

The study of the electroreduction of dioxygen to water has been the subject of an intense activity, in particular because of concern for the oxygen/hydrogen fuel-cell process. However, the recent renewal of the interest for this reaction, in the context of biomimetic chemistry, is undoubtedly due to the elucidation of the cytochrome c oxidase (CcO) structure. Indeed, this enzyme is able to promote in vivo, at physiological pH, the rupture of the O–O bond and the reduction of molecular oxygen to water. CcO has been called ‘the oxygen electrode of nature’s fuel-cell’ by Momenteau and Reed 〚1〛. A schematic representation of the binding site of CcO appears in Fig. 1. Until now, both the iron and the copper atoms were thought to be essential to the catalysis of the four-electron reduction of dioxygen. This assumption is based on two observations: (i) it has been shown that dimetallic porphyrins are efficient catalysts for the reduction of dioxygen to water, in acidic media 〚2,3〛; (ii) the copper atom, close to heme a3 (Fig. 1), could interact with the molecular oxygen coordinated to the iron centre. These two observations led to consider the formation of a μ-peroxo complex as a prerequisite to the O–O bond cleavage. More recently, it has been pointed out that the role of the copper atom in the enzyme could be different 〚4〛. This article reports our own contribution in this area to the approach with synthetic models.
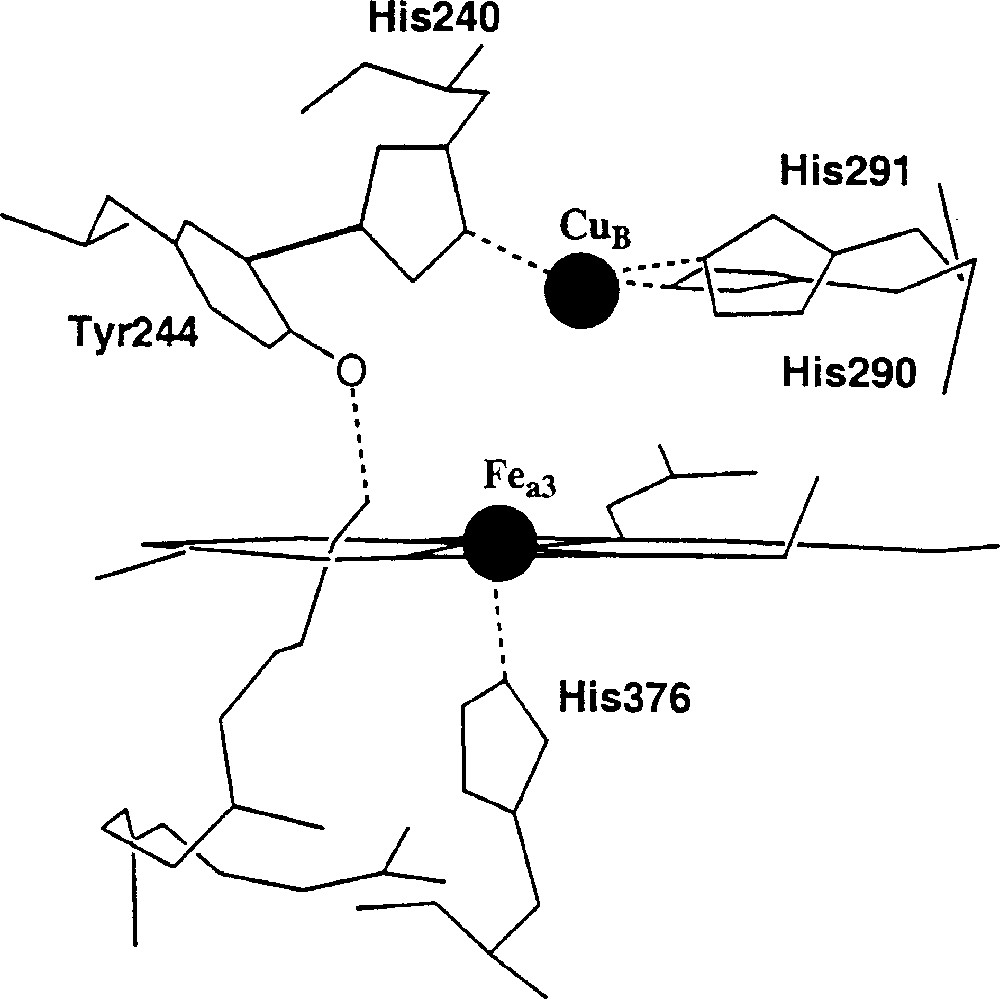
Simplified view of the dioxygen-binding site of cytochrome c oxidase. (For the sake of clarity, the double bonds and all the heteroatoms but one have been omitted).
