


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Numerous free-base and transition metal triarylcorroles bearing different meso- and 𝛽-substituents have been synthesized [1,2] and electrochemically examined [3], with particular emphasis being placed on elucidating the electronic configuration of those metal complexes which are known to act as air-stable radicals [4,5,6,7,8]. In 2010, Pierloot et al. [9] presented ab initio evidence for a noninnocent corrole ligand in the case of copper corroles, and since that time there have been many reports characterizing neutral copper corroles as having a reduced divalent central metal ion (CuII) and an oxidized macrocyclic ligand in its cation radical form (Cor∙2−) [5,6,7,10,11,12,13,14]. Evidence for this assignment has involved a wide variety of approaches, including structural analysis, spectroscopic measurements and electrochemical criteria to assess the noninnocent behavior of the corrole in question and a concise summary of what has been published in this area was recently reviewed by Ganguly and Ghosh [15]. In our own work we have suggested an electrochemical litmus test for evaluating the innocence or noninnocence of transition-metal corroles [3,5,10,11,13,16,17,18,19], and suggested that the presence of a facile reduction at potentials more positive than − 0.20 V versus SCE (Saturated Calomel Electrode) could be used as one electrochemical diagnostic criterion to establish ligand noninnocence in these systems.
Our recent studies on the electrochemistry of tetrapyrrole macrocycles has focused in part on cobalt corroles [16,17,18,19], some of which possessed redox-active meso-nitrophenyl groups [16,17] where ligand noninnocence or innocence was shown to be governed by the number and type of axial ligands bound to the formal cobalt(III) central metal ion. In one of these studies [19], we reported the effect of anions on the spectroscopic and electrochemical properties of noninnocent cobalt corroles and noted that the addition of cyanide anions (CN−) to solution led to the stepwise formation of a five- and six-coordinate cobalt(III) complex with an innocent corrole macrocyclic ligand, while the addition of other anions, such as fluoride (F−), led to a chemical reduction of the noninnocent corrole ligand giving an anionic [Cor3−CoII]− product. The reaction with F− was rationalized by the known ability of strong Lewis basic anions to reduce or form a solvent caged radical pair with certain π-acids (i.e. ligands with a low lying LUMO or SOMO-1) via anion induced electron transfer (AIET) [20,21,22].
There remains little doubt surrounding the assignment of ligand noninnocence in the case of copper corroles which are isolated as four-coordinate species and do not undergo axial ligation in solution and these [Cor∙2−CuII] systems therefore serve as ideal metallocorroles to explore the ability of AIET to occur in aprotic media. This is examined in the current study which reports synthesis and characterization of the five copper corroles (1–5) in Scheme 1, with one aim of this study being to elucidate the prevailing electrochemical and spectroscopic behavior of copper nitroaryl corroles in CH2Cl2 and the other to explore the possible reduction of these open-shell complexes by cyanide or fluoride anions added to solution in the form of tetra-n-butylammonium salts, TBACN or TBAF.
Structures and numbering of the investigated copper triarylcorroles 1–5 and the reference compound 0.
2. Experimental section
2.1. Material and instrumentation
All chemicals and solvents were of the highest electrochemical grade available and were used without further purification. NMR solvents were purchased from Eurisotope. DriSolve anhydrous dichloromethane (CH2Cl2, ⩾99.8%) was purchased from Sigma Chemical Co. Tetra-n-butyl-ammonium perchlorate (TBAP, ⩾99.0%), 95.0% tetra-n-butyl-ammonium cyanide (TBACN) and 98% tetra-n-butyl-ammonium fluoride hydrate (TBAF) were purchased from Sigma Chemical Co. and stored in a desiccator until used. Copper corrole 0 (see Scheme 1) was synthesized and characterized according to literature procedures [23].
1H NMR spectra were recorded on a Bruker Avance NEO 500 spectrometer operating at 500 MHz and available at the PACSMUB-WPCM technological platform, which relies on the “Institut de Chimie Moléculaire de l’Université de Bourgogne” and SATT SAYENS “TM”, a Burgundy University private subsidiary. All NMR shift values are expressed in ppm. 1H NMR spectra were calibrated using the residual peak of chloroform at 7.26 ppm and 19F NMR spectra were calibrated with an internal reference (CFCl3).
UV–visible spectra of the synthesized compounds were recorded on a Varian Cary 50 or Hewlett-Packard model 8453 diode array spectrophotometer. Quartz cells with optical path lengths of 10 mm were used. ESI mass spectra were recorded on a LTQ Orbitrap XL (THERMO) instrument for HR-MS spectra and on an AmaZon SL (Bruker) instrument for the LRMS spectra or on a Bruker Microflex LRF MALDI Tandem TOF mass spectrometer using dithranol as the matrix.
Thin-layer UV–vis spectroelectrochemical measurements were made using a commercially available thin-layer cell from Pine Instruments Inc. which had a platinum honeycomb working electrode consisting of 19 Pt-coated channels with each channel being 0.50 mm in diameter and a center-to-center distance of 0.75 mm. Potentials were applied and monitored with an EG&G PAR Model 173 potentiostat/galvanostat. High-purity argon from Matheson Trigas was used to deoxygenate the solution and a stream of inert gas was kept over the solution during each spectroelectrochemical experiment.
Electrochemical measurements were performed at 298 K using an EG&G Princeton Applied Research (PAR) Model 173 potentiostat/galvanostat, paired with a EG&G PAR Model 175 universal programmer and a Houston Instruments Omnigraphic 2000 XY Plotter. The three electrode system used for cyclic voltammetric measurements consisted of a glassy carbon working electrode, a platinum counter electrode and a saturated calomel reference electrode (SCE) which was separated from the bulk of the solution by a fritted glass bridge of low porosity. The bridge was purchased from Gamry Instruments and contained the solvent/supporting electrolyte mixture.
