



 CC-BY 4.0
CC-BY 4.0
1. Introduction
The creation of carbon–carbon (C–C) bonds with tight control of chemo-, regio-, and stereoselectivity is the key reaction to construct the carbon framework of organic molecules [1, 2]. Consequently, sophisticated methods for asymmetric synthesis as well as protection/deprotection strategies have to be considered together with eco-friendly conditions. In this context, enzymatic biocatalytic C–C coupling represents an efficient (asymmetric) alternative for the preparation of multifunctional products [3, 4, 5]. In the last few decades, several carboligases have been reported, each of them leading to a specific functional group or motif such as aldolases, Pictet–Spenglerases, oxidases, prenyltransferases, squalene/hopene cyclases, engineered hemoproteins, and thiamine diphosphate (ThDP) dependent enzymes. The last group is recognized as a powerful tool for the preparation by C–C bond formation of enantiopure α-hydroxyketones [6, 7, 8, 9], a structural motif present in a wide range of highly valuable compounds such as pharmaceuticals [10, 11, 12, 13, 14, 15], flavors [16], sweetners [17, 18], surfactants [19], and important synthetic intermediates for the preparation of diols or aminoalcohols.
ThDP-dependent enzymes display the formation of a C–C bond between two carbonylated compounds (aldehyde or ketone) according to a common mechanism involving activated ThDP I (Figure 1) to produce a highly reactive intermediate (III) via polarity reversal (umpolung reaction) characterized by two mesomeric forms (carbanion–enamine). The carbanion subsequently attacks the carbonyl group of a more electrophile (acceptor) substrate, leading to the formation of a C–C bond (IV) and to the release of the product (Figure 2). The stereocontrol of the new asymmetric carbon depends on the type of ThDP enzyme and the structure of substrates.
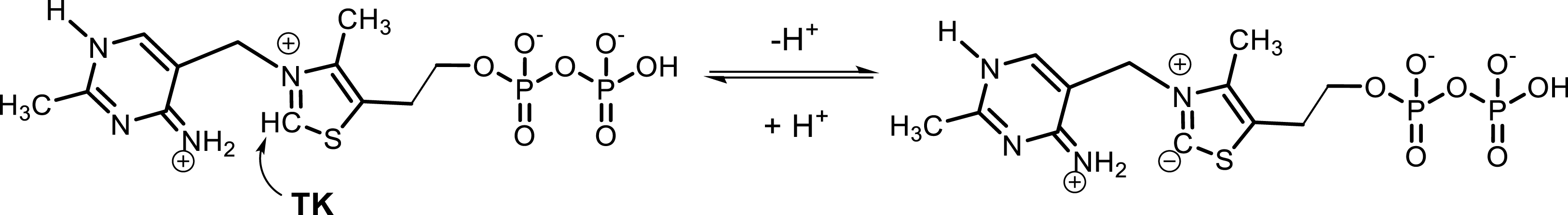
Activation of thiamine diphosphate.
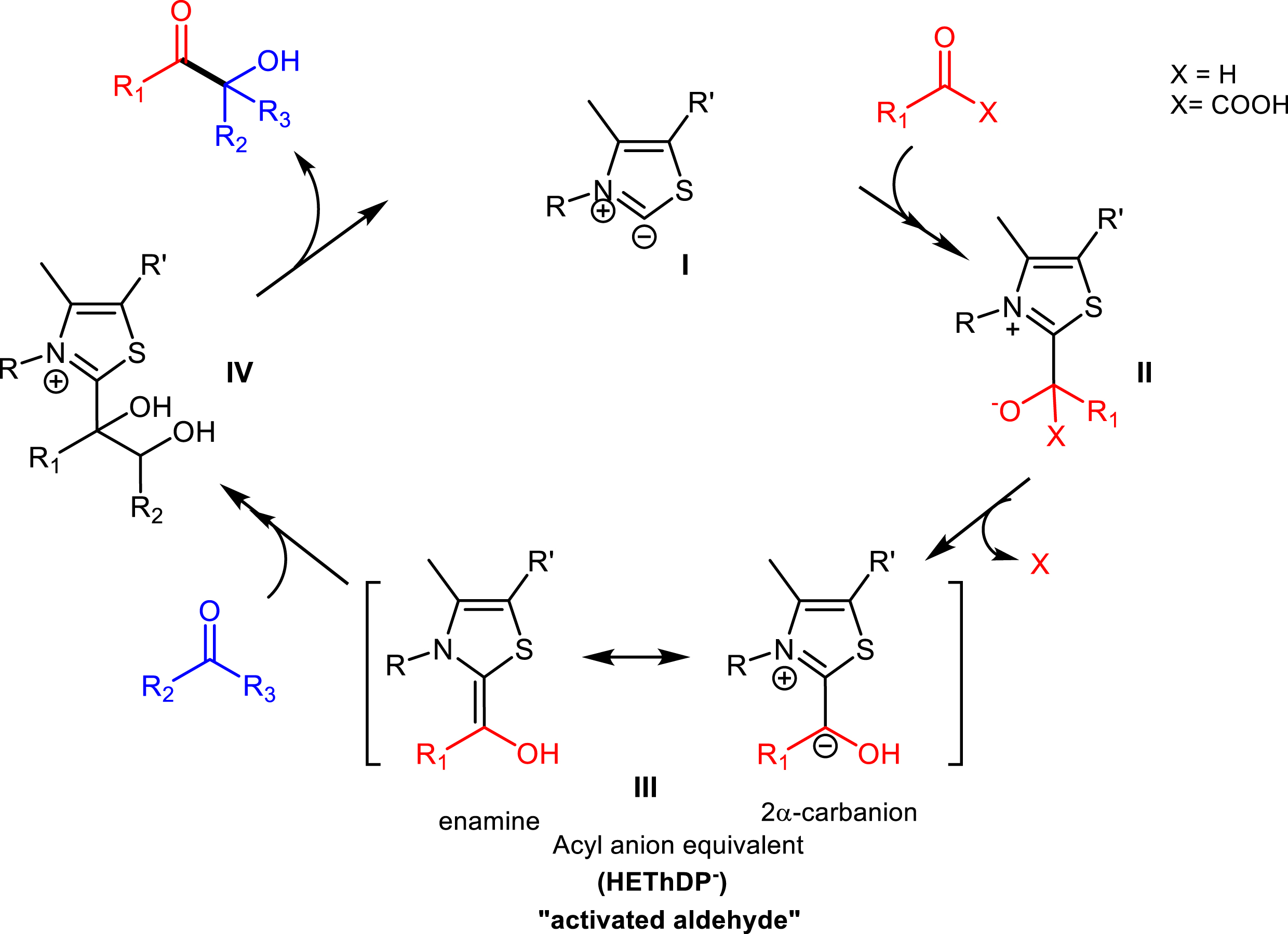
Catalytic cycle of ThDP-dependent carboligases.
Among ThDP enzymes, transketolase (TK, EC 2.2.1.1) has been largely studied and used for the synthesis of various enantiopure α-hydroxyketones considering its large scope of substrates. TKs are ubiquitous enzymes found in the cytoplasm of animal, plant, and microbial cells and in vivo catalyze the reversible transfer of a two-carbon ketol unit from d-xylulose 5-phosphate to d-ribose 5-phosphate or d-erythrose-4-phosphate to generate d-sedoheptulose-7-phosphate or d-fructose-6-phosphate and d-glyceraldehyde-3-phosphate (Figure 3).
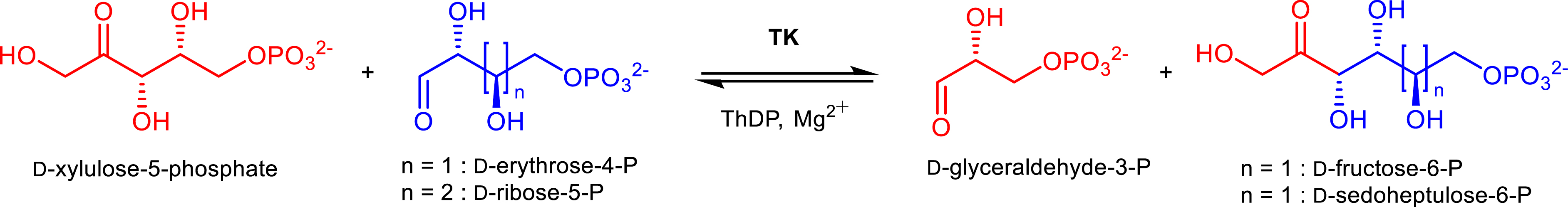
In vivo transketolase reaction.
With TK providing a link between the glycolysis and pentose phosphate pathway, this enzyme plays a key role in metabolic regulation in all living cells [20]. Particularly in humans, TK has been reported to be involved in neurodegenerative diseases Wernicke–Korsakoff syndrome and Alzheimer’s disease, and diabetes and cancers [21, 22, 23].
The first in vitro biocatalytic applications of this enzyme were described by Whitesides et al. in 1992 [24]. The reaction becomes more generally useful for synthetic purposes by using hydroxypyruvic acid (HPA) as a C2-ketol group donor, rendering the reaction almost irreversible due to the concomitant release of carbon dioxide as a by-product (Figure 4). The lithium salt of HPA is preferred as it is the commercially available cheaper form of this compound, and its synthesis is also described [25].
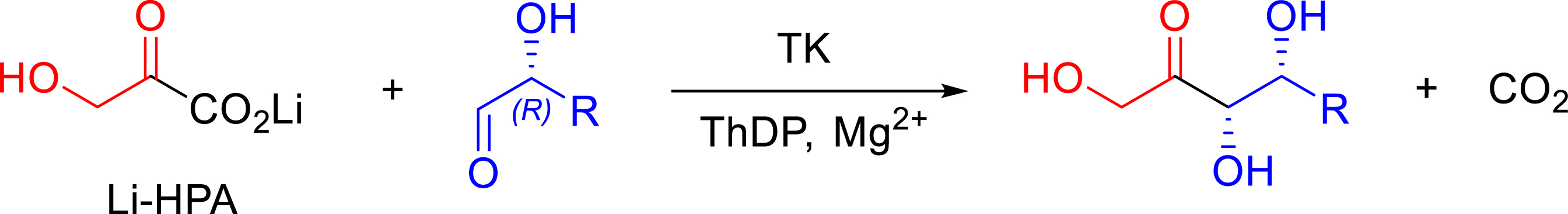
Irreversible reaction catalyzed by TKs in the presence of HPA.
Wild-type transketolases from spinach, S. cerevisiae, and E. coli were first used for the synthesis of α-hydroxyketones mostly polyhydroxylated such as natural and unnatural ketoses and analogues obtained from Li-HPA as donors and preferentially (R)-hydroxylated aldehydes as acceptors [26, 27, 28, 29, 30, 31, 32]. To extend the substrate spectra, the engineering of TKs by mutagenesis was developed together with appropriate screening assays providing efficient variants towards new α-ketoacids as donors (analogues of pyruvate) and aldehydes as acceptors (polyhydroxylated or not, arylated), leading to enantiopure α-hydroxyketones [33, 34, 35].
Another criterion to make biocatalytic applications more competitive is related to the thermostability of enzymes. Indeed, from the industrial point of view, enzymatic processes at elevated temperature offer many advantages such as higher reaction rate, improved solubility of organic substrates, and higher tolerance towards non-conventional media. In addition, for improvement by mutagenesis, thermostable enzymes also display better resistance against protein destabilizing factors [36]. For these reasons, this paper presents an overview of the advances obtained with a thermostable transketolase from Geobacillus stearothermophilus (TKgst) for the synthesis of various α-hydroxyketones in the context of other thermostable TKs having been isolated and used in biocatalysis but at a faster rate [37, 38].
2. Results and discussion
2.1. Production of TKgst
We have chosen Geobacillus stearothermophilus (formerly Bacillus stearothermophilus) as a potential source of a thermally stable TK enzyme [39]. This Gram-positive thermophilic bacterium is widely distributed in soil, hot springs, and ocean sediments, and grows in the temperature range of 30–758 °C. Many heat-stable enzymes, such as xylanase [40], l-arabinose isomerase [41], glycosidases [42], and α-amylases [43], have been isolated from this thermophilic bacterium. Before our discovery in 2013 in collaboration with de Berardinis et al. [39], no complete genome sequence was available in public databases for any G. stearothermophilus strain and no transketolase enzyme had hitherto been described. TKgst was expressed with a His6-tag at the N-terminus in E. coli BL21(DE3) and was purified using immobilized metal affinity chromatography. The expressed fusion proteins were applied to a Ni2+ chelating affinity column for purification. By this procedure, 160 mg each of the purified enzymes TKgst (0.6 U⋅mg−1) were effectively obtained from the cells grown in 1 L culture (5 g⋅L−1 of wet cells). We note that the enzyme can be lyophilized, producing a powder easy to use for chemists and enabling preservation over several months without loss of activity. TKgst also showed higher tolerance over time compared to other mesophilic TKs towards high-percentage cosolvents (up to 50%) such as DMF and ionic liquids such as BMIMCl, which is a useful property when substrates are not water soluble [44].
2.2. Thermostability of TKgst
As is characteristic of a microorganism, TKgst has an optimum temperature range of 60–70 °C, leading to an improvement of up to 10-fold when compared to the activity measured at 20 °C. TKgst retained 100% activity for 1 week at 50 °C and 3 days at 70 °C, and its activity decreased rapidly at 75 °C (half-life was about 15 min) [39]. The thermostability of TKgst was much higher than that of other characterized TKs from microbial sources commonly used in biocatalysis such as TKsce having an half-life of 35 min at 50 °C with immediate activity loss at 70 °C or TKeco more resistant (100% activity recovery at 50 °C for 90 min) but completely vanishing during 5 min exposure at 70 °C.
Given the TKgst thermostability, an alternative purification procedure by heat shock treatment at 65 °C for 45 min enabled an easy and rapid purification method, yielding 132 mg of purified enzyme from the cells grown in 1 L of culture [39].
2.3. Immobilization of transketolase from Geobacillus stearothermophilus
From an industrial point of view, the reusability of an enzyme is also a crucial point. For that reason, we investigated different supports for TKgst immobilization. First in collaboration with Forano et al., we showed that layered double hydroxides gave efficient adsorption/entrapment of TKgst with almost total immobilization, recovery of initial activity, and reusability over six cycles without loss of activity [44, 45]. The strategy is cheap, rapid, and eco-friendly. More recently in collaboration with Szymanska et al., TKgst was covalently immobilized on silica monolithic pellets. This strategy allowed performing the reaction in a basket-type reactor [46].