A new family of models of CcO built from two different moieties linked together and able to complex the iron and copper cations (Fig. 2) has been prepared. The tris(2-aminoethyl)-amine (tren) molecule was grafted to the porphyrin core either through four or three links of different lengths. This first study has shown that the bimetallic iron–copper complexes (Fig. 2, 1FeCu and 2FeCu) of these molecules indeed catalyse the reduction of dioxygen to water, but not selectively. On the other hand, the iron-only complexes (Fig. 2, 1Fe and 2Fe), are selective catalysts for the reduction of dioxygen through the four-electron process, being therefore functional models of CcO. This surprising result is detailed in Fig. 3. Curve a represents the O2 electroreduction mediated by the iron-porphyrin 1Fe adsorbed at the surface of a graphite disk rotating electrode 〚5,8〛: the disk current is almost equal to the one that would result from a real four-electron exchange. The production of H2O2 at the modified graphite disk is almost non-existent, as shown by the very weak current detected at the platinum ring of the rotating ring-disk electrode (RRDE), around the graphite disk. The two curves labelled EPGE in Fig. 3 represent the pure two-electron reduction of O2 at the unmodified graphite disk, and the re-oxidation of the hydrogen peroxide reaching the annular platinum electrode. It must be stressed on that identical results with the two different iron complexes 1Fe and 2Fe were obtained. This clearly means that the slight variations in the structure of the distal pocket, induced by the different number of links (3 or 4) is not sufficient to observe a significant difference. This shows that the second metallic site, and consequently the formation of a μ-peroxo complex between iron and copper, is not essential to the catalysis of the four-electron electroreduction of O2 at pH 7.
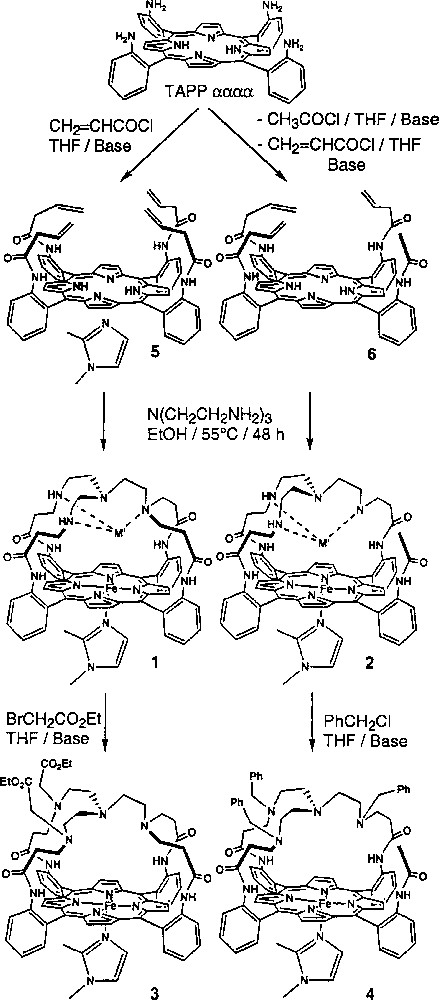
Synthetic route to different tren-capped porphyrins.
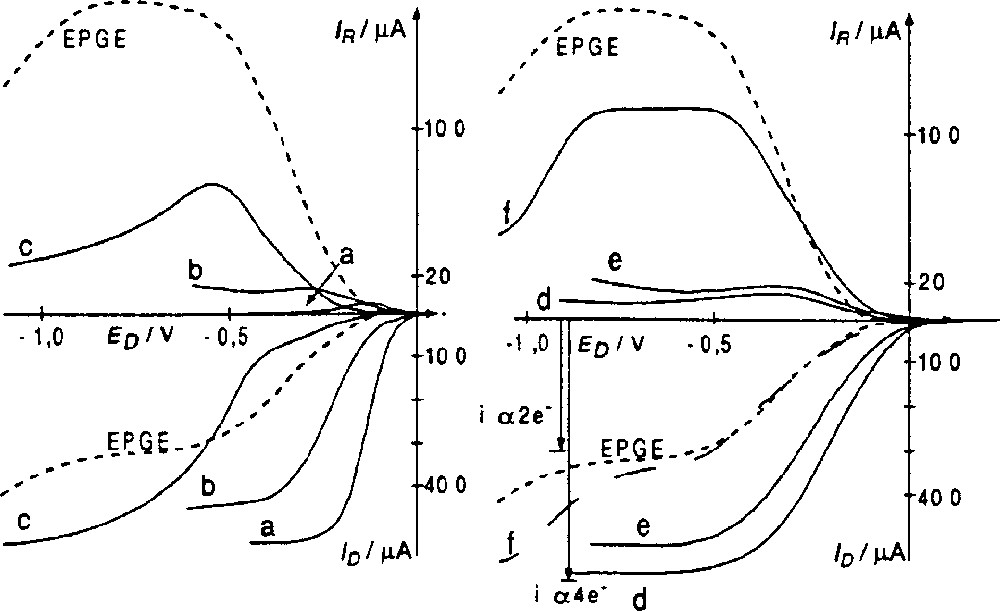
Rotating ring-disk voltammograms for O2 reduction; graphite disk impregnated with 1Fe (a), 3Fe (b), 4Fe (c) and 7Fe 1st scan (d), 7Fe 3rd scan (e), 7Co (f), bare graphite electrode (EPGE). pH = 6.86; Nr = 100 rpm; reference: SCE; pO2 = 1 atm; potential of the platinum ring-electrode: 0.8 V. At the disk: reduction of O2 to H2O or H2O2. At the ring: oxidation of H2O2 produced at disk.