2.2. Synthesis of 5,15-dimesityl-10- (2,4-dinitrophenyl)corrole, (Mes)2(2,4‐(NO2)2Ph)CorH3
In a round bottom flask, 5-mesityldipyrromethane (396.5 mg, 1.5 mmol) and 2,4-dinitrobenzaldehyde (147 mg, 0.75 mmol) were dissolved in 150 mL of CH3OH. Afterwards a solution of 3.8 mL of HClaq (36%w) in 75 mL of water was added and the crude material was stirred at room temperature for 2 h. The mixture was extracted with 80 mL of CHCl3 after which the organic phase was washed twice with 80 mL of water, dried on sodium sulfate, filtered and then diluted with 250 mL of CHCl3. p-Chloranil (369 mg, 1.5 mmol) was added and the mixture was stirred overnight at room temperature. The reaction mixture was evaporated to dryness and chromatographed on silica gel using a CHCl3-heptane (2:1, v:v) eluent. Yield 160.2 mg (0.23 mmol, 31%). UV–vis (CH2Cl2) 𝜆max [nm, 𝜀 × 103 (M−1⋅cm−1)]: 406 (106.2), 421 (86.9), 569 (20.5), 599 (14.8). 1H NMR (500 MHz, CDCl3), 𝛿 (ppm): 9.17 (d, 4 J = 2.5 Hz, 1H, HPh), 8.88 (d, 3 J = 4.5 Hz, 2H, H𝛽), 8.72 (d, 3 J = 8.0 Hz, 1H, HPh), 8.50 (d, 3 J = 5.0 Hz, 2H, H𝛽), 8.44 (d, 3 J = 8.0 Hz, 1H, HPh), 8.32 (d, 3 J = 4.5 Hz, 2H, H𝛽), 8.21 (d, 3 J = 4.5 Hz, 2H, H𝛽), 7.27–7.25 (s, 4H, mesityl, overlapped with CHCl3 deuterated solvent residual signal), 2.59 (s, 6H, CH3), 1.94 (s, 6H, CH3), 1.89 (s, 6H, CH3). LRMS (MALDI/TOF) [M+H]+: 701.13 (exp.), 701.29 (calcd). HRMS (ESI) [M+H]+: 701.2882 (exp.), 701.2871 (calcd). See Figures S1 and S2 for 1H NMR and ESI-MS data.
2.3. General synthetic procedures for copper corroles
2.3.1. Protocol A
Corrole (0.091 mmol) and Cu(acac)2 (191.0 mg, 0.73 mmol, 8.0 eq) were dissolved in dichloromethane (9 mL) and triethylamine (0.3 mmol). The mixture was stirred at room temperature for 20 min and then vacuum dried. The residue was purified by column chromatography as described below.
2.3.2. Protocol B
Corrole (0.041 mmol) and CuOAc2⋅H2O (66.4 mg, 0.33 mmol, 8.0 eq) were dissolved in dichloromethane (5 mL) and methanol (20 mL). The mixture was stirred at room temperature until the color changed to yellow-brown, after which the temperature was increased to 75 °C for 5 h. After evaporating to dryness, the residue was purified by column chromatography as described below.
2.4. (Mes)2(4‐NO2Ph)CorCu (1)
This compound was synthesized following Protocol B starting from 30.0 mg of free base corrole [24]. Purification was carried out on a silica gel column using a CH2Cl2/heptane (1:1, v:v) eluent. The synthesis of corrole 1 followed a modified synthetic procedure reported in the literature [25]. Yield: 31.2 mg (0.044 mmol, 95%). UV–vis (CH2Cl2) 𝜆max [nm, 𝜀 × 103 (M−1⋅cm−1)]: 396 (67.9), 537 (7.9), 597 (4.7). 1H NMR (500 MHz, CDCl3), 𝛿 (ppm): 8.36 (d, 3 J = 8.0 Hz, 2H, HPh), 7.99 (m, 2H, H𝛽), 7.77 (d, 3 J = 8.0 Hz, 2H, HPh), 7.36 (d, 3 J = 4.0 Hz, 2H, H𝛽), 7.21 (m, 2H, H𝛽), 7.03 (m, 6H, H𝛽, HMes), 2.40 (s, 6H), 2.06 (s, 12H). LRMS (MALDI/TOF) [M]+∙: 715.17 (exp.), 715.20 (calcd). See Figures S3 and S4 for 1H NMR and ESI-MS data.
2.5. (Mes)2(2,4‐(NO2)2Ph)CorCu (2)
This compound was synthesized following Protocol B starting from 30.5 mg of free base corrole (Mes)2(2,4‐(NO2)2Ph)CorH3. Purification was carried out on a silica gel column using a CH2Cl2/ heptane (1:1, v:v) eluent. Yield: 31.0 mg (0.0041 mmol, 94%). UV–vis (CH2Cl2) 𝜆max [nm, 𝜀 × 103 (M−1⋅cm−1)]: 397 (91.8), 601 (5.6). 1H NMR (500 MHz, CDCl3), 𝛿 (ppm): 8.99 (d, 4 J = 2.0 Hz, 1H, HPh), 8.58 (d, 3 J = 8.0 Hz, 1H, HPh), 8.05 (m, 2H, H𝛽), 7.86 (d, 3 J = 8.0 Hz, 1H, HPh), 7.36 (d, 3 J = 4.0 Hz, 2H, H𝛽) 7.23 (m, 2H, H𝛽), 7.04 (s, 2H, HMes), 7.03 (s, 2H, HMes), 6.82 (d, 3 J = 4.5 Hz, 2H, H𝛽), 2.40 (s, 6H, CH3), 2.07 (s, 6H, CH3), 2.05 (s, 6H, CH3). LRMS (MALDI/TOF) [M]+∙: 760.20 (exp.), 760.19 (calcd). HRMS (ESI) [M]+∙: 760.1887 (exp.), 760.1854 (calcd), [M+Na]+: 783.1753 (exp.), 783.1752 (calcd). See Figures S5 and S6 for 1H NMR and ESI-MS data.