2.4. Substrate scope of wild-type TKgst
2.4.1. Substrate scope of wild-type TKgst towards donor and acceptor substrates
As indicated before, the major advance for biocatalytic applications of TKs including TKgst was the use of HPA, rendering the reaction almost irreversible due to the release of carbon dioxide (Figure 5). We showed later that pyruvate can be accepted as a donor substrate by TKgst but with lower activity compared to HPA.
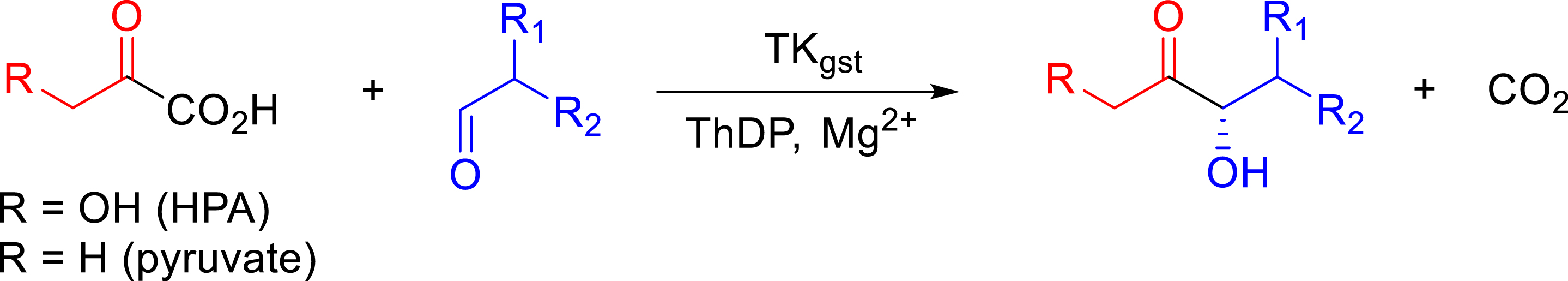
Irreversible reaction catalyzed by TKgst in the presence of α-ketoacid.
As expected, knowing the high homology of the TK active sites, we observed that the TKgst acceptor substrate spectrum in the presence of HPA as the donor was very similar to those of TKsce and TKeco already described (Figure 6).
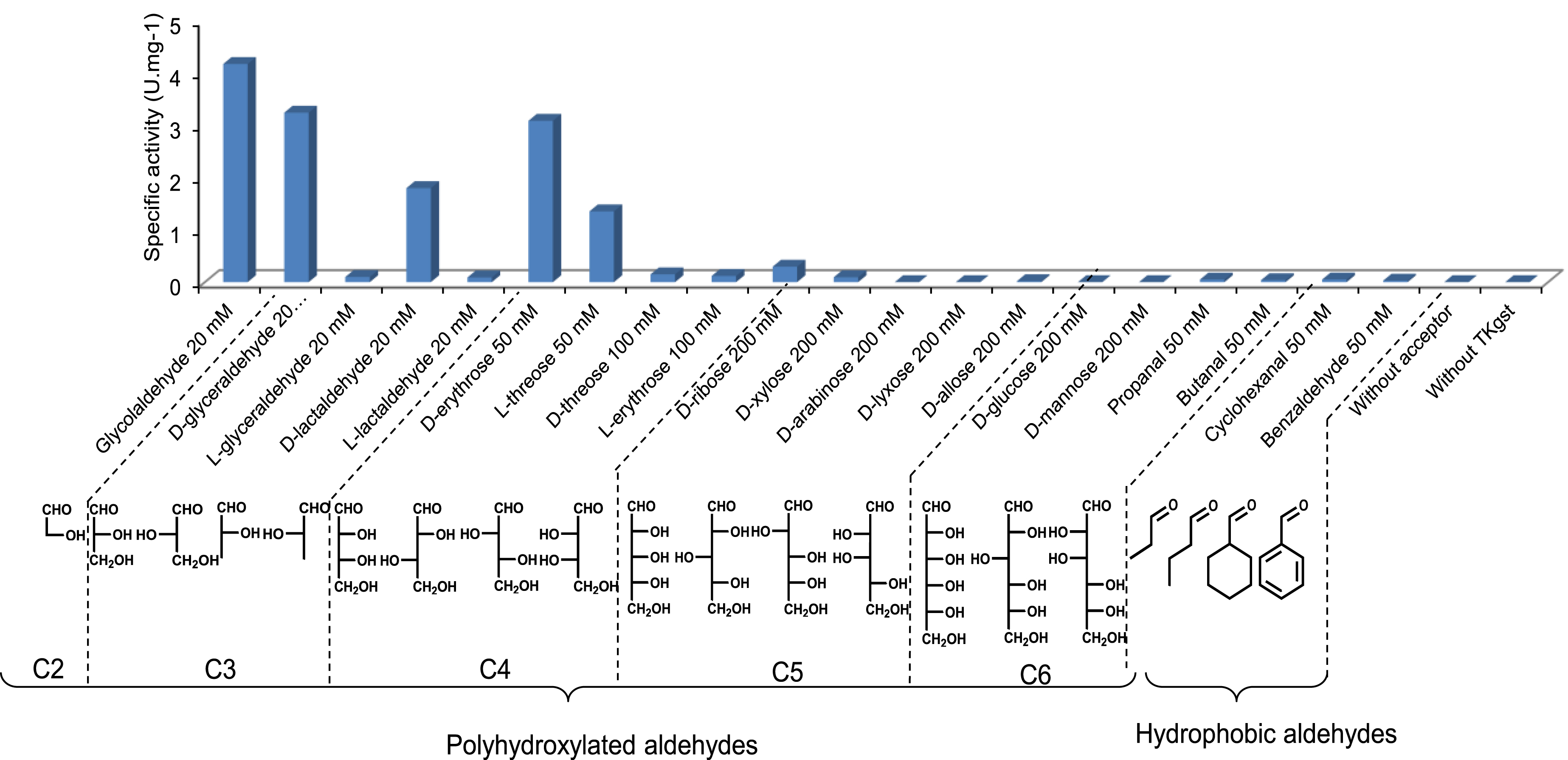
Specific activities of TKgst towards a panel of acceptor aldehydes (20–200 mM) were determined in the presence of Li-HPA at 25 °C.
TKgst showed high activity towards short-chain (2R) α-hydroxylated aldehydes (C2–C4). A significant decrease in TKgst activity was observed with aldehydes displaying a long carbon chain length (C5–C6) or without a hydroxyl group at the α or β position or hydrophobic and aromatic aldehydes.
In contrast with other described microbial TKs, TKgst showed significant activities towards (2S)-hydroxylated aldehydes with three and four carbon atoms [47] such as l-glyceraldehyde, l-lactaldehyde, l-erythrose, and d-threose. This is particularly interesting because these aldehydes can lead to the production of rare, high-value ketoses. However, the TKgst activities were much lower than those determined with their (2R) epimers (approximately 20 and 30 times lower in the case of lactaldehyde and glyceraldehyde, respectively). TKgst being thermostable, studies conducted at 60 °C showed an improvement in activities by a factor 4 to 5 towards C3 (l-glyceraldehyde and l-lactaldehyde) and C4 aldoses (l-erythrose and d-threose) compared to results obtained at 25 °C (Figure 7). These analytical results were significant because the activities measured at high temperatures were comparable to those obtained in the presence of their (2R) analogues at 25 °C.
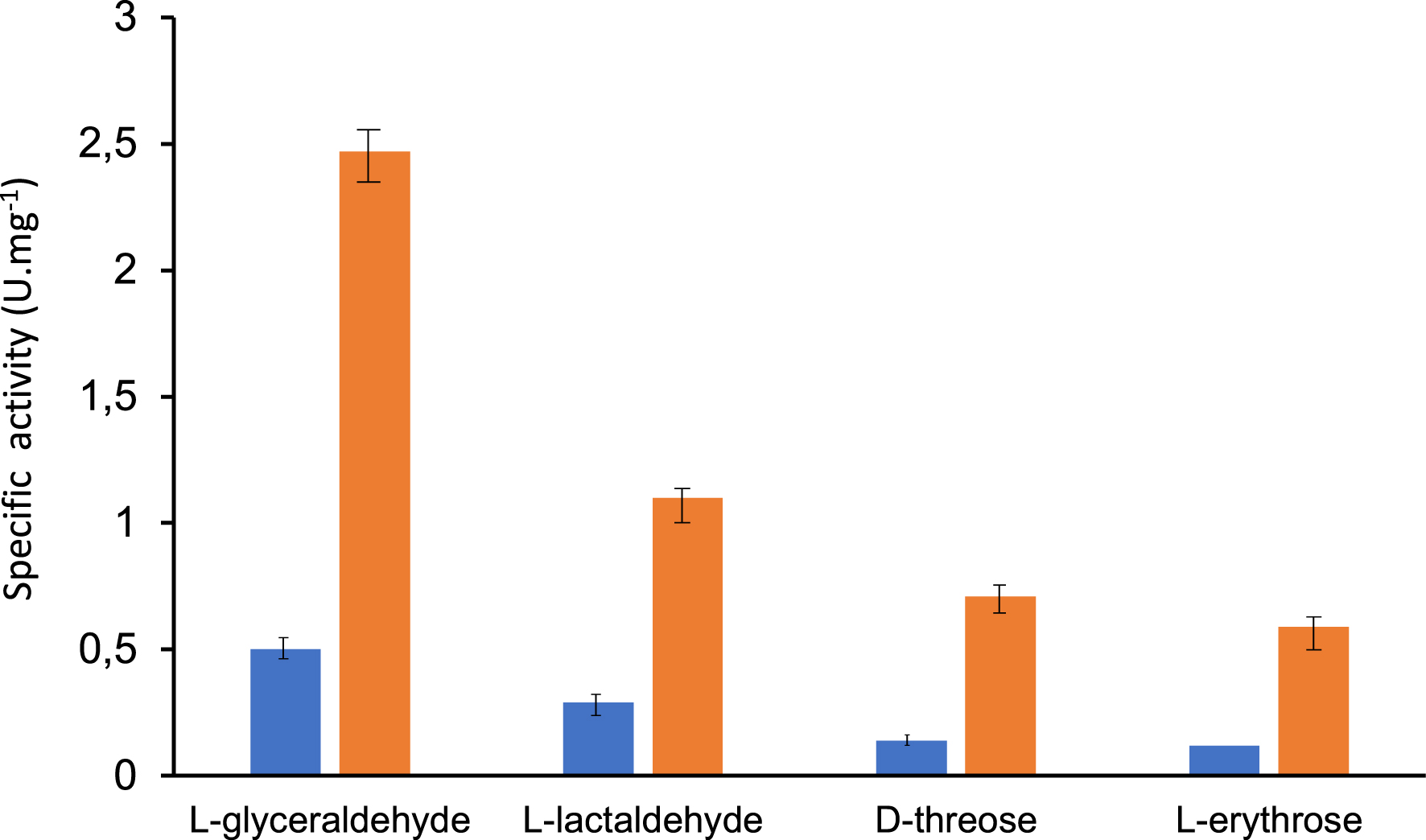
Specific activity of TKgst with (2S)-hydroxylated aldehydes and C3 (l-glyceraldehyde, l-lactaldehyde) and C4 aldoses (l-erythrose, d-threose) in the presence of Li-HPA at  25 °C and
25 °C and  60 °C.
60 °C.
Considering all these results, the improvement and broadening of the TKgst substrate scope particularly towards other α-ketoacids as donors and long carbon chain aldoses and hydrophobic and aromatic aldehydes as acceptors was investigated by mutagenesis strategies.
2.5. Improvement and modification of TKgst properties
2.5.1. Strategies for generating TKgst variant libraries
The strategies were based on the analysis of the active site to determine the key positions that should be mutated to improve TKgst activity towards the targeted substrates. The 3D structure of TKgst with its cofactors was recently determined showing as for the other TKs a dimeric enzyme with each monomer containing an identical active site [48]. However, as this structure was not yet available for the studies presented here, a model of active TKgst [49] was constructed by homology with TK from Bacillus anthracis (TKban) using a template already crystallized (PDB entry 3M49) belonging to the same microorganism species and having 74% identity. This model was validated by comparison with the 3D structure of TKgst.
It can be noted a high homology with the active sites of the other TK sources used in biocatalysis such as TKeco and TKyeast and also with the active sites of all other microbial TKs. The superimposition of TKeco, TKsce, and TKban active sites shows the same residues with a similar position to ThDP and divalent cation Mg2+ (Figure 8).