In light of this first series of experiments, it appeared interesting to probe the influence of the secondary amine groups of the tripod in the model molecules. Indeed, it has been proposed that in the enzyme, the protonated Tyr 244 could ease the O2 binding through the formation a μ-hydroperoxo complex. In the molecular models of the present study (Fig. 2), the amino groups of the tren could play such a role; if so, different substitutions of these groups should influence the catalytic activity. This hypothesis prompted us to synthesise two new tren-capped iron porphyrins, 3Fe and 4Fe. Their catalytic activities are illustrated by curves b and c of Fig. 3, respectively. Apparently, the catalyst is more efficient when it bears secondary amino groups instead of tertiary amines. Indeed, in the case of 3Fe the number of electrons exchanged is lower than in the case of 1Fe, the corresponding amount of hydrogen peroxide produced being slightly higher. Moreover, the potential at which the limiting current for O2 reduction is reached is more negative (–0.4 V versus –0.3 V). The situation is different for 4Fe, the complex in which the tertiary amine functions of tren is substituted by benzyl groups. As illustrated by curve c in Fig. 3, the iron complex 4Fe is a four-electron catalyst, even though a significant amount of H2O2 is detected at the onset of the dioxygen reduction. Although this catalyst is an efficient four-electron catalyst, the reduction occurs at much more negative potentials (–1.0 V) than with 3Fe, which bears two ethyl acetate groups.
It must be considered that the four iron complexes in the present work, as well as all the previously studied molecules could lead to the formation of intermolecular μ-oxo complexes, between two iron porphyrins. This possibility cannot be ruled out as long as the iron porphyrin site of the molecules has an open face, allowing the formation of the oxygen complex between two iron centres. In order to explore this hypothesis, the basket handle porphyrin 7 (Fig. 4) reported by Momenteau et al. 〚6〛 has been studied, because it cannot form a μ-oxo complex. Both the cobalt and the iron complexes of this porphyrin have been studied.
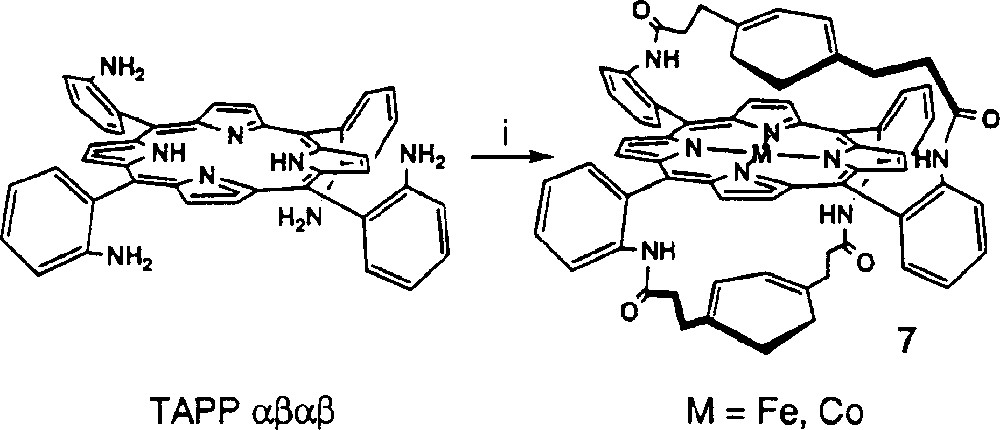
Synthesis of the basket-handle porphyrin 7. (i) benzene-1,4-dipropionic acyl chloride (2.1 equiv), NEt3, high dilution conditions.