2.6. (2,3,4,5‐F4Ph)2(2‐NO2thiophene)CorCu (3)
This compound was synthesized following Protocol B starting from 30.2 mg of free base corrole [26,27,28]. Purification was carried out on a silica gel column using a CH2Cl2/heptane (1:1, v:v) eluent. Yield: 30.3 mg (0.0039 mmol, 93%). UV–vis (CH2Cl2) 𝜆max [nm, 𝜀 × 103 (M−1⋅cm−1)]: 405 (73.2). 1H NMR (500 MHz, CDCl3), 𝛿 (ppm): 7.97 (d, 3 J = 4.0 Hz, 1H, Hthio), 7.93 (d, 3 J = 4.0 Hz, 2H, H𝛽), 7.43 (d, 3 J = 4.5 Hz, 2H, H𝛽), 7.40 (d, 3 J = 4.5 Hz, 2H, H𝛽) 7.36 (d, 3 J = 4.0 Hz, 1H, Hthio), 7.31–7.26 (m, 4H, HPh, H𝛽). 19F NMR (470 MHz, CDCl3), 𝛿 (ppm): − 137.55 to − 137.57 (m, 8F). LRMS (MALDI/TOF) [M]+∙: 780.98 (exp.), 780.99 (calcd). HRMS (ESI) [M]+∙: 780.9899 (exp.), 780.9875 (calcd). See Figures S7 and S8 for 1H NMR, 19F NMR and ESI-MS data.
2.7. (3‐NO2Ph)2(Ph)CorCu (4)
This compound was synthesized following Protocol B starting from 30.0 mg of free base corrole [29]. Purification was carried out on a silica gel column using a CH2Cl2/heptane (2:1, v:v) eluent. Synthesis of corrole 4 has already been reported in the literature [29]. Yield: 17.3 mg (0.026 mmol, 52%). UV–vis (CH2Cl2) 𝜆max [nm, 𝜀 × 103 (M−1⋅cm−1)]: 407 (80.6), 541 (6.3), 625 (4.3). 1H NMR (500 MHz, CDCl3), 𝛿 (ppm): 8.59 (m, 2H, HPh), 8.44 (d, 3 J = 8.0 Hz, 2H, HPh), 8.05 (d, 3 J = 8.0 Hz, 2H, HPh), 7.89 (m, 2H, H𝛽), 7.69 (t, 3 J = 8.0 Hz, 2H, HPh), 7.60 (m, 3H, HPh), 7.54 (d, 3 J = 5.0 Hz, 2H, H𝛽), 7.47 (m, 2H, HPh), 7.27 (d, 3 J = 4.5 Hz, 2H, H𝛽), 7.26 (m, 2H, H𝛽). LRMS (MALDI/TOF) [M]+∙: 676.06 (exp.), 676.09 (calcd). See Figures S9 and S10 for 1H NMR and ESI-MS data.
Half-wave potentials (V versus SCE) of copper triarylcorroles 0–5 in CH2Cl2 containing 0.1 M TBAP
| Cpd | E1∕2 (V versus SCE) | Ref. | ||||
|---|---|---|---|---|---|---|
| 2nd Ox | 1st Ox | 1st Red | 2nd Red (#e) | |||
| 0 | 1.46c | 0.78 | − 0.19 | − 1.94 (1)b | — | [10 ] |
| 1 | 1.40c | 0.80 | − 0.16 | − 1.16 (1)b | 0.95 | t.w |
| 2 | 1.43c | 0.83 | − 0.09 | − 0.92 (1)b | 1.00 | t.w |
| 3 | 1.57c | 1.08 | −0.19 | − 1.00 (1)b | 1.00 | t.w |
| 4 | 1.46c | 0.88 | − 0.08 | − 1.13 (2)b | 1.95 | t.w |
| 5 | 1.44 | 0.95 | −0.02 | − 1.10 (3)b | 2.94 | t.w |
Structures of the compounds are given in Scheme 1.
a ratio of cathodic peak current for 2nd Red (at the meso-nitroaryl group) over that of the first 1st Red (at the conjugated macrocycle).
b Obtained in CH2Cl2∕0.1 M TBAP at − 60 °C.
c Peak potential of irreversible process at scan rate of 0.1 V/s. t.w = this work.
2.8. (4‐NO2Ph)3CorCu (5)
This compound was synthesized following Protocol A starting from 60.0 mg of free base corrole [30]. The purification process consisted of a silica gel column using neat CH2Cl2 as the eluent. Synthesis of corrole 5 has already been reported in the literature [31]. Yield: 30.0 mg (0.042 mmol, 46%). UV–vis (CH2Cl2) 𝜆max [nm, 𝜀 × 103 (M−1⋅cm−1)]: 420 (37.5), 540 (5.0), 627 (3.5). 1H NMR (500 MHz, CDCl3), 𝛿 (ppm): 8.37 (m, 6H, HPh), 8.00 (m, 2H, H𝛽), 7.92 (d, 3 J = 8.5 Hz, 4H, HPh), 7.83 (d, 3 J = 8.5 Hz, 2H, HPh), 7.59 (d, 3 J = 4.5 Hz, 2H, H𝛽), 7.31 (m, 2H, H𝛽), 7.19 (d, 3 J = 4.5 Hz, 2H, H𝛽). LRMS (MALDI/TOF) [M]+∙: 721.06 (exp.), 721.08 (calcd). See Figures S11 and S12 for 1H NMR and ESI-MS data.