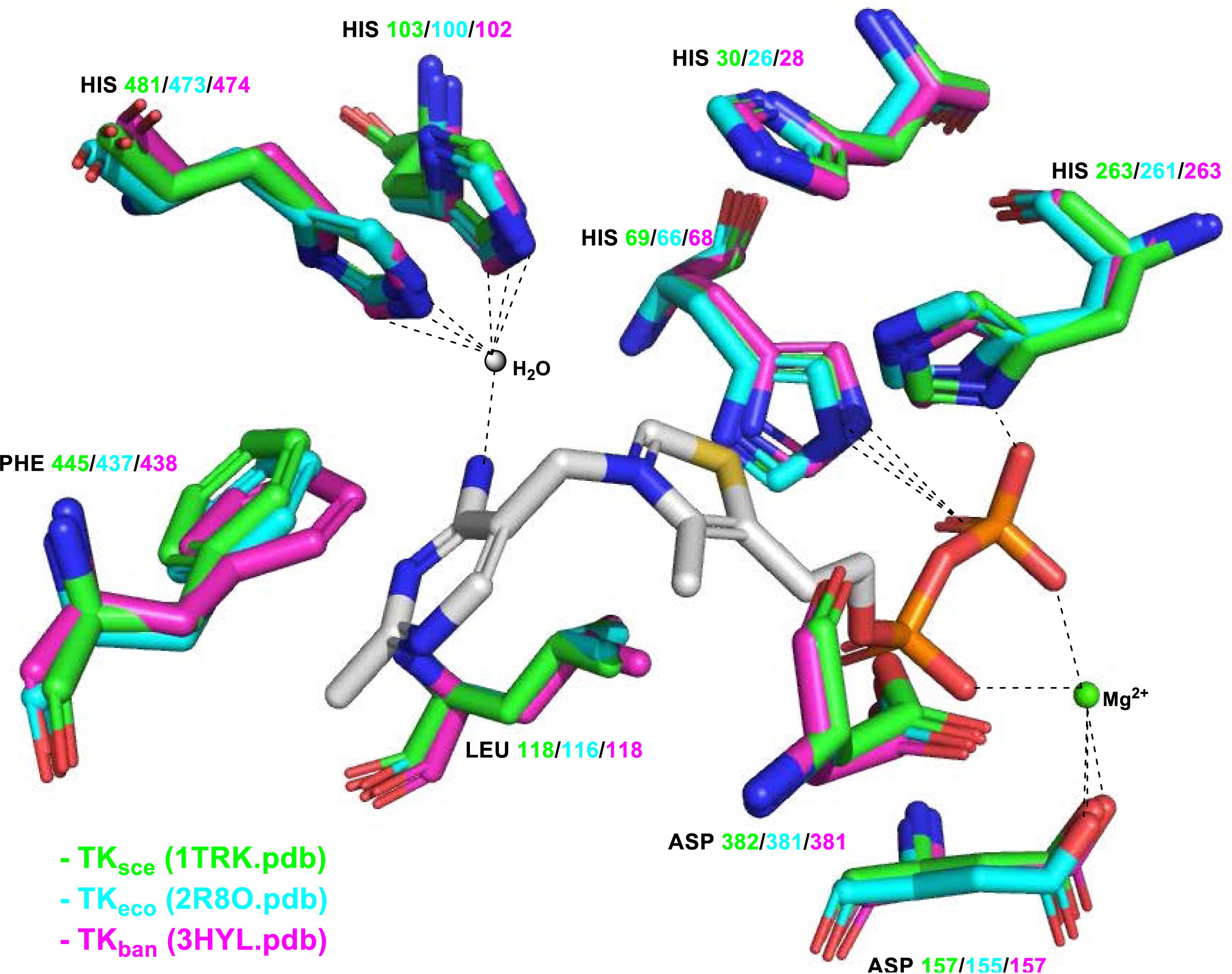
Comparison of ThDP binding motifs of TKsce (green 1TRK.pdb), TKeco (cyan 2R8O.pdb), and TKban (violet 3HYL.pdb).
After the identification of the targeted residues, each targeted amino acid of the wild-type sequence was replaced by the other 19 amino acids by site saturation mutagenesis (SSM) [50]. Different types of degenerate codons can be used such as NNS and NDT. In NNS, “N” stands for any nucleotide (A, T, G, or C) and “S” stands for G or C. NNS can encode 32 possible codons (4 × 4 × 2 = 32) including 20 codons for all amino acids plus 12 additional codons that encode some amino acids multiple times or encode stop codons. Alternative codons “NDT” [51] were designed to completely avoid stop codons while encoding several representative amino acids in each category: anionic, cationic, aliphatic, hydrophobic, hydrophilic, and aromatic. These “NDT” codons provide functional diversity without redundancy while reducing the number of enzyme variants compared to NNS and consequently the screening efforts. When more than two positions were investigated, NDT strategy was preferred in the studies presented here to reduce the size of the libraries.
2.5.2. Screening assays
The principle of the generic assay developed in liquid or solid phase was based on the TK-catalyzed decarboxylation of α-ketoacid and the release of carbon dioxide, inducing the variation in pH of the reaction medium monitored by a pH indicator. Indeed, during the reaction catalyzed by TK in the presence of α-ketoacid as the donor, a proton is consumed in each cycle, which releases an equivalent of
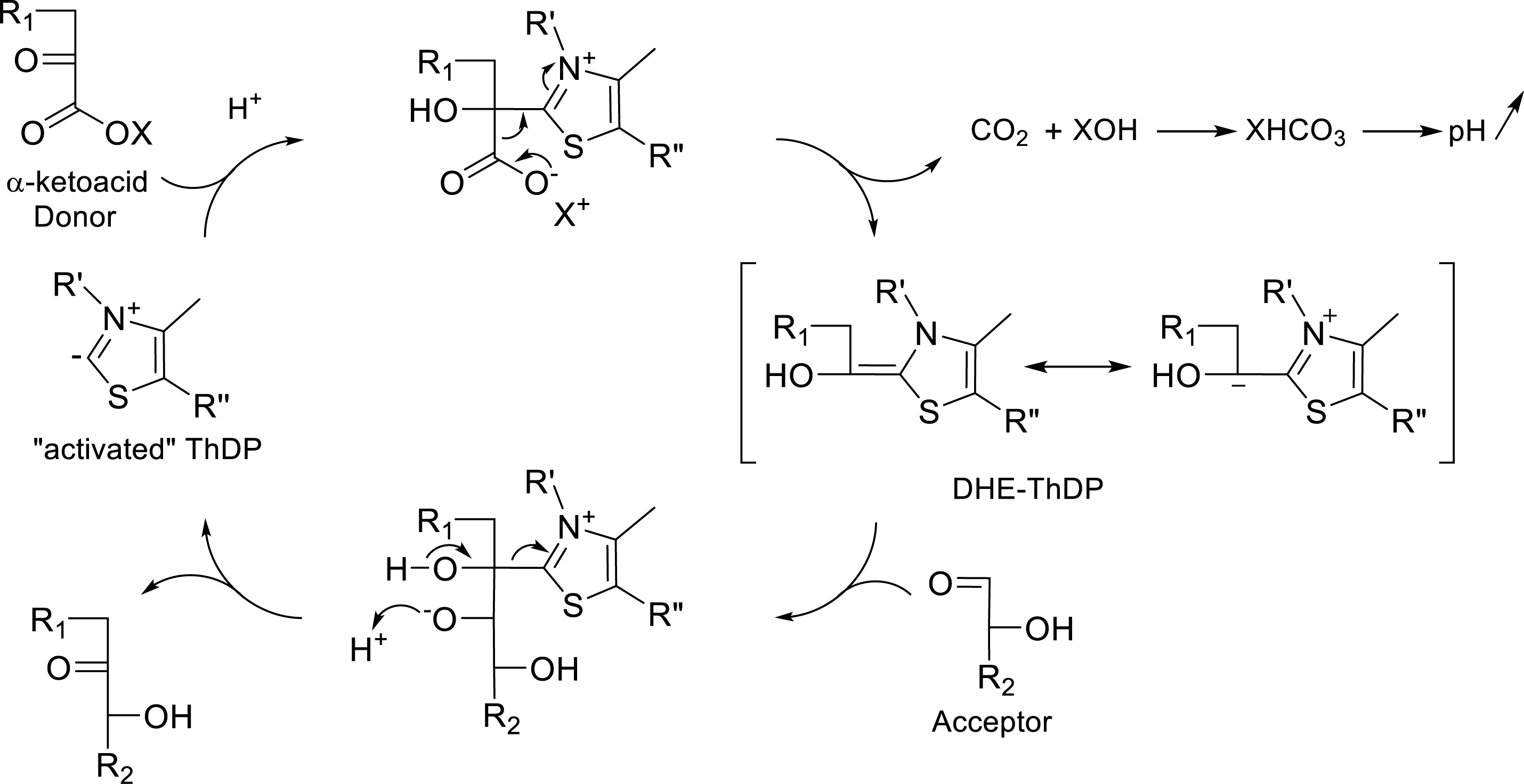
Catalytic cycle of TK in the presence of Li-HPA.
The bicarbonate ion is the dissociated form of carbonic acid (H2CO3) and is involved in a dynamic equilibrium of dissociation in water. This leads to a basification of the solution due to the partial formation of hydroxide ion raising the pH of the medium, which is approximately 7.5. Based on this pH variation in reaction medium, we first developed in collaboration with Fessner et al. a colorimetric screening assay in the liquid phase with purified variants [52]. More recently, the same principle was applied to a semi-solid phase, allowing the direct detection of clones expressing active TKgst variants [53].
For the qualitative liquid screening assay [52], upon release of carbon dioxide from α-ketoacid, the pH increase in the reaction mixture can be determined photometrically by the color change of phenol red. This pH indicator was selected for its pKa of 7.4 and its turning zone (6.8 < pH < 8.2) being compatible with the optimal pH of the reaction catalyzed by TK, which is approximately 7.5 [54]. Phenol red changes from bright yellow at acidic pH to bright pink at basic pH, allowing easy visualization of the progress of the reaction (Figure 10). In addition, phenol red in its deprotonated form has a high molar absorption coefficient: ε = 56,000 M−1⋅cm−1 at λmax = 557 nm, detected by spectrophotometry with high sensitivity. At low buffer concentration (2 mM of triethanolamine, pH 7.5), this generic, cheap, rapid method allowed continuous monitoring for quantitative determination of kinetic parameters [52].
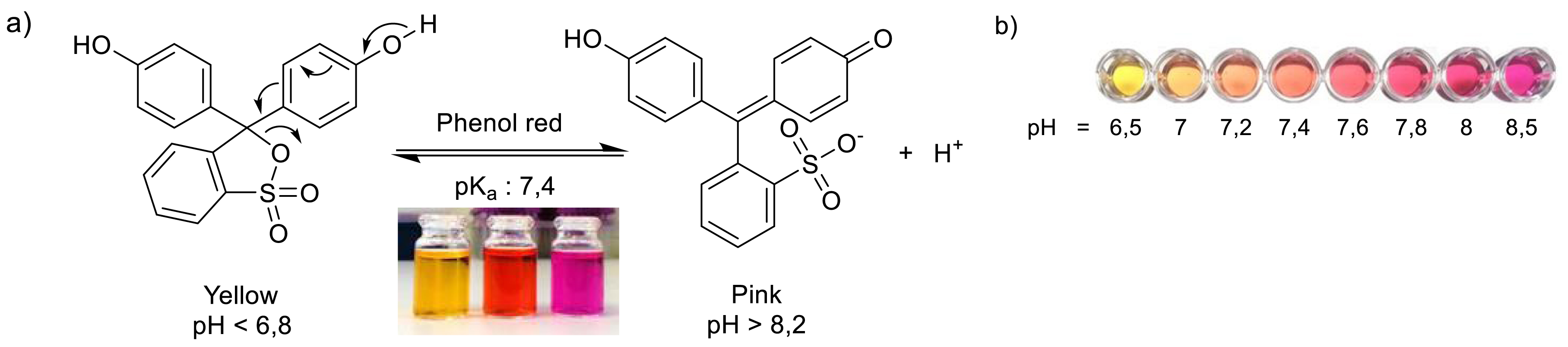
TK pH-metric activity test based on the use of phenol red as the colored indicator.
The qualitative solid phase assay procedure [53] was based on the strategies already developed with other enzymes by Turner et al. [55] and Bornscheuer et al. [56], allowing one to detect the color variation directly in clones expressing the targeted variant. In our case, the pH indicator used to detect the variation in the reaction medium was bromothymol blue offering higher contrast than phenol red described previously in the liquid phase. The libraries of clones expressing TKgst variants were first cultured on Petri dishes and then transferred on a nitrocellulose membrane, which was placed on a semi-solid medium containing the substrates, cofactors, and a pH indicator bromothymol blue. The colonies became blue if TK variants were active towards the substrates (Figure 11).
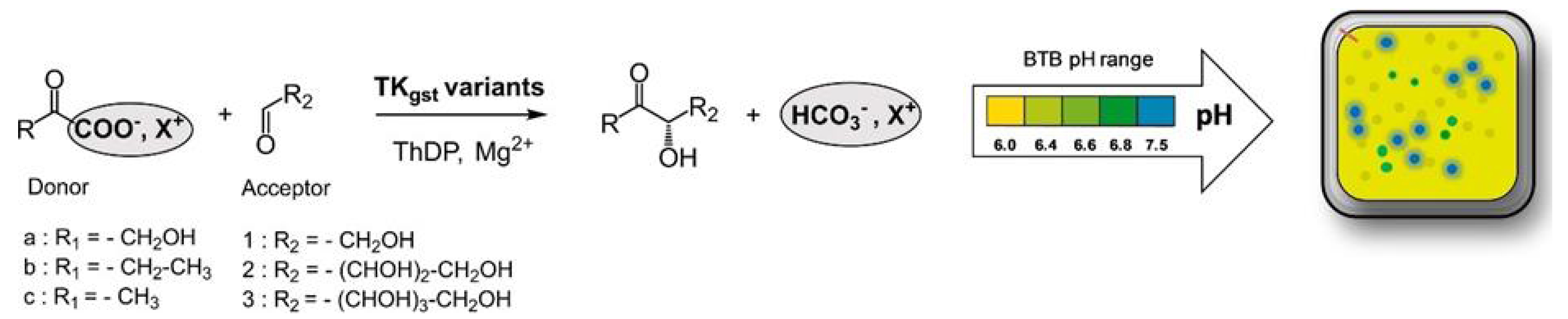
pH-metric activity test based on semi-solid medium containing bromothymol blue as the colored indicator.
2.5.3. Extension of TKgst substrate spectra
Enhancement of TKgst activity towards (2S or 2R) α-hydroxyaldehydes (C3–C6) as acceptors with Li-HPA as donor
Due to their scarcity in nature and complex synthesis, natural and unnatural ketoses particularly with long carbon chains have not been widely assayed for their potential properties on different biological targets, but recent studies have started considering these forgotten compounds. For these reasons, the improvement of TKgst towards the appropriate substrates for obtaining such ketoses and analogues with different types of stereochemistry is of great interest.
Our strategy was based on the analysis of the wild-type TKgst active site with the physiological acceptor substrate d-erythrose-4-phosphate (Figure 12). We applied SSM to key positions in direct interaction of this substrate depending on the structure of the novel targeted aldehyde as the substrate.
