine Albouy from LPS in Paris-Saclay for in situ X-ray measurements under stretching, Cédric Lorthioir for conducting solid-state NMR analyses, Benoît Le Rossignol from the Hutchinson Research Centre (Montargis, France) for fruitful collaboration, and Ilias Iliopoulos and Ludwik Leibler for their continuous encouragement and support.