Cyclic voltammograms of copper corroles 1–5 in CH2Cl2 containing 0.1 M TBAP. Scan rate = 0.1 V/s. Potentials for the meso-nitroaryl reduction are shown in blue, the number of electrons transferred in the second reduction step is indicated above the half-wave potential and peak potentials for chemically generated products formed after oxidation are given in red.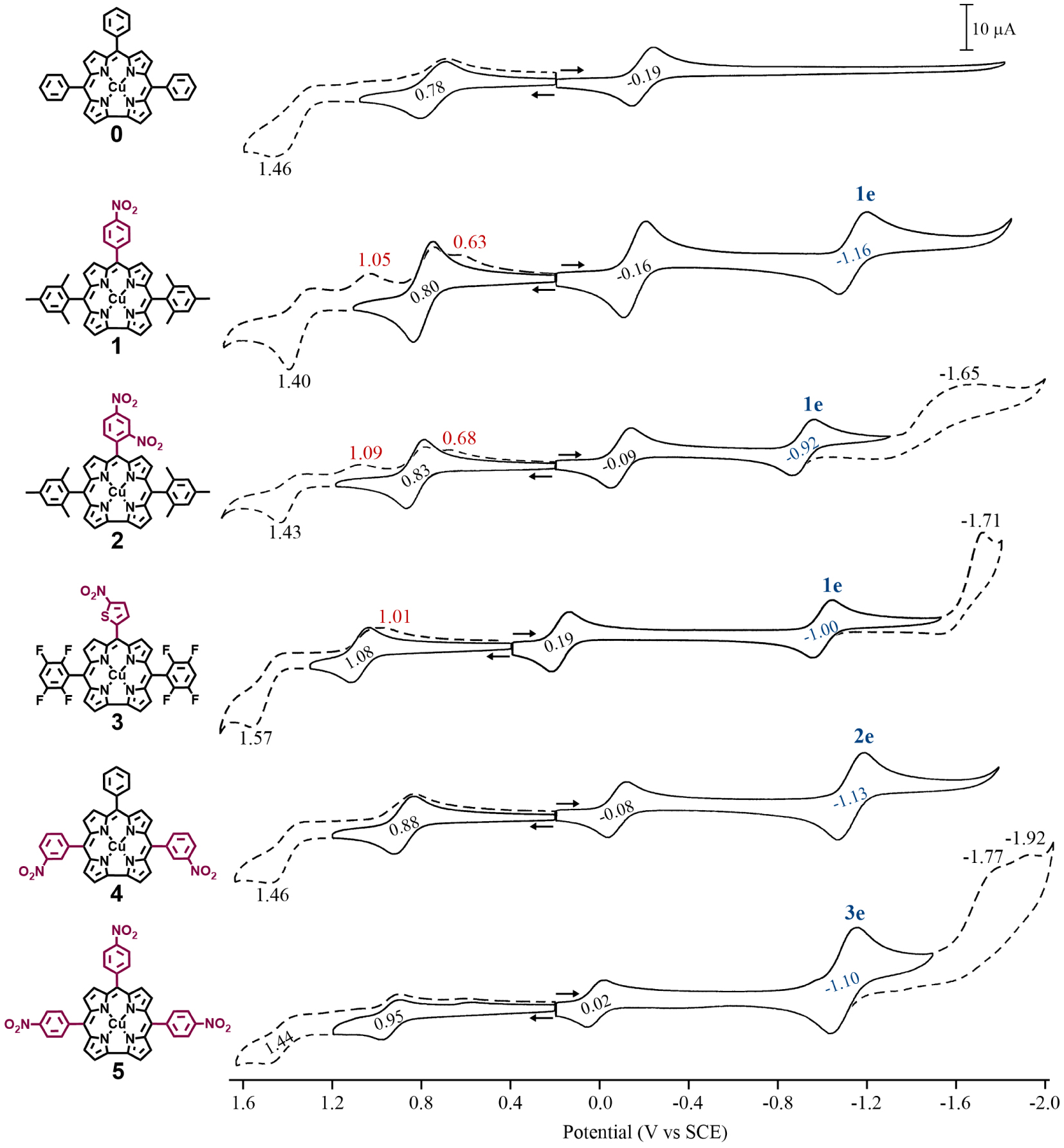
3. Results and discussion
3.1. Electrochemistry
Each copper corrole was electrochemically examined in CH2Cl2 containing 0.1 M TBAP at room temperature. Examples of cyclic voltammograms for 1–5 are given in Figure 1 and half-wave potentials are summarized in Table 1 which also includes data for the parent copper triphenylcorrole (0 in Scheme 1) under the same solution conditions [10].
As seen in Figure 1, the nitroaryl corroles 1–5 exhibit two reversible reductions and at least one reversible oxidation while the parent compound 0 exhibits only one reversible reduction and one reversible oxidation within the solvent potential window. Half-wave potentials for the first reduction of the corroles are located between 0.19 and − 0.19 V versus SCE and are assigned to occur at the conjugated macrocycle as given in (1).
| (1) |
This assignment of electron transfer site is consistent with earlier assignments for numerous copper [5,11,12,13,32] and other noninnocent metallocorroles [5,15,16,17,18,19,33,34] and fits with the Kadish electrochemical diagnostic criterion [18] that noninnocent macrocycles will undergo a facile reduction in nonaqueous media (vide supra). As seen in Figure 1 and Table 1, the most facile one-electron addition occurs for 3 ( V), consistent with the electron withdrawing properties of both the meso-nitrothiophene group and the two meso-tetrafluorophenyl substituents while corrole 1 bearing two mesityl and one para-nitrophenyl meso-group exhibits the most difficult ligand centered reduction of the investigated nitroaryl corroles at E1∕2 = −0.16 V. All of the nitroarylcorroles are easier to reduce than the parent compound, 0 (E1∕2 = −0.19 V), an expected result due to the highly electron-withdrawing NO2 substituent.
The second reduction of compounds 1–5 ranges from E1∕2 = −1.16 (for 1) to − 0.92 V (for 2) and these values can be compared to − 1.94 V for the second reduction of corrole 0 when the measurement was carried out at − 60 C [10]. This large difference in potential for the second reduction of the five copper corroles bearing one or more nitroaryl substituents is consistent with a change in the site of electron transfer, from π-ring centered in the case of 0 to the electroactive meso-nitroaryl ring(s) in the case of compounds 1–5. This assignment of electron transfer site in 1–5 is consistent with the known electrochemistry of nitrobenzene [35], which is reduced in two steps, the first being reversible and located at E1∕2 = −1.08 V versus SCE in CH2Cl2 [36]. The half-wave potential for the second reduction of 1 (E1∕2 = −1.16 V) is close to that of free nitrobenzene while the second reversible reduction of corroles 2 (E1∕2 = −0.92 V) and 3 (E1∕2 = −1.00 V) are located at potentials close to reported E1∕2 values in nonaqueous media for reduction of m-dinitrobenzene [37] and 2-nitrothiophene [38], respectively. Additional evidence for reduction of the nitroaryl groups is given by , defined as the ratio of cathodic peak current (ipc) for the second electroreduction over the ipc value for the first reduction of the same compound. As seen in Table 1, this ratio is approximately 1.0 for compounds 1–3, 2.0 for compound 4, and 3.0 for compound 5, values consistent with the number of nitroaryl substituents on the examined corrole and indicating a single one-electron reduction of this group in each case. Reductions occurring at the meso-NO2Ph groups of iron [39,40], cobalt [16,17,19,40] and copper [31] corroles have previously been reported while similar meso-substituent-centered reductions have also been shown to occur for porphyrins bearing meso-nitrophenyl groups [40,41].