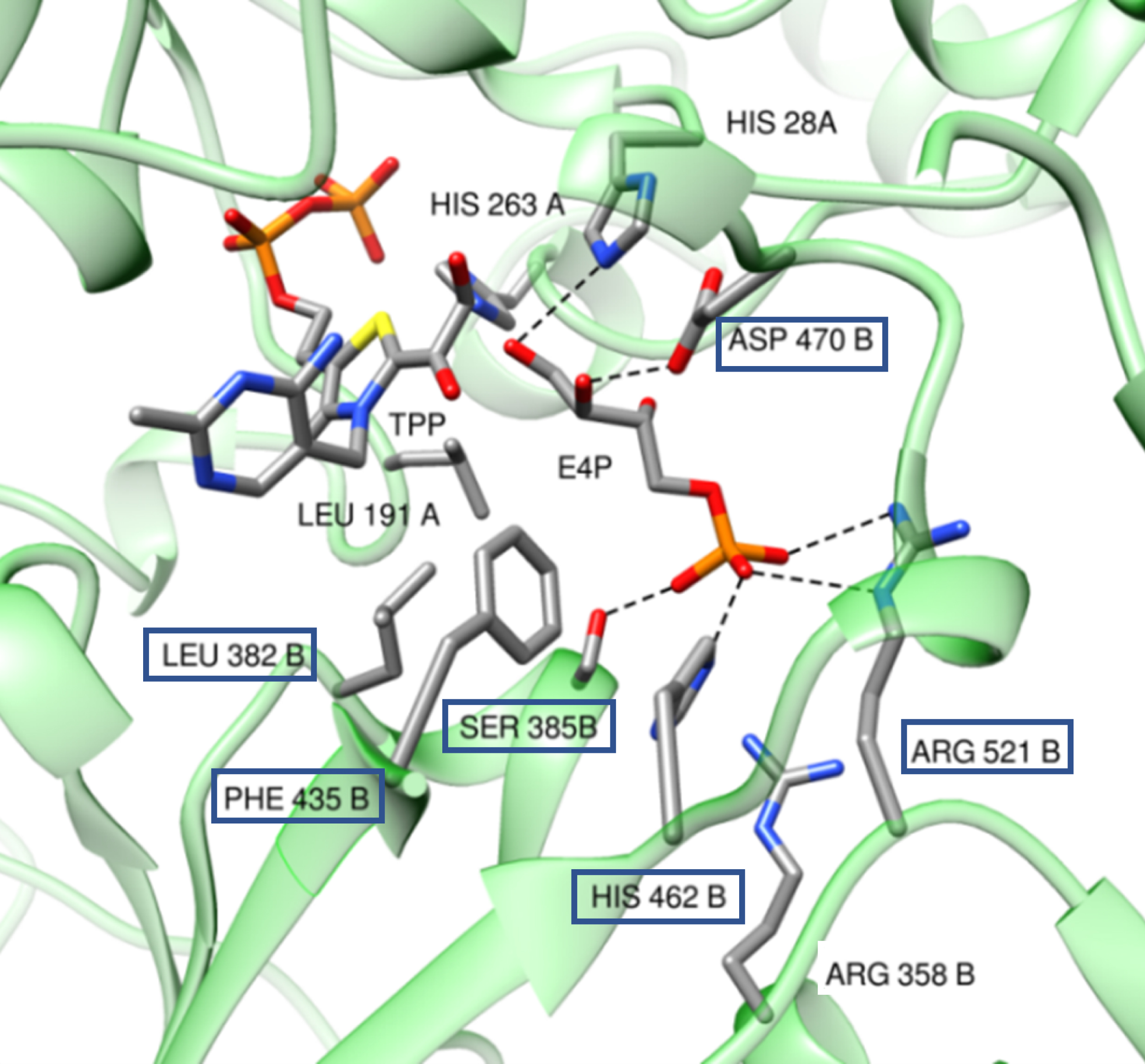
Model of wild-type TKgst based on the X-ray crystal structure of TKban (PDB entry 3M49) with the physiological acceptor substrate d-erythrose-4-phosphate (E4P).
Some examples of ketoses obtained with specific variants selected from TKgst libraries are presented in Table 1.
Synthesis of ketoses (C5–C8) from aldoses (C3–C6) in the presence of the most efficient TKgst variants
| Cn aldose acceptors | Cn+2 ketose product | TKgst variant | Reaction progress (%) | Reaction time | Isolated yield (%) | de (%) |
|---|---|---|---|---|---|---|
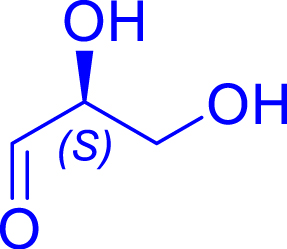 |
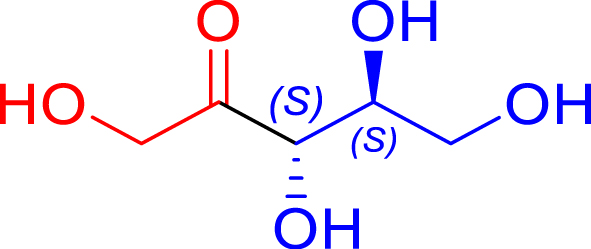 |
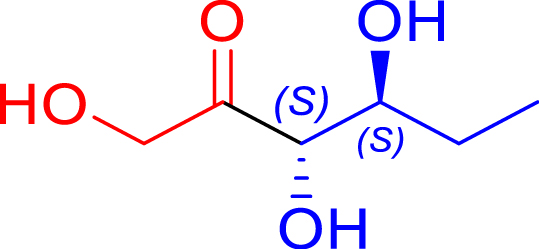
L382D/D470S | 98 | 24 h | 63 | >95 |
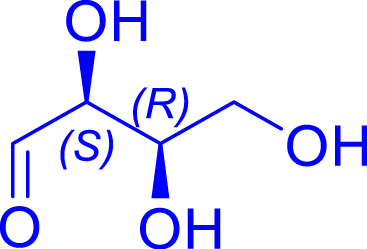
| l-glyceraldehyde C3 | l-ribulose C5 | |||||
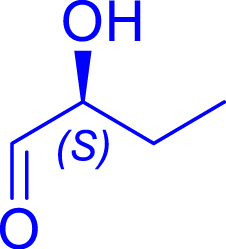 |
 |
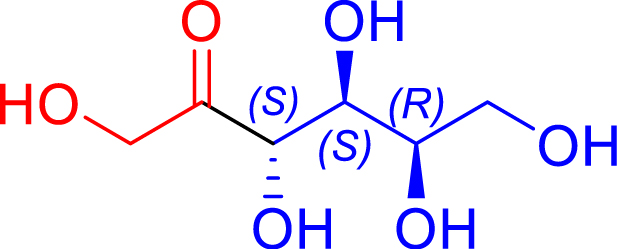
L382D/D470S | 98 | 24 h | 55 | >95 |
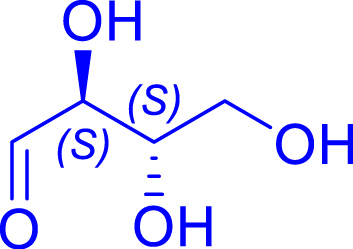
| l-lactaldehyde C3 | 5-deoxy-l-ribulose C5 | |||||
 |
 |
L382F/F435Y | >95 | 48 h | 62 | >95 |
| d-threose C4 | d-tagatose C6 | |||||
 |
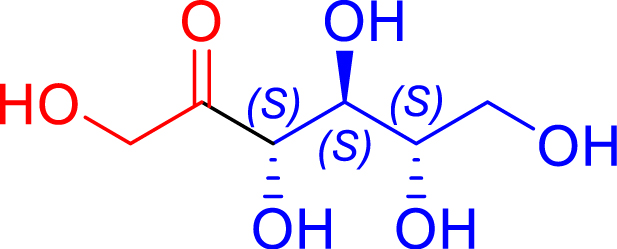 |
L382F/F435Y | >95 | 72 h | 84 | >95 |
| l-erythrose C4 | l-psicose C6 | |||||
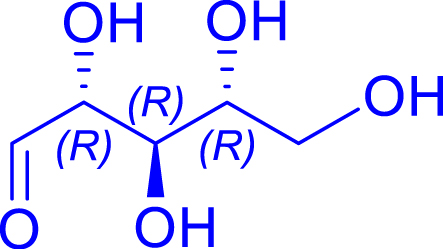 |
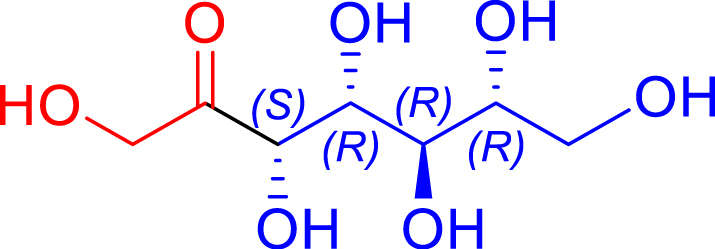 |
H462N/R521Y | >95 | 32 h | 67 | >95 |
| d-ribose C5 | d-altro-heptulose C7 | |||||
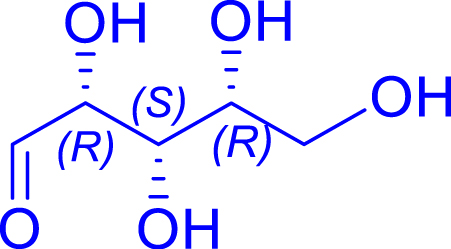 |
 |
H462N/R521Y | >95 | 96 h | 59 | >95 |
| d-xylose C5 | d-ido-heptulose C7 | |||||
 |
 |
S385D/H462S/ R521V | 93 | 7 days | 70 | >95 |
| d-allose C6 | d-glycero-d-altro-octulose C8 |
As we reported that wild-type TKgst tolerates at 60 °C (2S)-hydroxylated aldehyde as an acceptor substrate contrary to the other TKs [48], we first investigated extending these activities towards short carbon chain aldehydes (C3) such as (2S)-lactaldehyde and (2S)-glyceraldehyde through SSM at two key positions: D470, which interacts with the C2(R) hydroxyl group of the physiological erythrose-4-phosphate (DE4P) in the wild-type enzyme, and L382, located at the opposite side of the C2 aldehyde (Figure 12). The best variant, L382D/D470S, increased activity by factors 4 and 5 towards (2S)-lactaldehyde and (2S)-glyceraldehyde, respectively, forming the corresponding products 5-deoxy-l-ribulose and l-ribulose with 63% and 55% isolated yields, respectively.
Based on these first results, a second generation of TKgst variants was investigated to extend TKgst activity towards longer carbon chain aldoses (C3–C6) by targeting four other positions S385, F435, H462, and R521 near, or by direct interaction with, the phosphate group of the physiological acceptor substrate d-E4P (Figure 12) [57, 58]. Three efficient TKgst variants were identified following the screening of TKgst variant libraries. The L382F/F435Y variant showed an improvement by factors 3.7 and 4 compared to the wild-type TKgst towards the (2S)-tetroses d-threose and l-erythrose, respectively, producing the corresponding Cn+2 ketoses d-tagatose and l-psicose, respectively. The double variant H462N/R521Y demonstrated an increase in conversion by factors 3.5 and 4 towards the pentoses d-ribose and d-xylose compared to the wild-type enzyme, forming d-altro- and d-ido-heptulose, respectively, with 67% and 59% yields. The triple variant S385D/H462S/R521V allowed the synthesis of d-glycero-d-altro-octulose from d-allose while this was impossible with wild-type TKgst.
In conclusion, the aldoses (C3–C6) were almost totally converted into the expected ketose products with good to excellent isolated yields (62%–80%) and in reasonable reaction time (24 h–72 h) except the hexose d-allose requiring 7 days of reaction time with the double variant H462S/R521V. Globally, the mutations of variants on targeted positions near the phosphate group of the physiological substrate gave higher or de novo conversions compared to the wild-type enzyme. In addition, all compounds were obtained with excellent diastereoselectivities, one stereoisomer being obtained by in situ nuclear magnetic resonance (NMR) analysis of the reaction mixture. In all cases, the (S)-configuration of the C3 ketose product was confirmed by NMR spectrum comparison with the already described product.
Among the ketoses listed in Table 1, some of them have been already described for their valuable biological properties. Hexoses include d-tagatose, which is a sweetener and antidiabetic compound [17, 18], and l-psicose, which is used for the synthesis of l-fructose, a precursor of an inhibitor of glucosidases [59, 60]. The heptulose d-altro-heptulose is a marker of sugar metabolism disorders such as cystinosis [61, 62]. The d-ido-heptulose has been reported to be a precursor of valiolamine and N-substituted derivatives, glucosidase inhibitors useful for the treatment of hyperglycemic symptoms [63]. Among octuloses, d-glycero-d-altro-octulose has been identified in the plant Spinacia oleracea, and its (5S)-epimer d-glycero-d-ido-octulose, which is very abundant in Craterostigma plantagineum, has been described as a plant antioxidant involved in desiccation tolerance and could be a potential reactive oxygen species scavenger with applications in nutrition and healthcare [64, 65, 66]. Recent studies have also shown the essential role of octuloses in the metabolism of parasites such as Trypanosoma and various Leishmania species, opening the way for potential octulose analogue-based inhibitors to treat the diseases caused by these parasites [67].
Enhancement of TKgst activity towards aromatic aldehydes with Li-HPA as donor
In collaboration with the Fessner group, we investigated novel electrophiles ortho-, meta-, and para-substituted nitrosoarenes, which had not been previously investigated with TKs [68, 69]. These substrates led to the formation of corresponding aromatic hydroxamic acids, which have a wide range of medical applications (antifungal, antibacterial, antioxidant, anti-inflammatory) due to their chelating properties (Table 2) [70, 71].
Panel of nitroarenes (ortho-, meta-, and para-substituted) used as acceptor substrates for TKgst variant in the presence of Li-HPA to obtain the corresponding hydroxamic acids
| Aromatic group (X)a,b | Acyl group (R) | Yield (%) |
|---|---|---|
| H | CH2OH | 41/50 |
| 4-Br | CH2OH | 28/54 |
| 4-Cl | CH2OH | 41 |
| 3-Cl | CH2OH | 20 |
| 3,4-Cl2 | CH2OH | 10 |
| 4-CH3 | CH2OH | 49 |
| 3-CH3 | CH2OH | 29 |
| 4-Cl, 3-CH3 | CH2OH | 37 |
| 3-Cl, 4-CH3 | CH2OH | 32 |
| 4-CF3 | CH2OH | 9 |
| 3-CF3 | CH2OH | 17 |
| 4-N(Me)2 | CH2OH | 0 |
| H | CH3 | 5 |
a Substituent position relative to the nitroso group.
b DMSO as cosolvent for X=H and 4-Br and acetone as cosolvent for the other aromatic group.