The catalytic activity of the iron complex 7Fe is presented in Fig. 3 (curves d, e). It is obvious that this catalyst reduces dioxygen through a four-electron mechanism with almost no production of hydrogen peroxide, as shown by the curves at the platinum ring. This observation is of importance, as this is a definite proof that an iron porphyrin, by itself, is an efficient four-electron catalyst, at least for the electroreduction of O2. This clearly excludes the formation of a μ-oxo complex as a prerequisite to the four-electron reduction. Additionally, the electrodes modified by adsorption of this catalyst are quite stable, as shown by the small loss of activity between the first and third scan.
When the same experiment is achieved with the cobalt complex 7Co (curve f), only the two-electron reduction of dioxygen occurs. This observation establishes that, at this pH, the Fe and Co derivatives behave differently for the O2 reduction. A Co porphyrin must have the possibility to form an intermolecular μ-oxo complex, in order to reduce O2 to water; this is not the case for the iron derivative.
In the present work, a general synthetic pathway led to a large family of tren-capped porphyrins, and to different iron and iron–copper complexes. Several catalysts, differing by the substitution of the amino groups of the tren cap, have been evaluated for their capacity to reduce dioxygen to water. It has been shown that an iron-only complex can be a four-electron catalyst, an observation that means that the formation of a bimetallic intramolecular μ-peroxo complex is not necessary. Moreover, from the study with a bis-strapped porphyrin, it has been concluded that an iron porphyrin catalyses the reduction of dioxygen to water without any intermolecular interaction, which is not true for cobalt porphyrins.
Typical experimental procedure
α-5,10,15,20-Tetrakis(o-acrylamidophenyl) porphyrin (5). Prepared according to the described method 〚7〛.
α-5,10,15,20-{o-〚3,3’,3’’,3’’’-(N,N,N’,N’’-Tris(2-aminoethyl)amine) tetrapropionamido〛 tetraphenyl}porphyrin (1). Under argon, a 100 ml two-neck round bottom flask was charged with the Michael acceptor (5) (0.45 mmol, 400 mg) and MeOH (40 ml) and the mixture was heated at 55 °C for 45 min. Tren (0.45 mmol, 72 μl) was then directly added with a Hamilton syringe and heating continued for 48 h. The mixture was cooled, concentrated and directly poured onto a 15-μm silica gel column (5 × 20 cm). The desired product was eluted with 4% MeOH/CH2Cl2. After evaporation to dryness, 234 mg of a purple powder were collected (yield = 43%). 1H NMR (500 MHz, 〚d5〛pyridine, 50 °C): δ = 11.42 (s, 2H, –NHCO), 9.17 (s, 2H, –NHCO), 9.08 (d, 3J(H,H) = 4.5 Hz, 2H, Hβ-pyr), 9.06 (m, 2H, Haro), 9.02 (d, 3J(H,H) = 5.0 Hz, 2H, Hβ-pyr), 8.96 (s, 2H, Hβ-pyr), 8.81 (m, 4H, Haro + Hβ-pyr), 8.09 (d, 3J(H,H) = 6.5 Hz, 2H, Haro), 7.87 (t, 3J(H,H) = 7.5 Hz, 2H, Haro), 7.83 (t, 3J(H,H) = 7.5 Hz, 2H, Haro), 7.77 (d, 3J (H,H) = 7.5 Hz, 2H, Haro), 7.55 (t, 3J (H,H) = 7.0 Hz, 2H, Haro), 7.46 (t, 3J (H,H) = 7.0 Hz, 2H, Haro), 2.35–2.12 (m, 10H), 2.09–1.99 (m, 4H), 1.86–1.80 (m, 2H), 1.09 (m, 2H), 0.84 (m, 2H), 0.70 (m, 2H), 0.47 (m, 2H), 0.01 (m, 2H), –0.13 (m, 2H), –1.57 (m, 2H), –2.29 (s, 2H, –NHpyr). 13C NMR (125 MHz, 〚d5〛pyridine, 50 °C): δ; = 139.0, 137.0, 135.0, 131.4, 131.2, 126.0, 125.0, 124.5, 81.0, 56.0, 50.5, 45.0, 44.8, 43.5, 36.5, 35.5, 34.2, 27.0, 26.8. UV/vis (CH2Cl2): λmax nm (10–3 ϵ; dm3 mol–1 cm–1) = 421 (180.4), 515 (9.0), 548 (3.1), 588 (3.4), 644 (1.8). HRMS (LSIMS): m/z (%): 1 037.493 9 calculated for C62H61N12O4 〚M+–H〛, found 1 037.492 9 (100); anal. calculated for C62H60N12O4·2 H2O: C, 69.38, H, 6.01, N, 15.66, found: C, 68.81, H, 5.54, N, 15.79; iron complex (1Fe): HRMS (LSIMS): m/z (%): 1 091.413 2 calculated for C62H59N12O4Fe 〚M+–H〛, found 1 091.416 0 (100).