As mentioned above, nitrobenzene [35] and nitrothiophene [38] are both characterized by two reductions in nonaqueous media, the second of which is irreversible. The same is seen for the copper corroles 2, 3 and 5 in CH2Cl2 (Figure 1) where the second and third reductions are assigned to occur at the meso-nitroaryl group. It is worth noting that the copper corrole 2 bearing both a ortho- and para-NO2 substituent on one meso-phenyl ring of the compound displays only a single one-electron reduction at − 0.97 V while m-dinitrobenzene displays two closely spaced reversible reductions [37,42] located at − 0.90 and − 1.25 V versus SCE when measured in acetonitrile containing tetra-n-propylammonoium perchlorate [37].
The first oxidation of corroles 1–5 is reversible and located at potentials ranging from 0.80 to 1.08 V versus SCE. On the basis of literature assignments for compound 0 and related copper corroles [5,10,11,12,43], this process in the current study is assigned as a ligand-centered oxidation, resulting in a cationic product with a divalent CuII metal center and a doubly oxidized corrole macrocycle according to (2).
| (2) |
A plot of the first oxidation potential versus the first reduction potential for compounds 0–5 in CH2Cl2 containing 0.1 M TBAP is shown in Figure 2 and reveals a characteristic of these noninnocent corrole systems. The plot in the figure is linear with a high correlation coefficient of , thus suggesting that the site of both electroreduction and electrooxidation remains the same throughout the series of compounds, and supporting the assignments given in (1) and (2). Inductive effects of meso-substituents on corroles, porphyrins and related macrocycles are known to govern redox potentials [3,19,41,44,45,46,47,48] and it was expected that a linear plot would be obtained between the two redox potentials in Figure 2 (assuming that the sites of electron transfer remain the same throughout the series); however the magnitude of the slope in the plot of Figure 2 provides insight into how the electron withdrawing substituents influence differently the oxidation and reduction potentials of noninnocent corroles in the currently investigated series of compounds. As seen in the figure, the slope of the linear regression analysis is 0.804 with a standard error (or deviation) of the slope (Sb) of 0.050. The obtained slope of less than 1.0 indicates that reduction of the noninnocent copper triarylcorroles (or the LUMO/SOMO-1) is more affected by electron withdrawing substituents on the meso-aryl groups than is the oxidation (or HOMO energies) by a factor of ∼1.25.
Plots of E1∕2 for first oxidation versus E1∕2 for first reduction of corroles 0–5 in CH2Cl2 containing 0.1 M TBAP.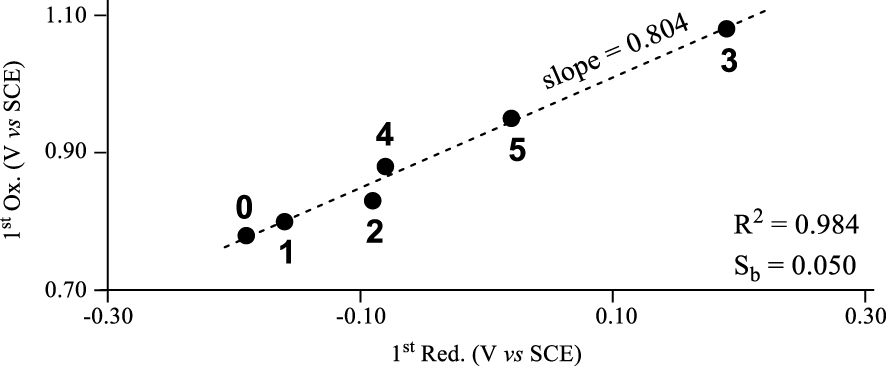
Each investigated corrole also undergoes a second oxidation which is irreversible for 1–4 and located at an anodic peak potential (Epa) between 1.40 and 1.57 V (see Figure 1). A chemical reaction follows this electron transfer and leads to a new electrooxidation product which is reduced at potentials between Epc = 1.01 and 1.05 V for 1–3 on the reverse scan. Interestingly, corrole 5 displays a reversible second oxidation at E1∕2 = 1.44 V indicating the absence of a coupled chemical reaction on the cyclic voltammetry timescale, but plots of Epc values for this process in compounds 1–5 versus E1∕2 for the first reduction or first oxidation are linear, suggesting that the site of the second electron abstraction does not change in the series of investigated corroles.