A key aspect of these reactions was the formation of a carbon–nitrogen bond instead of the typical carbon–carbon bond formed by TKs with other aldehydes as electrophiles. The TKgst variant L382N/D470S (positions previously identified for improved activity towards hydrophobic aldehydes) exhibited the highest activity towards these substrates, with a sevenfold increase in activity compared to wild-type TKgst. The replacement of aspartate at position 470 with serine, a non-charged but polar amino acid, seems to enhance interaction during the carboligation step. This mutation had been described in a previous study with non-hydroxylated acceptor substrates [72]. Additionally, this variant has a melting temperature (Tm) of 73.9 °C (compared to 75.5 °C for wild-type TKgst) and remains highly stable in the presence of cosolvents, retaining 80% to 100% residual activity in DMF, acetone, and acetonitrile.
Enhancement of TKgst activity towards novel hydrophobic α-ketoacids as donors and different aldehydes as acceptors
In 2017, a challenging work developed jointly with Fessner et al. was conducted to expand the donor substrate spectrum of TKgst to α-ketoacids with hydrophobic side chains, which had not previously been considered for TKs [72]. The selection of key positions in the active site was guided by a structural study based on the model of TKgst with HPA as the donor substrate, constructed from the crystal structure of TKban (Figure 13).
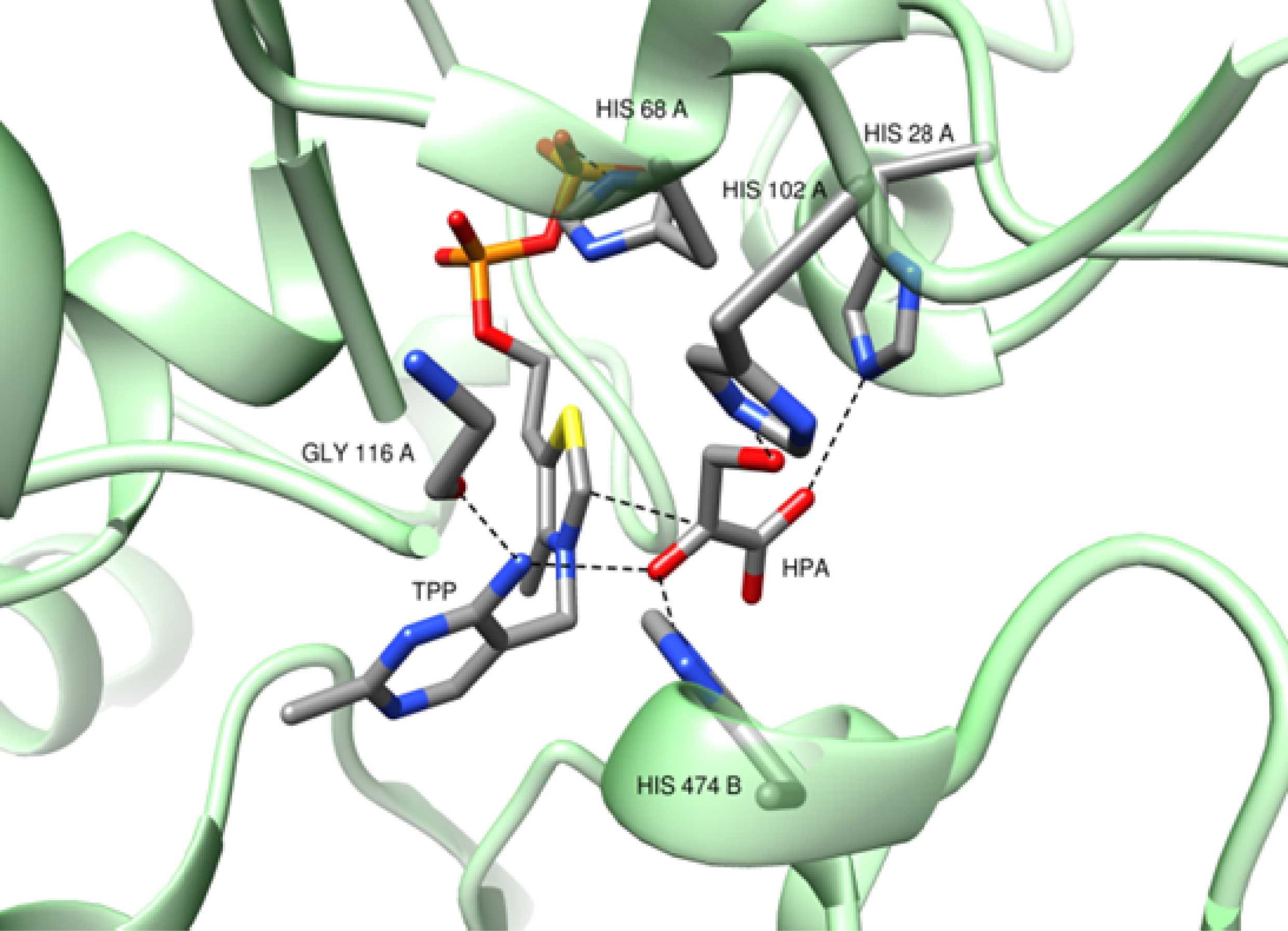
Model of wild-type TKgst based on the X-ray crystal structure of TKban (PDB entry 3M49) with hydroxypyruvate (HPA) as donor. The model was built using Modeller 9.14 and Chimera.
The active site residues interacting with the hydroxyl group of HPA, namely H68, H102, G116, and H474, were targeted. After screening libraries created by the SSM technique at positions H102 and H474, the variant H102L/H474S showed relative rate increases by factors of 4.4–32 with pyruvate, 2-oxobutyrate, and 3-methyl-2-oxobutyrate (Figure 14).
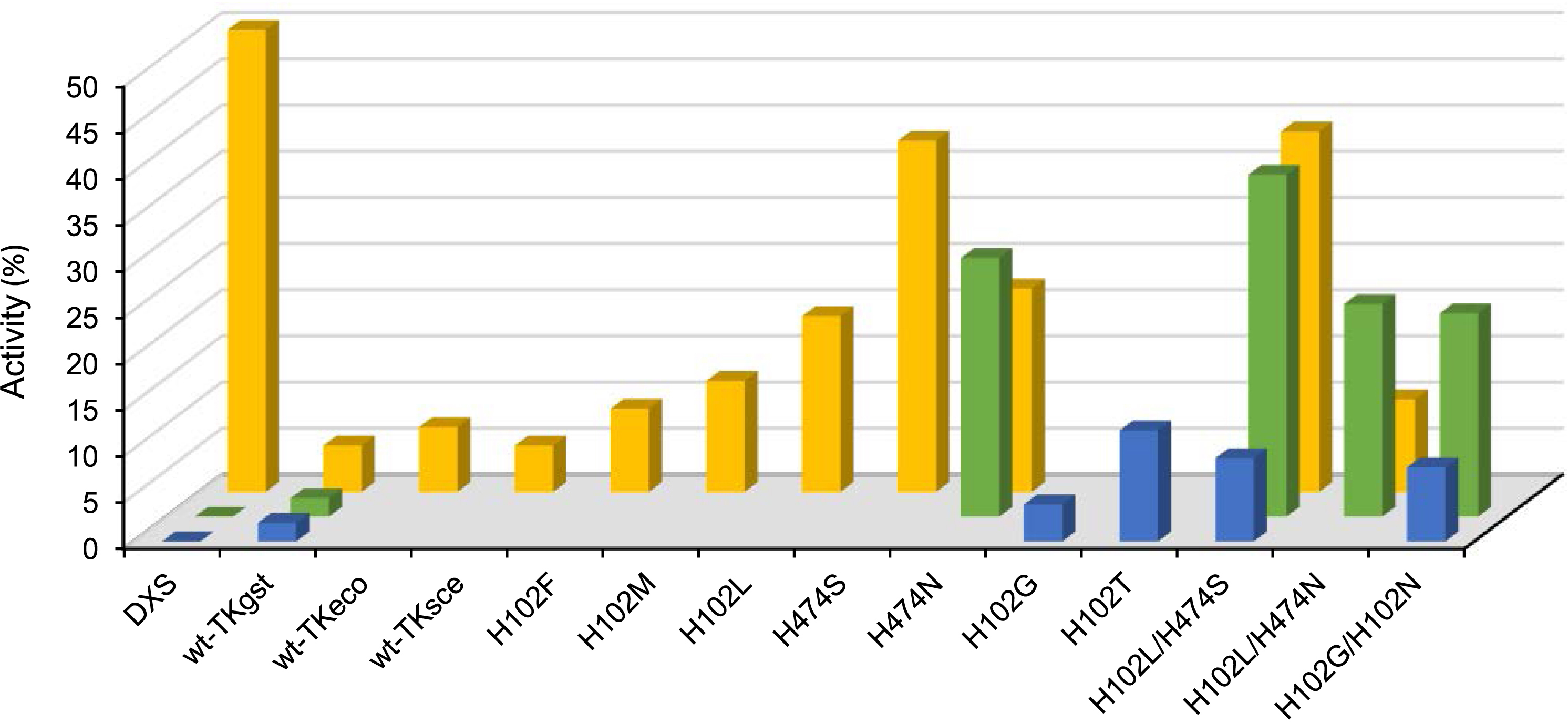
Activity of TKgst variants (kcat) with glycolaldehyde as acceptor against non-natural nucleophile substrates relative to wt-1-deoxy-d-xylulose-5-phosphate-synthase (DXSeco), wt-TKgst, and wt-TKeco ( pyruvate
pyruvate  2-oxobutanoate
2-oxobutanoate  3-methyl-2-oxobutanoate).
3-methyl-2-oxobutanoate).
In position His474, it seems to be essential to preserve a hydrogen bond with the carbonyl group, which was attained by the replacement by serine, providing a slightly reduced size. The replacement of His102 by a non-polar leucine residue not only provided extra space but also improved the binding of the hydrophobic alkyl chain of the substrate in the otherwise initially highly polar active site of wild-type TKgst. The best variants using glycolaldehyde as the acceptor were H102L with pyruvate giving a 6-fold increase, H102L/474S with 2-oxobutyrate giving a 30-fold increase, and H102T with 3-methyl-2-oxobutyrate giving a 11.4-fold increase while wild-type TKgst produced any activity with 2-oxobutyrate and 3-methyl-2-oxobutyrate and slight activity with pyruvate (Figure 16). We note that the H102T activity towards pyruvate reached 80% of that obtained with 1-deoxy-d-xylulose-5-phosphate-synthase (DXSeco) when using pyruvate as the physiological substrate.
These results offered interesting prospects for the combination of these new donors with hydroxylated or non-hydroxylated aldehydes as acceptors.
Enhancement of TKgst activity towards hydrophobic α-ketoacid as donors and hydrophobic aldehyde as acceptors
The hydrophobic donors particularly 2-oxobutyrate were investigated with hydrophobic aldehyde as acceptors to produce the corresponding unsymmetrical α-hydroxyketones (acyloins), which are valuable as flavors or non-ionic surfactants when compounds display long carbon chains (Figure 15) [16, 19].
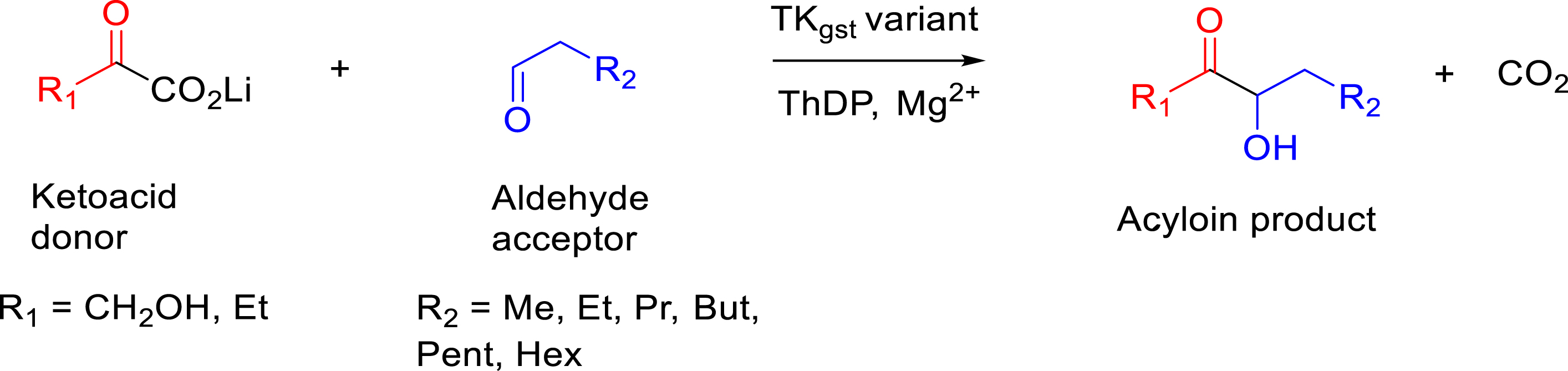
Synthesis of monohydroxylated α-hydroxyketones from hydrophobic α-ketoacid as donors and hydrophobic aldehyde as acceptors.
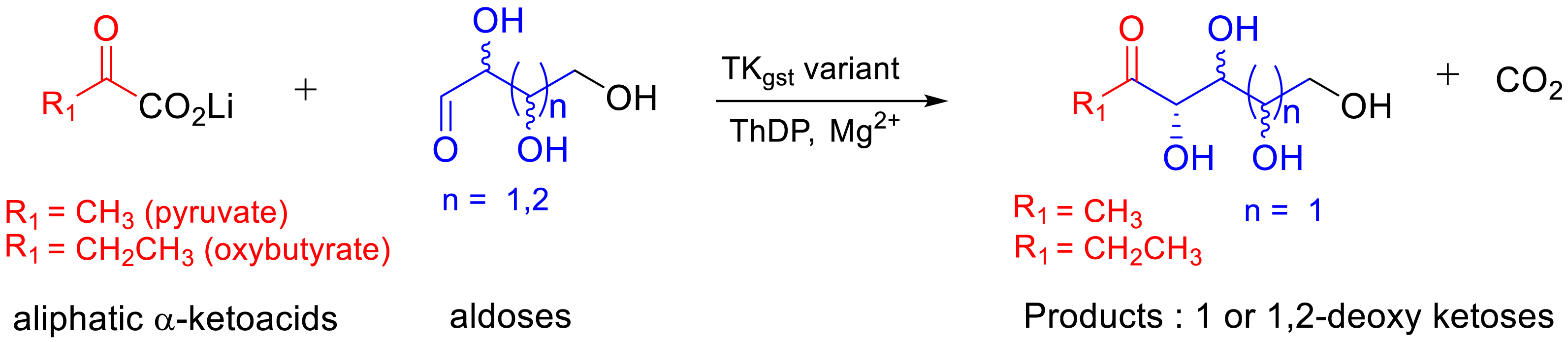
Synthesis of 1- and 1,2-deoxy-ketoses from aliphatic α-ketoacids combined with aldoses (C4–C5).
