

1 Introduction
Functional-group transposition within an organic molecule is an important technique for synthetic work. The isomerisation of allylic cyanide is a major industrial process in the adiponitrile synthesis 〚1〛. Another example is the isomerisation of 1,4-dichloro-2-butene, which is an intermediate in the synthesis of chloroprene, a monomer for oil-resistant rubbers 〚2〛. The rearrangement of allylic alcohols, which converts one isomer to another by a 1,3-transposition of the hydroxy group with simultaneous displacement of the double bond, is also an important process. A typical example is illustrated in Fig. 1. Conversions to the isomerised alcohol are often less than 100%, due to the reverse reaction.

Typical example of rearrangement of allylic alcohols.
Such a process needs the presence of a catalyst. Pioneering investigations have shown that an acid can catalyse the rearrangement 〚3〛. For example, in the early studies of the synthesis of vitamin A, sulfuric acid was used to convert one allylic alcohol to its isomer 〚4, 5〛. However, the yields were moderate in part because of side-reactions. In this process, an allyl cation is generated as intermediate, which gives side-products, typically dienes, ethers or skeletal rearrangement products.
2 VO(OR)3, first efficient catalyst for the isomerisation of allylic alcohols
In the 70’s, new catalysts for this reaction were reported in the literature. Chabardes et al. (from Rhône-Poulenc) found that trialkyl vanadates could undergo the isomerisation of allylic alcohols at 150–160 °C with a good selectivity 〚6, 7〛. This chemistry is applied commercially in fragrance industry 〚8〛. When linalool is heated at 160 °C with tris(triphenylsilyl)vanadate as catalyst, the mixture at equilibrium contains approximately 30% of the two primary alcohols, geraniol and nerol, along with the unchanged linalool (Fig. 2). Subsequently, Kane has improved the process: in order to shift the equilibrium in favour of the terminal alcohols, the linalool is first converted to a borate ester 〚9〛. In that case, equilibration of the borate esters yields 75–80% of the primary alcohols.
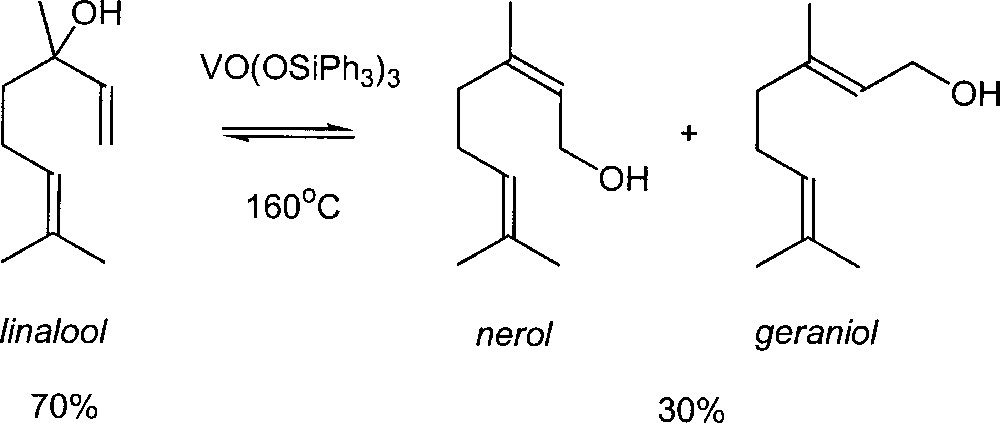
Industrial synthesis of nerol and geraniol using a vanadium oxo complex as catalyst.
Chabardes et al. proposed a mechanism involving the vanadium–oxo bond V=O (Fig. 3) 〚6, 10〛. The first step is an exchange of the substrate with an alkoxo ligand of the trialkyl vanadate catalyst (step I). A migration of the allyl group to the metal oxo unit then takes place (step II). The mechanism involves a cyclic transition state, which closely resembles a Claisen rearrangement. The product is then released after an exchange step between the new alkoxo complex and the substrate (step III). All steps are reversible; hence, the composition of the equilibrated product mixture is determined by the relative thermodynamic stabilities of the products.