UV–vis spectral changes for compounds 1–5 (a) during an applied reducing potential (∼10−4 M) in CH2Cl2 containing 0.1 M TBAP and (b) upon addition of TBACN (0.02 M) to ∼10−5 M corrole in CH2Cl2 where the changes were recorded as a function of time.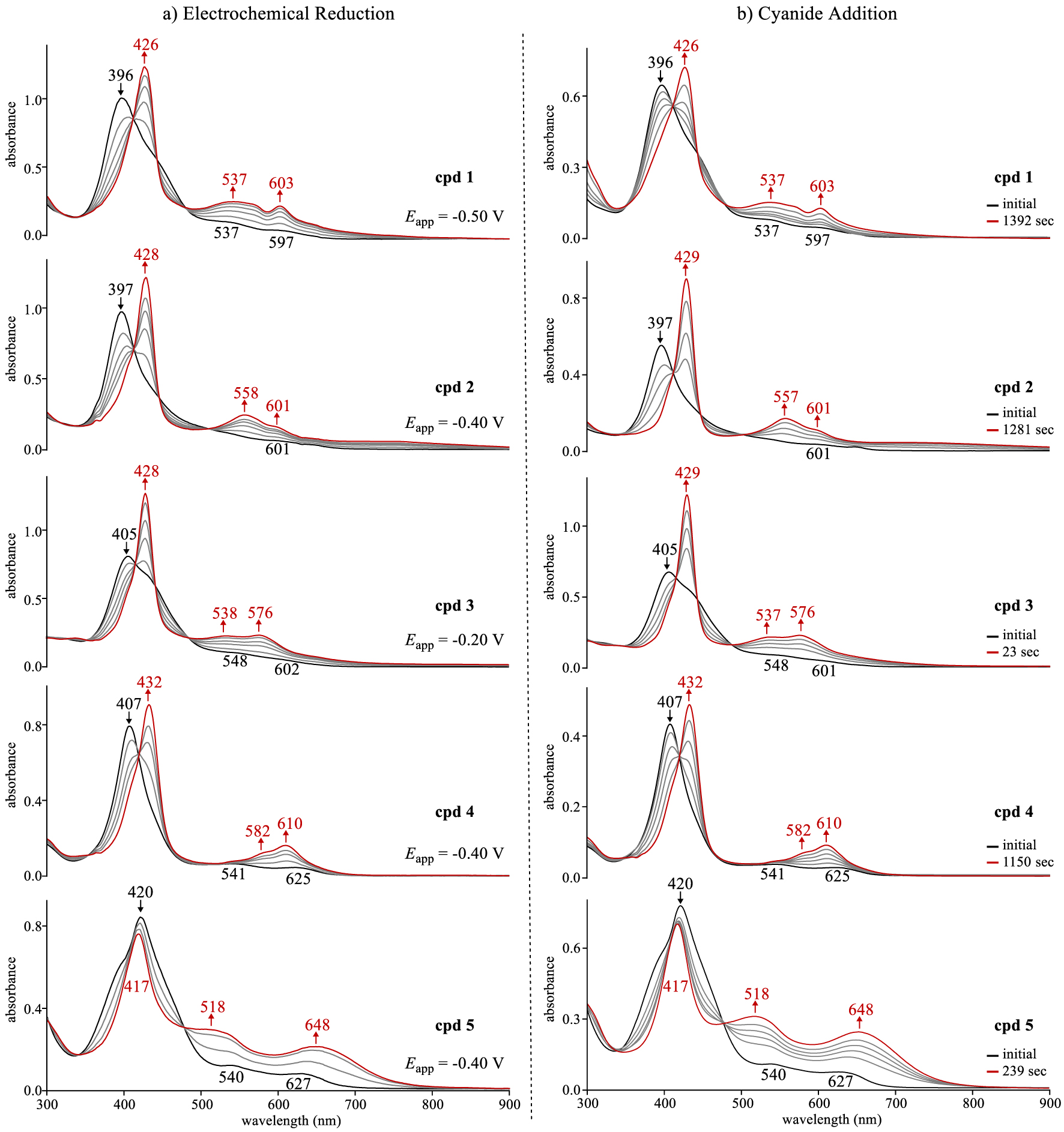
3.2. Electrochemical or chemical reduction via anion induced electron transfer (AIET)
To characterize the one-electron reduction product(s) of 1–5, thin-layer UV–vis spectroelectrochemical studies were carried out in CH2Cl2 containing 0.1 M TBAP. The spectral changes obtained under the influence of an applied reducing potential in the thin layer cell are presented in Figure 3a and the data for the neutral and singly reduced corroles in the CH2Cl2 solvent containing 0.1 M TBAP are summarized in Table 2.
UV–vis spectral data of neutral and singly reduced copper corroles 0–5 in CH2Cl2 containing 0.1 M TBAP
| Cpd | 𝜆max, nm (𝜀 × 10−4 M−1⋅cm−1) | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Neutral | Singly reduced | ||||||
| Soret | Q-Band(s) | Soret | Q-Band(s) | ||||
| 0 | 410 (7.2) | 540 (0.4) | 633 (0.2) | 432 (9.0) | 578 (1.3) | 607 (1.8) | [10] |
| 1 | 396 (6.8) | 537 (0.8) | 597 (0.5) | 426 (8.5) | 537 (1.8) | 603 (1.6) | t.w |
| 2 | 397 (9.2) | 601 (0.6) | 428 (11.9) | 558 (2.4) | 601 (1.6) | t.w | |
| 3 | 405 (7.3) | 548 (0.9) | 602 (0.6) | 428 (11.6) | 538 (1.9) | 576 (2.0) | t.w |
| 4 | 407 (8.1) | 541 (0.6) | 625 (0.4) | 432 (8.3) | 582 (1.2) | 610 (1.5) | t.w |
| 5 | 420 (3.8) | 540 (0.5) | 627 (0.4) | 417 (3.4) | 518 (1.3) | 648 (1.0) | t.w |
Structures of the compounds are given in Scheme 1.
t.w = this work.
The neutral corroles 1–5 have a well-defined Soret band located between 396 and 420 nm and broad ill-defined Q-bands as seen in Figure 3a. Similar spectra have been reported for 0 under the same solution conditions [5,10]. Upon reduction of corroles 1–4, the Soret band at decreases in intensity as a new red-shifted Soret band of higher intensity grows in at 426–432 nm along with two new Q-bands located at 537–582 nm and 576 to 610 nm. There are no near-IR bands characteristic of a corrole π-anion radical and the spectral changes in Figure 3a for 1–4 are consistent with (1) which specifies generation of [CorCuII]−, a compound which possesses an intact conjugated macrocycle (i.e. Cor3−).
Copper corrole 5 exhibits slightly different behavior upon electroreduction. As seen in Figure 3a, the Soret band of 5 shifts by only 3 nm to higher energy wavelengths while the corresponding band for compounds 1–4 shifts by 23–31 nm to the red after reduction. Singly reduced 5 also has two broad and intense Q-bands at 518 and 648 nm, a spectral pattern quite different than is seen for singly reduced 0–4. This difference between the UV–vis spectrum of singly reduced 5 and the singly reduced corroles 0–4 (as well as various other copper corroles [10,12,13]) might result from a change in the site of electron transfer, but the plot in Figure 2 does not lend credence to this explanation. Nonetheless, the final spectrum of singly reduced 5 possesses broad Q-bands in the visible region which appear similar to hyperporphyrin-type spectra reported for peripherally protonated free base tris(p-aminophenyl)corrole isomers [49], a phenomenon which might arise from a mesomeric effect of the para-nitro substituents which is known to enhance delocalization of the conjugated π electrons, thus stabilizing the single negative charge on the molecule via resonance [50,51,52].