New TKgst variants were created by combining the most beneficial mutations identified previously with hydrophobic donors (H102L/H474S) with those identified earlier for hydrophobic aldehydes (F435I) [73]. The triple variant H102L/H474S/F435I was able to transfer the acyl group from 2-oxobutyrate to aliphatic aldehydes, enabling the synthesis of the corresponding monohydroxylated α-hydroxyketones (C3–C7) while except with propanal, any activity of the wild-type TKgst was observed with the longer carbon chain aldehydes. Even if the yields and enantiomeric excess (ee) were globally lower than those obtained with HPA, the products obtained with oxobutyrate as the donor are not easy to obtain with other enzymatic or chemical routes and were isolated and purified with accepted yields except for C8 (Table 3) [74].
Combination of hydrophobic donor ketoacids and aldehydes for the synthesis of monohydroxylated α-hydroxyketones in the presence of the triple variant H102L/H474S/F435I
| Ketoacid donor | Aldehyde acceptor | TKgst variant | Fold increase for TKgst wt | Reaction time (h) | Isolated yield (%) | ee (%) |
|---|---|---|---|---|---|---|
| HPA R1 = –CH2OH |
R2 = Me | L382F | 4 | 24 | 56 | 94 |
| R2 = Et | L382F | 6 | 24 | 43 | 99 | |
| R2 = Pr | L382F | 0.5 | 24 | 44 | 99 | |
| R2 = But | L382F | 2 | 24 | 41 | 98 | |
| R2 = Pent | L382F | ∞ | 24 | 21 | 87 | |
| R2 = Hex | L382F | 6 | 24 | 7 | 91 | |
| Oxobutyrate R1 = −Et |
R2 = Me | H102L/H474S/F435I | 20 | 24 | 25 | 12 |
| R2 = Et | H102L/H474S/F435I | ∞ | 24 | 47 | 6 | |
| R2 = But | H102L/H474S/F435I | ∞ | 24 | 20 | 31 | |
| R2 = Pent | H102L/H474S/F435I | ∞ | 24 | 25 | 33 | |
| R2 = Hex | H102L/H474S/F435I | ∞ | 24 | <5 | nd |
nd = not determined.
Enhancement of TKgst activity towards hydrophobic α-ketoacid as donors and hydroxylated aldehyde as acceptors
The objective was to produce 1- or 1,2-deoxy-ketoses when using pyruvate or 2-oxobutyrate, respectively, as donors appropriately polyhydroxylated with increased carbon chain length aldoses such as two tetroses (d-erythrose (C4) and l-threose (C4)) and two pentoses (d-ribose (C5) and d-xylose (C5)) (Figure 16, Table 4) [75, 76].
Synthesis of 1- and 1,2-deoxy-ketoses at 50 °C using TKgst variants in the presence of 2-oxobutyrate and pyruvate as donors and aldoses as acceptors (d-erythrose, l-threose, d-ribose, d-xylose)
| Donor | Acceptor | Product | Time (h) | TKgst variant | TKgst (mg/mL) | In situ yielda (%) | Isolated yield (%)a,b |
|---|---|---|---|---|---|---|---|
| R1 = Et Oxobutyrate |
 |
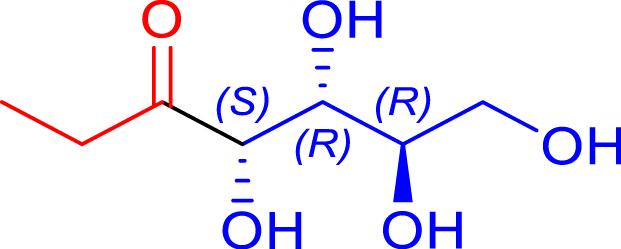 |
24 | H102L/L118I | 0.5 | 85 | 71 |
| d-erythrose | 1,2-deoxy-d-arabino-hept-3-ulose | ||||||
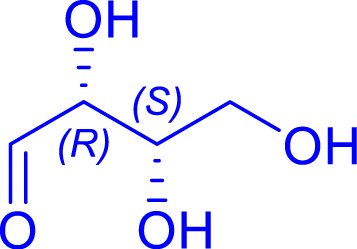 |
 |
6 | H102L/L118/H474S | 0.5 | 89 | 86 | |
| l-threose | 1,2-deoxy-d-ido-hept-3-ulose | ||||||
 |
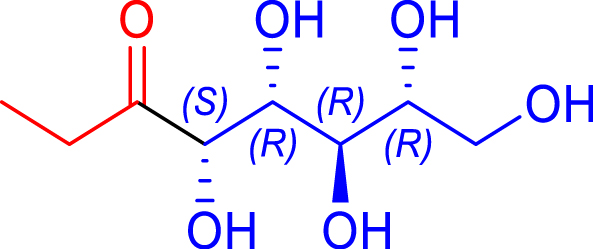 |
72 | H102L/L118I | 3 | 68 | 68 | |
| d-ribose | 1,2-deoxy-d-altro-oct-3-ulose | ||||||
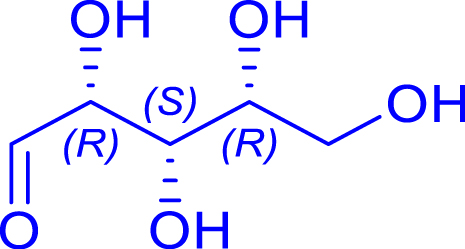 |
 |
48 | H102L/L118I | 3 | 82 | 61 | |
| d-xylose | 1,2-deoxy-d-ido-oct-3-ulose | ||||||
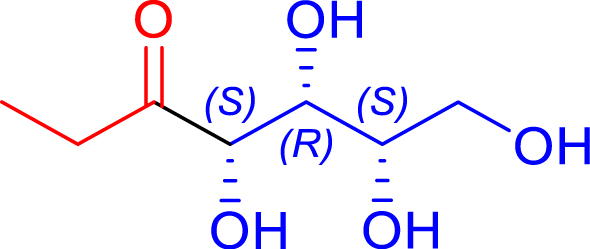
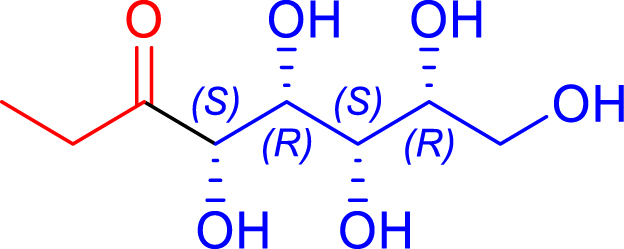
| R1 = Me Pyruvate |
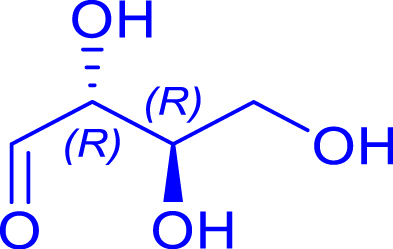 |
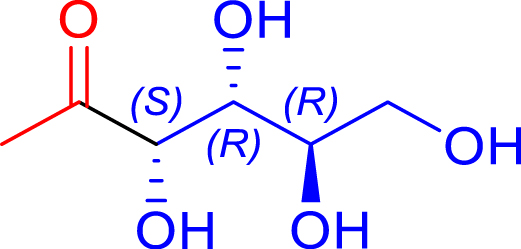 |
24 | H102L/L118/H474S | 1 | 74 | 68 |
| d-erythrose | 1-deoxy-d-fructose | ||||||
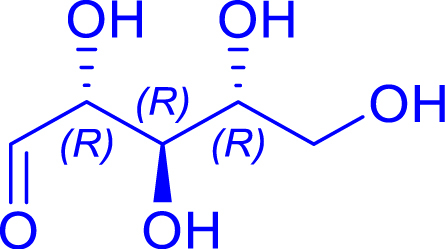
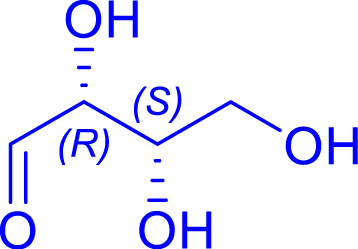 |
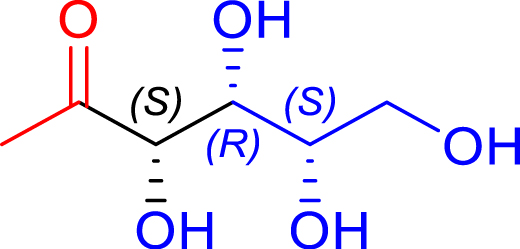 |
24 | H102L/L118/H474S | 1 | 81 | 60 | |
| l-threose | 1-deoxy-l-sorbose |
a Reactions were carried out with TKgst (0.5–3 mg), ThDP (0.1 mM), MgCl2 (1 mM), donors (50–60 mM), and α-hydroxyaldehydes (50 mM) at pH 7 and 50 °C.
b Determined by in situ 1H NMR analysis.
Among the previous TKgst variants, we combined the best mutations identified earlier, H102L and H474S, for hydrophobic α-ketoacids (pyruvate and oxobutyrate) with a new mutation on L118I, which is involved in the stabilization of the thiazolium cycle of ThDP and which could also stabilize the aliphatic donor substrates when histidine replaced isoleucine by interacting with an appropriate longer carbon chain residue.
Even if the use of higher enzyme quantities was required when using pentoses as acceptors, both donors 2-oxobutyrate and pyruvate formed the expected 1,2-deoxy-hexoses or 1,2-heptuloses and 1-deoxyhexoses, respectively, in pure form (one stereoisomer was identified in the reaction mixture by NMR) with good to excellent isolated yields. Some of them have been already described as displaying interesting properties; for example, 1-deoxy-d-fructose is a potential metabolic inhibitor and antimetabolite.
2.6. Multienzymatic synthesis strategies involving TKs
We applied the strategy of cascade reaction for in situ generation of TKgst donor and acceptor substrates, which were unstable or costly or not commercially available. To compete with the productivity of traditional methods, the use of two or even more enzymes in cascade can considerably improve the efficiency of a multistage synthesis by obviating the isolation of intermediates, thus saving time, resources, reagents, and energy while reducing waste [77, 78]. Cascade reactions can be performed along a simultaneous one-pot strategy when all the enzyme requirements are compatible. To meet limitations, such as substrate/product/reagent inhibition or incompatibility of reaction conditions (pH, temperature), a telescoped, sequential one-pot procedure can be used.
Knowing that Li-HPA was obtained from toxic bromopyruvate with modest yields [20] and its instability at higher temperatures than 25 °C [25, 47], a different strategy for HPA in situ generation was investigated. We first reported a procedure with l-α-transaminase from Thermosinus carboxydivorans (TAtca) able to produce HPA from l-serine, developed in collaboration with de Berardinis et al. (Figure 17) [54].
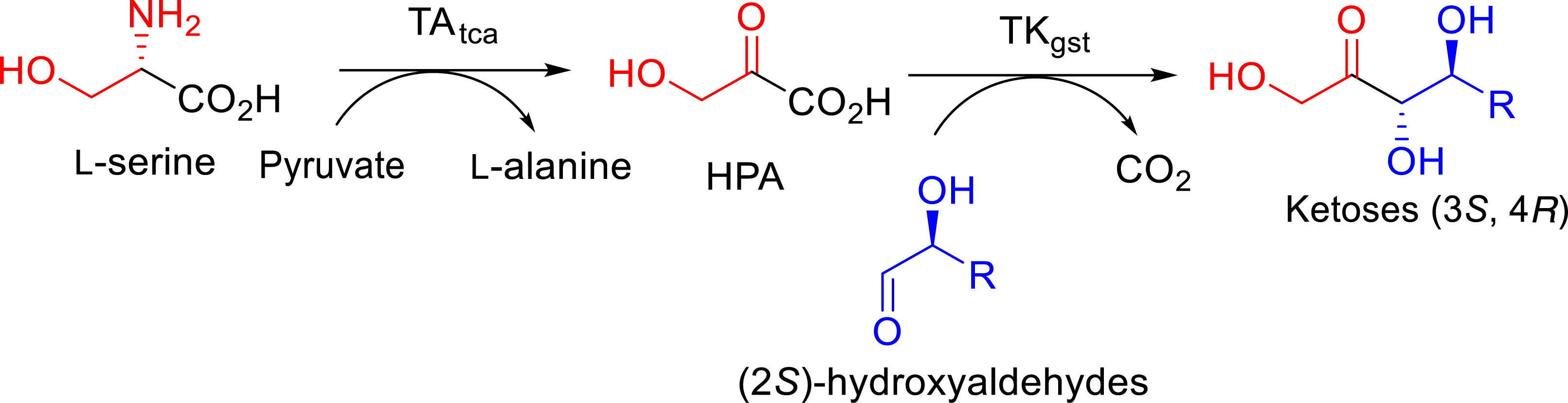
In situ generation of HPA catalyzed by TAtca coupled with TKgst for the synthesis of (3S, 4S) ketoses.
This is the first example of TA–TK coupling at high temperature cited in the literature. The TAtca being thermostable, it could be coupled in a “one-pot” at 60 °C with the thermostable TKgst. This approach was applied to the synthesis of rare l-erythro-ketoses (3S, 4S) in the presence of (2S)-aldehydes for which TKgst has low activity at 20 °C. The reaction products were obtained in good to excellent yields (51%–98%) with no accumulation of HPA in the medium and complete conversion of l-serine.
Ward et al. has since discovered more efficient thermostable TAs within metagenomic libraries [37] from Thermobifida fusca (TAtfu) and Geobacillus stearothermophilus (TAgst), which showed opposite stereoselectivities. The two thermostable TAs retain approximately 70% of their activity after 24 h of incubation at 60 °C. They were selected to generate HPA from a racemic mixture of serine.