α-5,10,15,20-{o-〚3,3’,3’’,3’’’-(N,N,N’,N’’-Tris(2-aminoethyl)amine) (N’,N’’-diethoxycarbonylmethyl)tetrapropionamido〛tetraphenyl}porphyrin (3). In a 250-ml two-neck round bottom flask under argon, 200 mg (0.19 mmol) of (1) and 80 μl (0.76 mmol) of 1,8-diazabicyclo〚5.4.0〛undec-7-ene (DBU) were dissolved in 100 ml of freshly distilled THF. The solution was heated at 55 °C. With a syringe, 430 μl (0.38 mmol) of ethyl-2-bromo acetate were slowly added. Stirring was maintained during 24 h and then, the mixture was dried under vacuum. The residue was dissolved in methylene chloride and washed twice with 10 ml of aqueous NaOH (5%). The organic phase was concentrated by rotary evaporation and poured onto a 15-μm silica gel column (3 × 10 cm). The desired product was eluted with 1.5% MeOH/CH2Cl2. After evaporation to dryness, 120 mg of powder were collected (yield = 50%). 1H NMR (500 MHz, CDCl3, 50 °C): δ = 9.76 (s, 2H, –NHCO), 8.92 (d, 3J (H,H) = 5.0 Hz, 2H, Hβ-pyr), 8.89 (d, 3J (H,H) = 4.7 Hz, 2H, Hβ-pyr), 8.84 (s, 2H, Hβ-pyr), 8.70 (s, 2H, Hβ-pyr), 8.66 (d, 3J (H,H) = 8.5 Hz, 2H, Haro), 8.57 (d, 3J (H,H) = 8.5 Hz, 2H, Haro), 8.07 (s, 2H, –NHCO), 8.02 (d, 3J (H,H) = 7.5 Hz, 4J (H,H) = 1.0 Hz, 2H, Haro), 7.86 (t, 3J (H,H) = 8.0 Hz, 4J (H,H) = 1.5 Hz, 2H, Haro), 7.81 (t, 3J (H,H) = 8.0 Hz, 4J (H,H) = 1.5 Hz, 2H, Haro), 7.52 (m, 4H, Haro), 7.37 (t, 3J (H,H) = 7.5 Hz, 4J (H,H) = 1.0 Hz, 2H, Haro), 3.92 (q, 3J (H,H) = 7.0 Hz, 4H, –CH2 ester), 2.58 (d, 2J (H,H) = 16.5 Hz, 2H, –CH2), 2.51 (m, 2H), 2.37 (m, 2H), 2.12 (m, 4H), 2.01 (m, 2H), 1.86 (m, 2H), 1.70 (m, 4H), 1.10 (t, 3J (H,H) = 7.5 Hz, 6H, –CH3), 0.91 (m, 2H), 0.66 (m, 4H), 0.55 (m, 4H), –1.72 (m, 4H), –2.55 (s, 2H, –NHpyr). 13C NMR (125MHz, 〚d5〛pyridine, 50 °C): δ = 171.4, 171.0, 170.7, 137.3, 136.7, 133.7, 131.2, 130.0, 129.6, 124.4, 124.0, 123.6, 123.3, 60.8, 56.0, 52.4, 51.6, 51.3, 51.0, 49.4, 47.5, 35.5, 32.6, 14.3; UV/vis (CH2Cl2): λmax nm (10–3 ϵ; dm3 mol–1 ·cm–1) = 421 (271.1), 514 (16.7), 547 (5.1), 588 (6.0), 645 (2.2), MS (FAB): m/z (%): 1 209.9 (100) 〚M+–H〛; anal. calculated for C70H72N12O4: C, 69.52, H, 6.00, N, 13.90, found: C, 69.21, H, 6.07, N, 13.81; iron complex (3Fe): MS (MALDI/TOF, linear mode): m/z (%): 1 263.8 (100) 〚M+–H〛.
The ring-disk voltammetry studies were performed following well-documented methods 〚8〛.
Acknowledgements
The authors thank the ‘Région Bourgogne’ and the CNRS for financial support. D.R. and A.D. gratefully acknowledge the French Ministry of Education, Research and Technology for their grant.