The effect of cyanide anions (CN−) on UV–vis spectra of the copper corroles was also examined and the time dependent spectral changes observed upon addition of an aliquot to give a 0.02 M TBACN/CH2Cl2 solution of 1–5 (∼10−5 M) is shown in Figure 3b. In each case, the spectral changes seen upon the addition of CN− are strikingly similar to what is seen for the same corroles under the application of an applied reducing potential (Figure 3a). Moreover, the final spectrum of each corrole after electrochemical or chemical conversion to its monoanionic form are identical to each other, thus providing clear evidence for the ability of cyanide anions to reduce the open-shell noninnocent Cor∙CuII to a [CorCuII]− product according to (1) via anion induced electron transfer (AIET).
The time elapsed for conversion of the neutral copper corrole to its final chemically reduced form ranged from 23 s for corrole 3 to 1392 s for compound 1. Moreover, a monotonic increase in the elapsed time needed to complete the AIET processes was observed with decrease in the first reduction potential of the compound (i.e. a more negative E1∕2 value). This trend is consistent with the thermodynamically driven electron transfer (ET) event from the HOMO of cyanide anion to the LUMO/SOMO-1 of the corrole “π-acid” complex [20,21,22] where the relative energies of the corrole LUMO/SOMO-1 are indirectly determined by the first reduction potential.
A simple diagram for this AIET process is given in Scheme 2, where the HOMO of the anion, in this case cyanide, lies at an energy level well above that of the open-shell copper corrole orbitals as detailed by Ghosh, Solomon and coworkers [6] as well as Nocera and coworkers [53].
Schematic energy diagram for the HOMOs of anions (X−) and the LUMO/SOMO-1 of the open-shell copper corrole depicting thermal electron transfer (ET).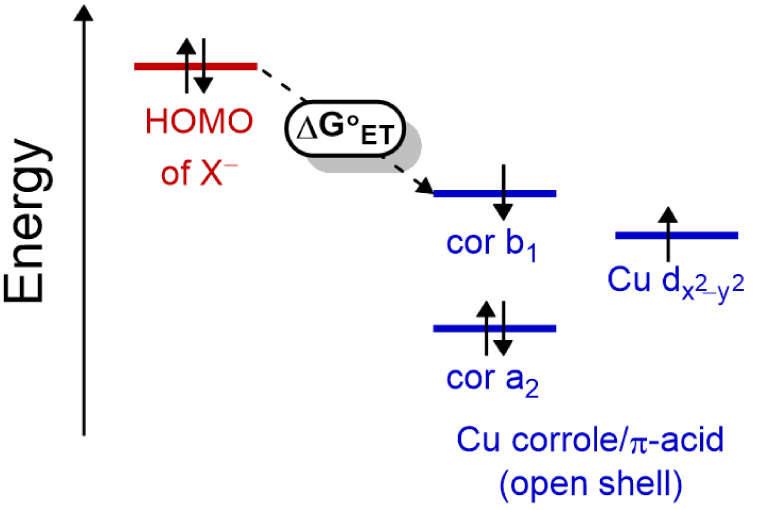
UV–vis spectral changes for compounds 3–5 upon addition of TBAF (0.05 M) to ∼10−5 M corrole in CH2Cl2 where the changes are recorded as a function of time.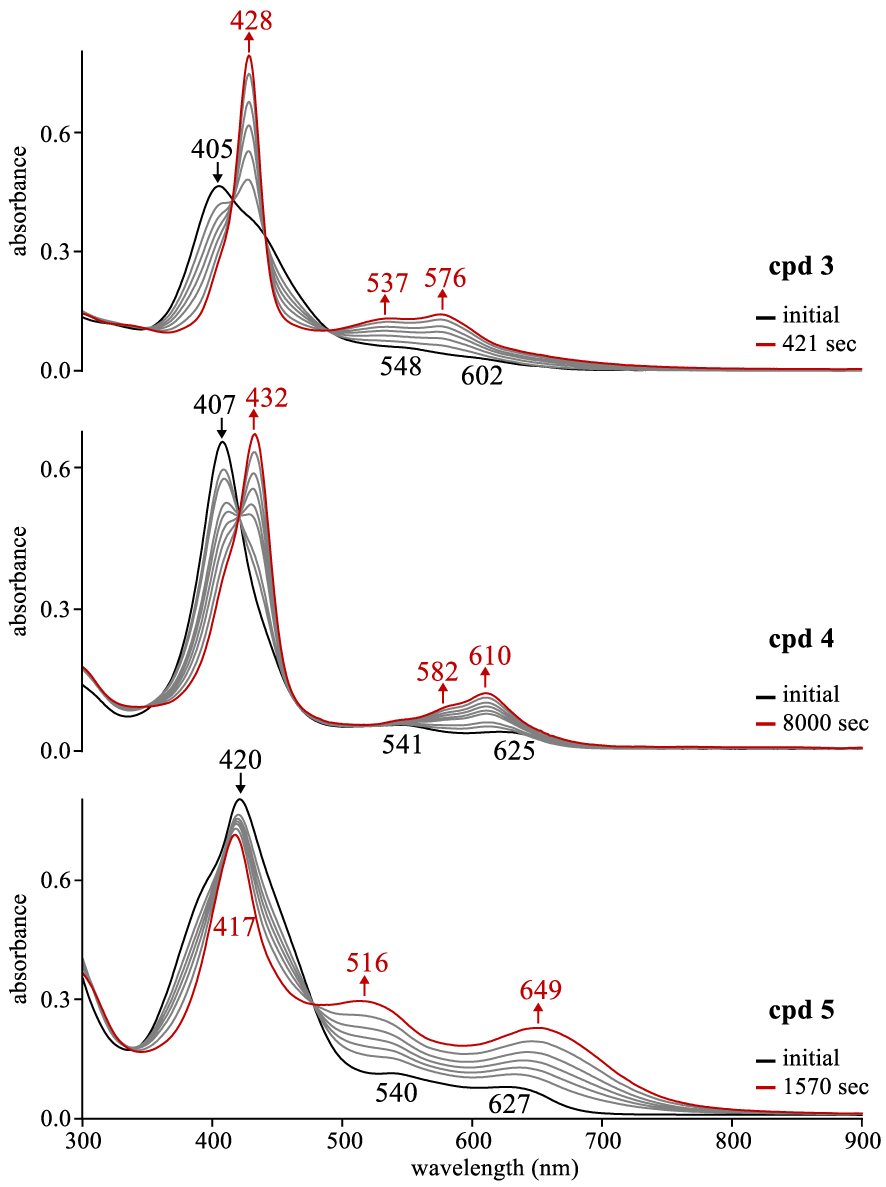