Based on our bi-enzymatic cascade reaction, an additional third enzymatic step was investigated to generate (2S)-aldehydes such as l-glyceraldehyde, d-threose, and l-erythrose in collaboration with Clapes et al. [79] The precursors of α-hydroxylated aldehydes (2S) are glycolaldehyde and formaldehyde, which are inexpensive and achiral in the presence d-fructose-6-phosphate aldolase from E. coli (FSAeco) (Figure 18).
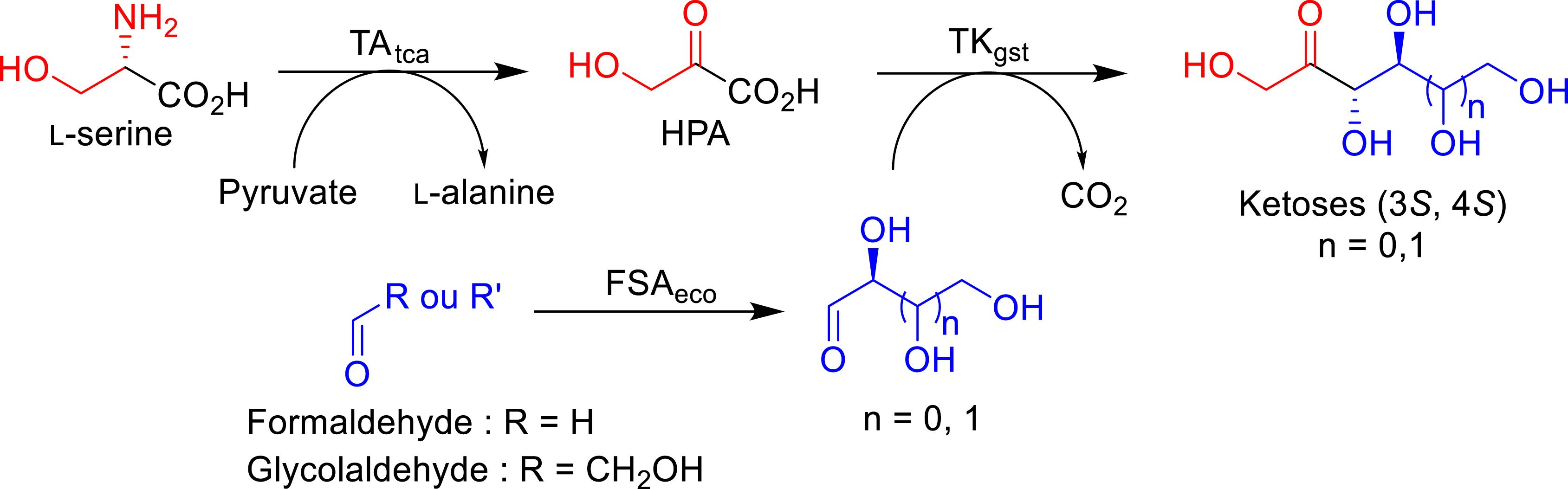
In situ generation of aldehydes (2S) with FSAeco and HPA with TAtca coupled with TKgst in a one-pot sequential cascade.
The simultaneous coupling of FSAeco and TKgst in a one-pot was not possible since glycolaldehyde and formaldehyde would react directly with HPA in the presence of TKgst to form l-erythrulose and DHA, respectively. Therefore, a sequential process was developed in which the α-hydroxylated (2S) aldehyde is produced in the first step catalyzed by FSAeco and then used in solution without intermediate purification as the acceptor substrate of TKgst. The synthesis of the corresponding l-erythro-ketoses (3S, 4S) was carried out from HPA also in situ generated from l-serine in the presence of the thermostable TAtca described previously. The simultaneous coupling of TAtca and TKgst makes it possible to carry out the synthesis at 60 °C and thus to increase the low affinity of TKgst for (2S)-hydroxylated aldehydes.
This process made possible the synthesis of three rare ketoses (3S, 4S)—l-ribulose from l-glyceraldehyde, d-tagatose from d-threose, and l-psicose from l-erythrose—which are compounds with high added value, with diastereomeric excess (de) greater than 95% and product isolated yields of 53, 55, and 49%, respectively.
The main disadvantage of these TA strategies for HPA generation is the release of alanine as the by-product, which is not in favor of atom economy. Another enzymatic strategy avoiding the use of cosubstrate has been considered via serine, allowing the oxidation of the amine function by a microbial amino acid oxidase (AAO, EC 1.4.3.3) in the presence of O2 and catalase (EC 1.11.1.6) to dismutate the hydrogen peroxide formed during the oxidation stage. We turned to dAAOrg [80], already reported by Pollegioni et al. [81, 82], to accept the polar amino acid d-serine with higher specific activity compared to dAAO from pig kidney. But dAAOrg has never been investigated for producing α-ketoacid HPA from d-serine, especially at the preparative scale. In a sequential reaction, we also investigated producing d-serine in collaboration with Protéus by Seqens from achiral glycine and formaldehyde as precursors with E. coli cells expressing threonine aldolase [83]. After total conversion of the substrate, the crude extract containing d-serine was used as the precursor of HPA (Figure 19).
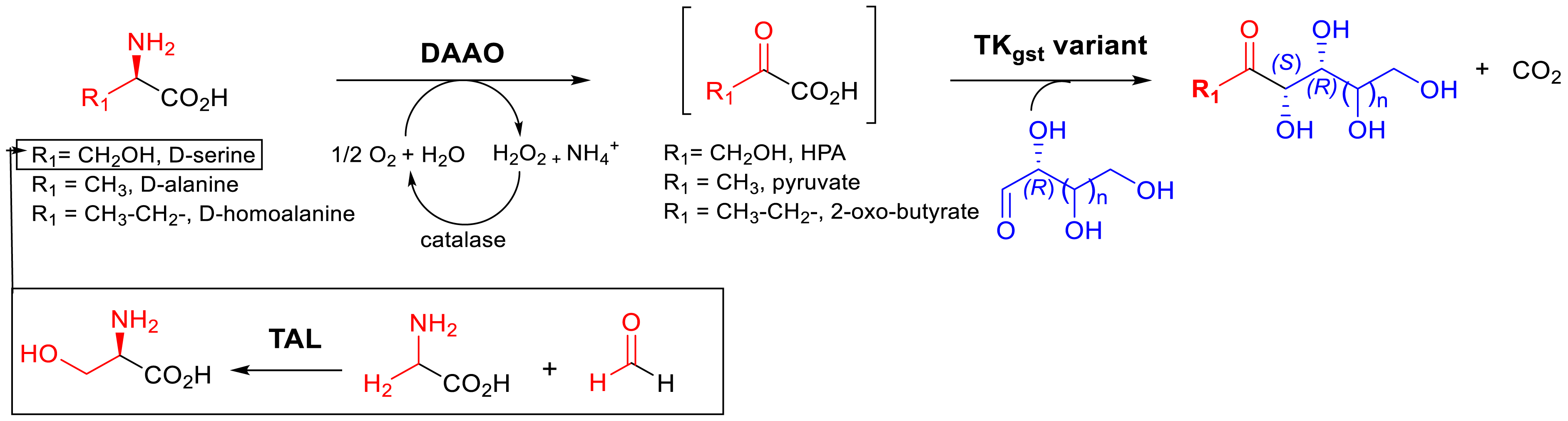
In situ generation of α-ketoacids from dAAO (dAAOrg for HPA and dAAO4536 for pyruvate and oxobutyrate) coupled with TKgst for the synthesis of ketoses and analogues.
In parallel, we discovered in collaboration with Prozomix a thermostable dAAO4536 from metagenomic library screening not applicable for producing HPA but pyruvate and oxobutyrate from d-alanine and d-homoalanine, respectively (Figure 21) [74].
Finally, dAAOrg (or dAAO4536) was coupled with the TKgst variant in a one-pot, two-step simultaneous or sequential cascade sequence with different aldehydes (hydrophobic or polyhydroxylated with increased carbon chain length) as TKgst acceptor substrates introduced into the reaction mixture at the same concentration as d-amino acids. A complete conversion of all substrates was observed, and targeted compounds were recovered with high enantio- and diastereoselectivities. These approaches appeared particularly appropriate for expensive, unstable, and commercially unavailable a-ketoacid synthesis.
2.7. Promiscuous cross-acyloin condensation reaction catalyzed by TKgst
Previous studies conducted with propanal showed that the variant H102L/L118I/H474S was capable of catalyzing the self-acyloin condensation of propanal to yield propioin (Figure 20) [84].
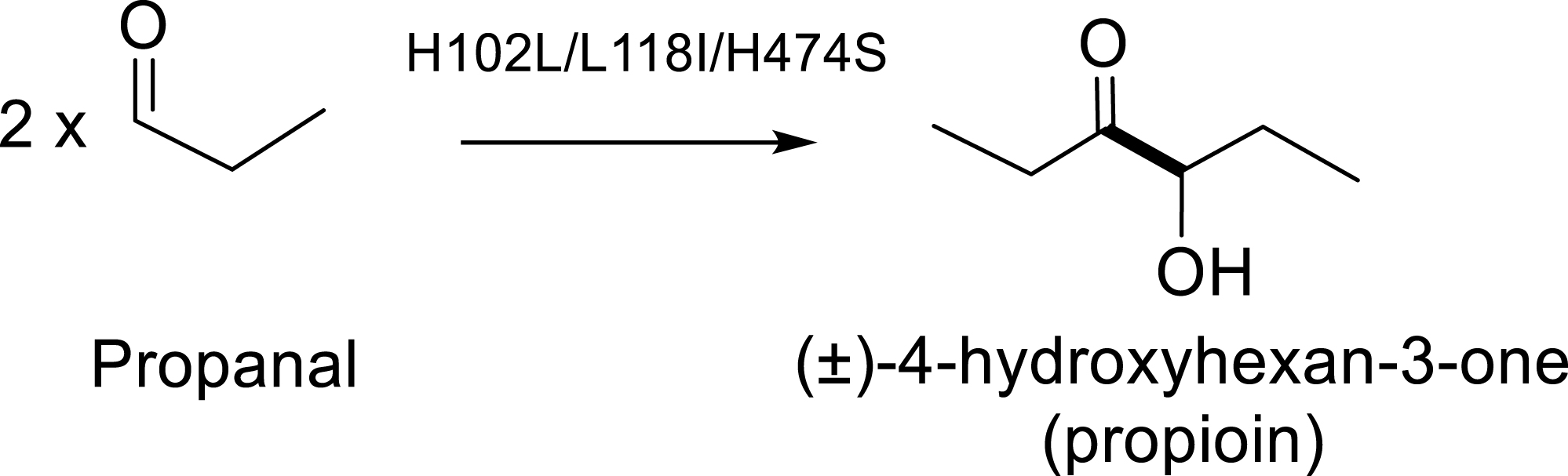
Self-condensation of two propanal molecules giving (±)-4-hydroxyhexan-3-one (propioin) catalyzed by TKgst variant H102L/L118I/H474S.
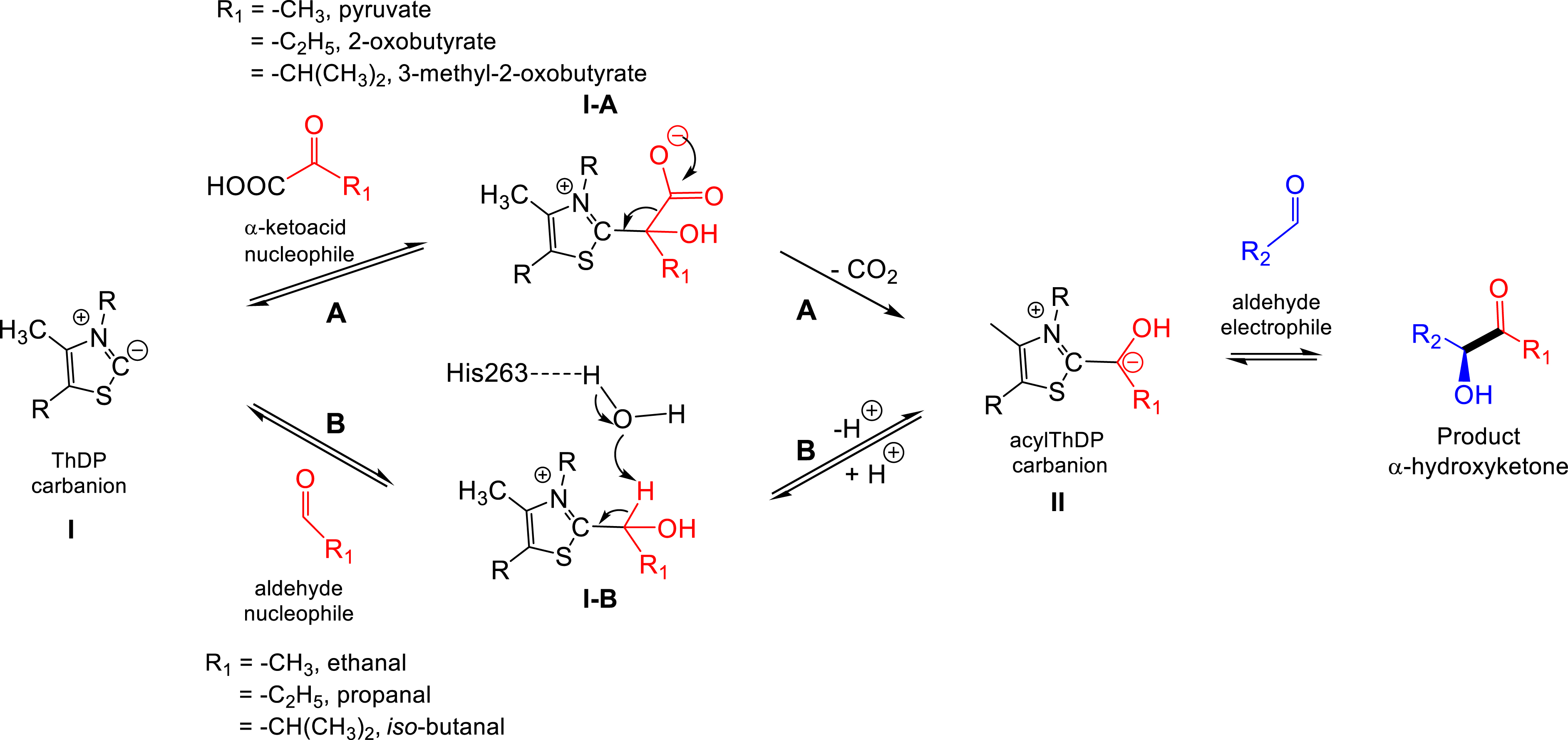
Proposed mechanism for acyloin condensation according to pathway B compared to pathway A giving the common α-hydroxy-β-(polyhydroxy)alkylthiamine diphosphate (acylThDP) carbanion II generated from aliphatic α-ketoacids (pathway A) or from corresponding aliphatic aldehydes (pathway B).
According to these results, a possible mechanism for the acyloin condensation reaction was investigated in the presence of two aldehydes as TKgst substrates by in silico studies using H102L/L118I/H474G(S) TKgst variant active site models [85]. The presence of leucine in place of histidine in the 102 position (H102L) may contribute to the stabilization of the first aldehyde molecule in the active site. Then, an attack of ThDP carbanion I on the carbonyl of the first molecule of aldehyde acting as a nucleophile followed by a proton transfer of I–B via a water molecule to His263 led to acylThDP carbanion II (Figure 21). In the last step, acylThDP carbanion II attacked the carbonyl group of the second aldehyde molecule acting as the electrophile to form the acyloin product. In this mechanism, the aldehyde acting as the nucleophile can replace the corresponding acyl group of corresponding α-ketoacids but avoids decarboxylation and the release of carbon dioxide. We proved that both pathways led to the same acylThDP carbanion II and products [85].
To extend this reaction to the more challenging cross-acyloin condensation, we used three aldehydes’ (ethanal, propanal, and iso-butanal) analogues of α-ketoacids (pyruvate, 2-oxobutyrate, and 3-methylbutyrate used previously) combined with hydroxylated aldehydes (C2–C4; glycolaldehyde, d-glyceraldehyde, and d-erythrose) in the presence of TKgst variants H102L/L118I/H474S(G) identified earlier (Figure 22) [85].
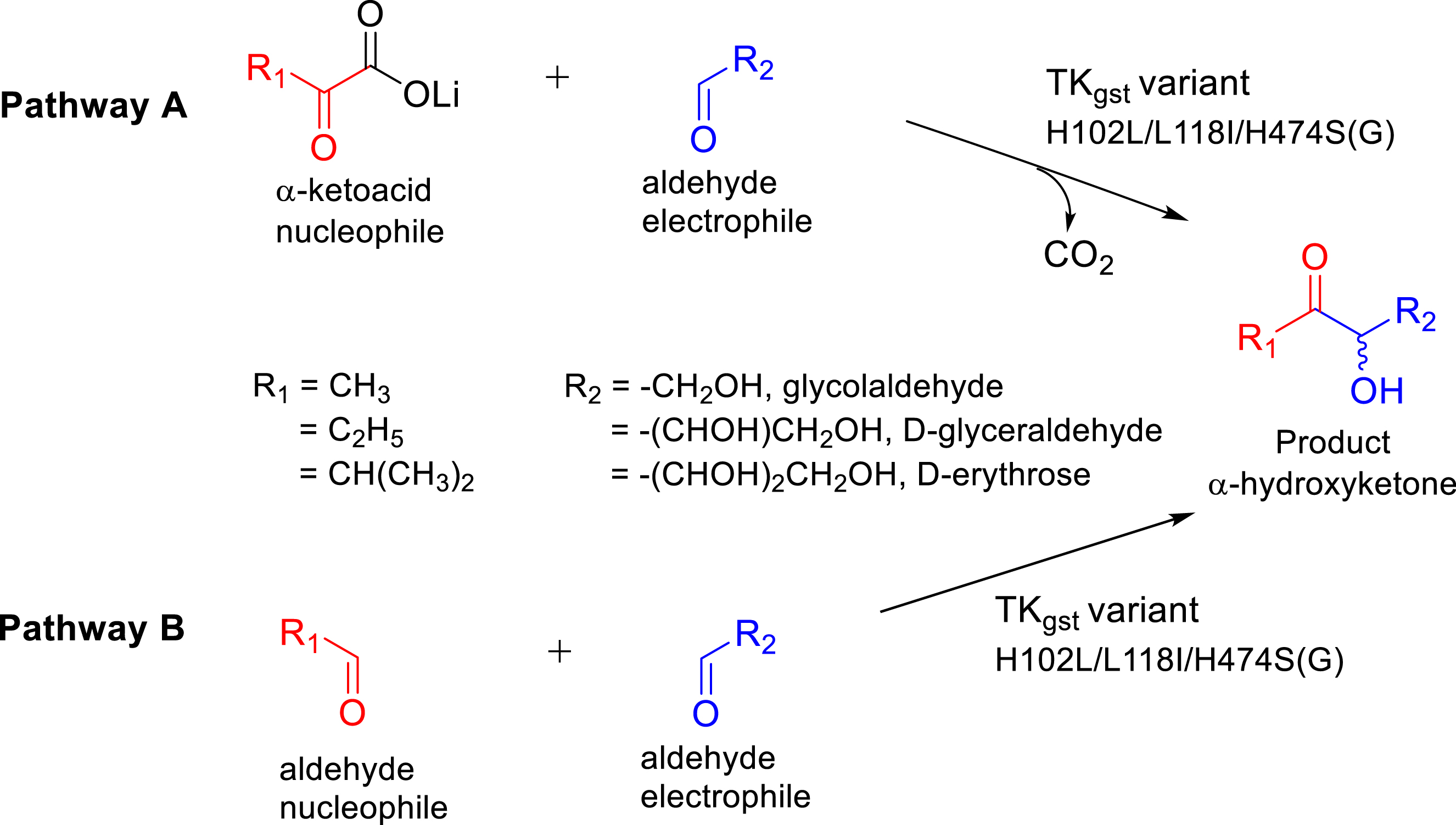
Synthesis of α-hydroxyketones catalyzed by TKgst variants from two possible nucleophiles, aliphatic α-ketoacids (pyruvate, 2-oxobutyrate, 3-methyl-2-oxobutyrate; pathway A) and the corresponding aliphatic aldehydes (propanal, butanal, iso-butanal; pathway B) in the presence of hydroxylated aldehydes as electrophiles.
For each synthesis with stoichiometric amounts of both different aldehydes (Table 5), the in situ analysis of the reaction medium by NMR showed the formation of only one α-hydroxyketone, highlighting the selectivity of the variants. Indeed, the uncontrolled cross-acyloin condensation can lead to four products. All α-hydroxyketones were obtained with good isolated yields comparable to those obtained by pathway A from the carboxylation of α-ketoacids (Table 5) [85].
Synthesis of α-hydroxyketones using TKgst variants in the presence of ethanal, propanal, and iso-butanal as nucleophiles (50 mM) and hydroxylated aldehydes (glycolaldehyde, d-glyceraldehyde, d-erythrose) as electrophiles (50 mM). Reactions were performed at 37 °C in phosphate buffer (50 mM) at pH 7.0
| Donor | Acceptor | Product | Time (h) | TKgst variant | In situ yield (%)a | Isolated yield (%) | deb (%) |
|---|---|---|---|---|---|---|---|
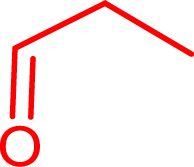
propanal |
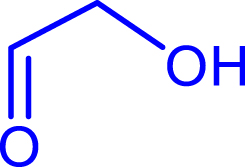 |
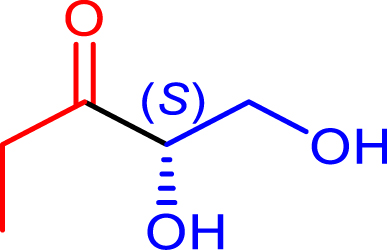 |
72 | H102L/L118I/H474G | 80 | 50 | 69 (S) |
| glycolaldehyde | |||||||
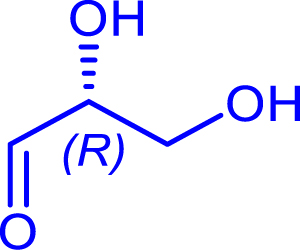 |
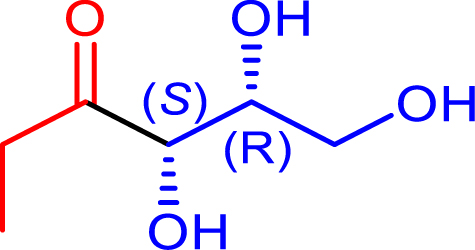 |
72 | H102L/L118I/H474S | 48 | 31 | >95 | |
| d-glyceraldehyde | |||||||
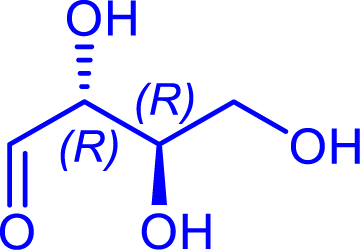 |
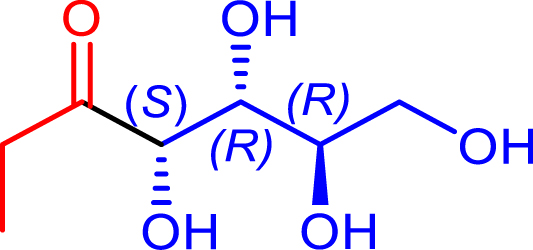 |
48 | H102L/L118I/H474S | 75 | 50 | >95 | |
| d-erythrose | |||||||
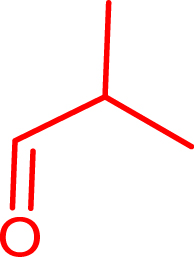
Iso-butanal |
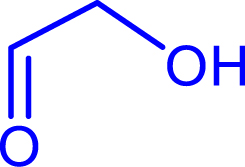 |
 |
72 | H102L/L118I/H474G | 82 | 60 | 34 (S) |
| glycolaldehyde | |||||||
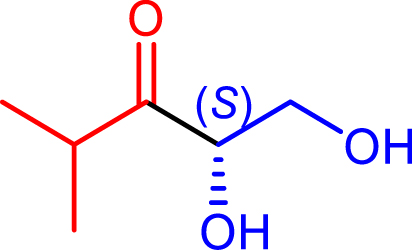
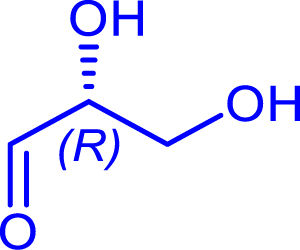 |
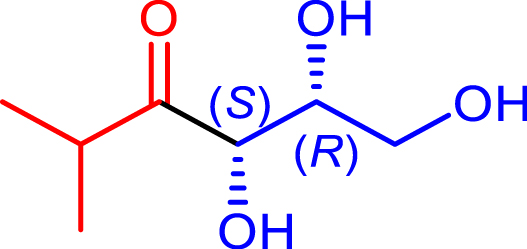 |
48 | H102L/L118I/H474S | 39 | 31 | >95 | |
| d-glyceraldehyde |
a Determined by in situ 1H NMR using TSP-d4 as an internal standard and calculated based on in situ product formation.
b Determined by chiral GC-MS analysis after derivatization.
The formation of the targeted α-hydroxyketones by cross-acyloin condensation offers the great advantage of ensuring atom economy by avoiding the release of carbon dioxide generated by the decarboxylation of α-ketoacids. This novel enzymatic acyloin condensation reaction catalyzed by TK should broaden the synthetic toolkit for creating unsymmetrical α-hydroxyketones and enhance the efficiency of previous enzymatic and chemical approaches in terms of mass metrics.
3. Conclusion
Wild-type TKs in cells exclusively transfer a ketol unit from a ketose phosphate to an aldose phosphate Cn leading to a Cn+2 ketose phosphate by a reversible reaction. The results reported in this paper showed that TKgst engineering allowed the synthesis of a wide range of non-phosphorylated α-hydroxyketones (aliphatic, hydroxylated, or aromatic) in one step with excellent yields and stereoselectivities. The noteworthy evolvability of thermostable TKgst was mostly based on its robust 3D structure against destabilizing mutagenesis factors. The best variants selected from libraries created by rational design based on active site analysis enhanced the wild-type TKgst towards targeted nucleophiles, analogues of pyruvate, coupled with electrophiles such as aliphatic, increased carbon chain length polyhydroxylated, and aromatic aldehydes. An essential tool to select the best variants was the development of a generic rapid and cheap pH-shift-based screening assay applied in the liquid or solid phase. The latter enabled visual detection of clones expressing active TKgst by their specific coloration.
The best efficient TKgst variants were first used to catalyze the usual TK reaction from the α-ketoacid and aldehyde as the nucleophile and electrophile, respectively. As the main hurdle for biocatalytic application is the instability/cost of α-ketoacids, one-pot strategies were investigated for in situ generation of α-ketoacids from corresponding l- or d-amino acids with l-transaminase or d-amino acid oxidase. The novel promising approach was based on a promiscuous TKgst reaction, allowing selective cross-acyloin condensation of two aldehydes, one playing the role of the nucleophile in place of α-ketoacid and the other aldehyde acting as the electrophile. This original TKgst catalyzed reaction provides atom economy while avoiding carbon dioxide release and achieving similar efficiency compared to the commonly used pathway.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
This work was funded by the Fonds Régional Innovation Laboratoire (grant DOS00494484/00 to LH), Pack Ambition Recherche ID 1701105201-61617, the Agence Nationale de la Recherche (grants ANR-09-BLAN-0424-CSD3 and ANR-22-CE07-0038-01), ERA CoBioTech TRALAMINOL—ID: 64 (grant to LH), and MSCA-ITN-ETN-2020 CC-TOP—ID: 956931.
Acknowledgments
The authors thank Lionel Nauton and Vincent Thery for molecular modeling studies, Muriel Joly for TKgst variant production, Martin Leremboure for GC-MS analysis, PhD students (Juliane Abdoul-Zabar, Marion Lorillière, Nazim Ocal, Giuseppe Arbia, and Camille Gadona), and postdocs (Ghina Ali, Thomas Moreau, Sandrine Sorel, Romain Dumoulin, Mélanie L’Enfant, Hubert Casajus, and Aurélie Lagarde).