UV–vis spectral changes for reference corrole 0 (a) during an applied reducing potential (∼10−4 M) in CH2Cl2 containing 0.1 M TBAP, (b) upon addition of TBACN (0.02 M) to ∼10−5 M corrole in CH2Cl2 and (c) upon addition of TBAF (0.5 M) to ∼10−5 M corrole in CH2Cl2 where the changes are recorded as a function of time.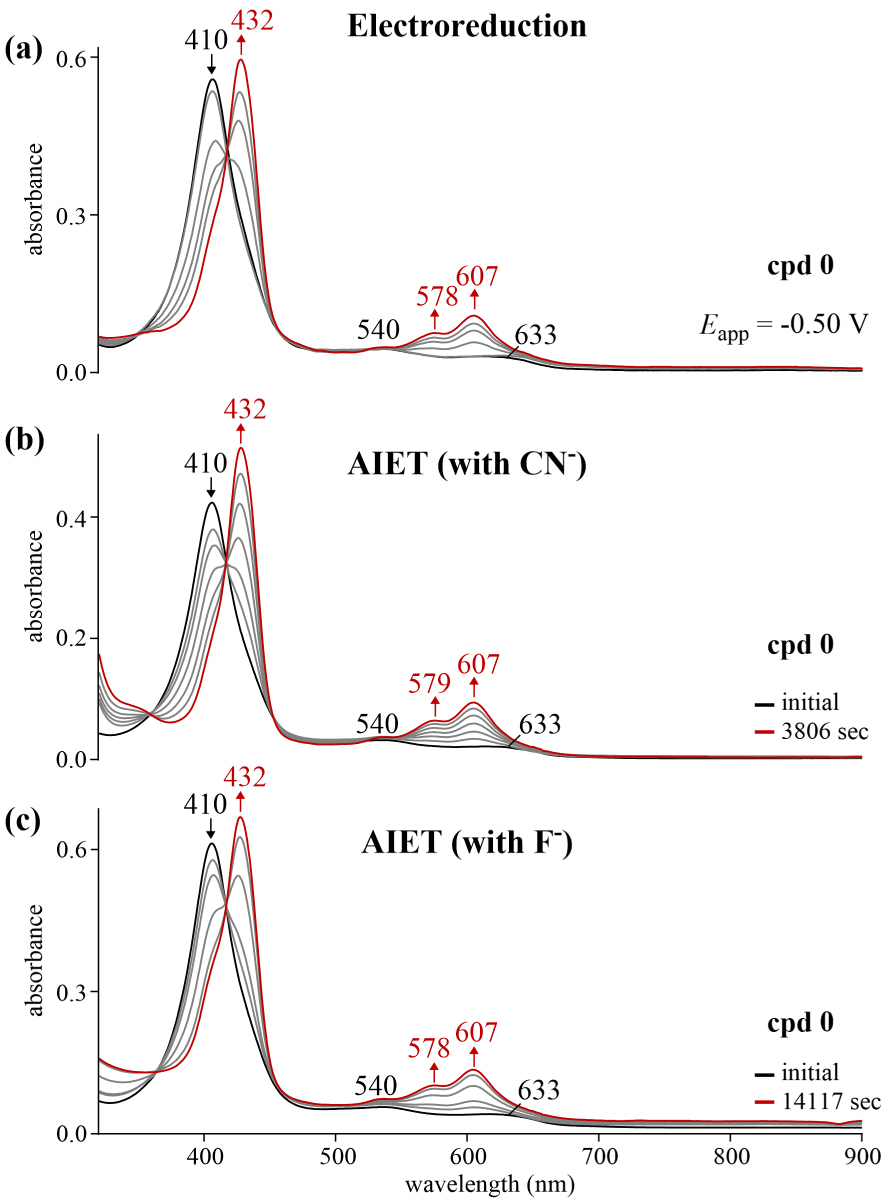
Like in the case of cyanide anion, F− has also been shown to reduce π-acids [22] and was previously shown to reduce noninnocent cobalt corroles [19]. Thus, the ability of fluoride anions to reduce open-shell noninnocent copper corroles by the addition of TBAF to solution was also investigated in the current study. As seen in Figure 4, the addition of TBAF (0.05 M) to a CH2Cl2 solution of corroles 3–5 led to exactly the same pattern of spectral changes as seen for either electroreduction in a thin layer cell or chemical reduction via CN− (Figure 3). However, The time to complete reduction of these corroles by anion induced electron transfer was slower with F− than with CN− and this is shown by a comparison of the data for compounds 3–5 in Figure 3 with that which was observed in Figure 4 where the rates decreased by a factor of ∼18 to 6.5 depending upon the substitution pattern.
Identical UV–visible were also observed for the electrochemically and chemically reduced parent compound 0 (Figure 5) but the time for reduction by anion induced electron transfer was slower for F− than CN− despite the ten-fold increase in the concentration of fluoride as compared to cyanide.
4. Conclusion
The mounting evidence for the ability of Lewis basic anions to reduce noninnocent corroles as described in the current study on copper corroles and also in our previous report for cobalt corroles [19] suggests that the occurrence of an anion induced electron transfer (AIET) event in aprotic media can serve as an additional probe for provisionally assigning the noninnocence or innocence of metallocorroles, particularly where there is a more cryptic noninnocent behavior as in the case of silver corrole derivatives [4,5].