Proposed mechanistic model of the vanadium-catalysed isomerisation of allylic alcohols (all steps are reversible).
3 Group 5 and 6 transition metals catalysts
In 1982, Fujita and collaborators reported a tungsten-based catalyst, which is far more active than the known vanadium catalyst 〚11〛. The linalool can be isomerised in the presence of WO(OR)4.pyridine at 200 °C with a selectivity of 92–95% (Fig. 4). According to them, the superiority of the tungsten catalyst under its industrial application lies in separation of the catalyst from the reaction mixture by flash distillation. The shortcoming of VO(OR)3 is that a trace of the catalyst is also distilled off to cause reverse isomerisation in the rectification process.
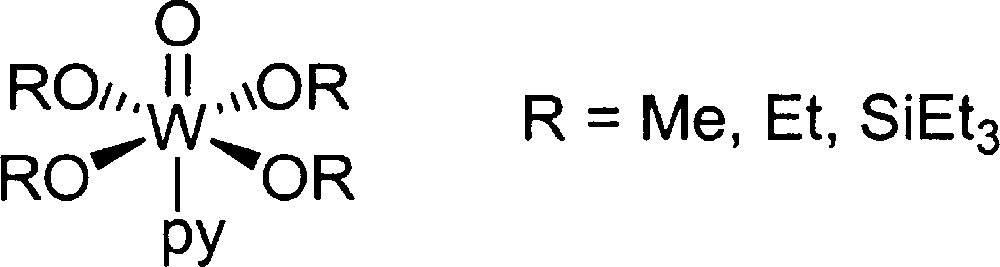
Tungsten catalyst reported by Fujita et al.
Takai et al. discovered a new oxo catalyst for the isomerisation of allylic alcohols, which is active at room temperature 〚12〛. They reported in 1985 that VO(acac)2 or MoO2(acac)2 activated with Me3SiOOSiMe3 effectively induces the isomerisation with good selectivity (Fig. 5). The active species has not been characterised; however, an IR analysis of a mixture of Me3SiOOSiMe3 and VO(acac)2 suggested the presence of a Me3SiOO– group attached on the vanadium atom.

Isomerisation of allylic alcohols in the presence of the catalyst prepared in situ from VO(acac)2 or MoO2(acac)2 and Me3SiOOSiMe3.
They also investigated the mechanism of the isomerisation using those systems (Fig. 6). Treatment of (S) 3-methyl-2-cyclohexen-1-ol (40% ee) with a VO(acac)2- Me3SiOOSiMe3 catalyst gave (R) 1-methyl-2-cyclohexen-1-ol (29% ee) and the unchanged starting material (S, 38% ee). Therefore, the hydroxy group of the substrate resides mainly on the same face of the olefinic double bond. This strongly suggests that the isomerisation does not proceed via a free allylic cation and supports the original mechanism proposed by Chabardes et al.
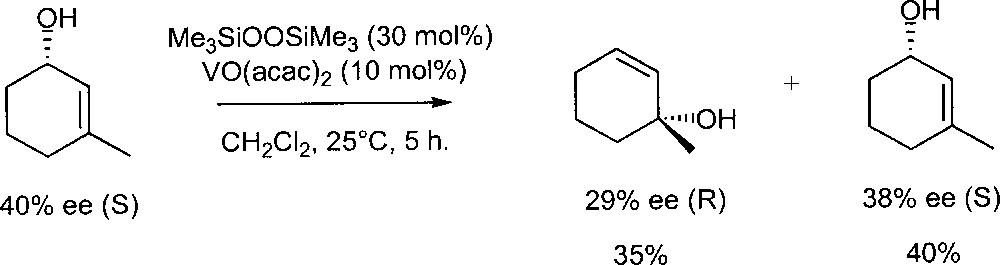
Isomerisation of (S) 3-methyl-2-cyclohexen-1-ol in the presence of the catalyst prepared in situ from VO(acac)2 and Me3SiOOSiMe3.
Osborn and co-workers found that the complex MoO2(OCMe2CH=CH2)2(CD3CN)2 (1) in CD3CN is converted into (2) containing the rearranged –OCH2CH=CMe2 ligand (Fig. 7) 〚13〛. The analogous imido complexes MoO(NR)(OCMe2CH=CH2)2 have also been synthesised and shown to isomerise similarly, although significantly more slowly. Kinetic experiments support the proposed pericyclic transition state TS1: the reduced rate of this process when one oxo ligand is replaced by a more electron donating imido ligand indicates that in the transition state some accumulation of negative charges takes place on the metal, with a cationic allyl group.
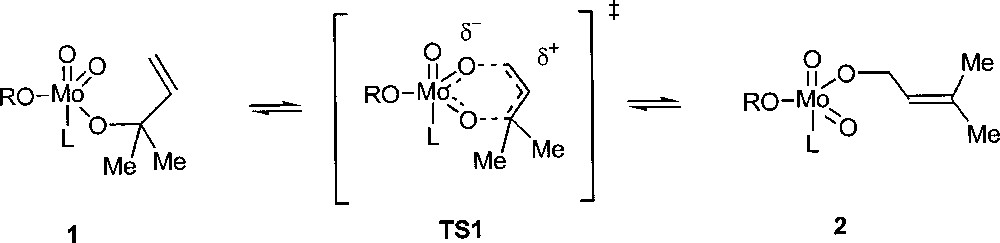
Allylic rearrangement at a molybdenum-oxo centre.
Based on these observations, they found that MoO2(OtBu)2 could catalyse the rearrangement of allylic alcohols at 25 °C. However, they found that slow reduction on the molybdenum (VI) centre takes place, causing a loss of catalytic activity with time.
Chromium (VI) has not been used as catalyst for this reaction, obviously because of its strongly oxidising character. However, Muzart et al. recently found that oxidation of allylic alcohols by CrO3 affords a mixture of isomers (Fig. 8) 〚14〛. An intramolecular rearrangement of the allylic alcohol involving an oxo ligand of the metal has been postulated.

Chromium oxide-catalysed oxidation of allylic alcohol by TBHP.
4 Going from group 6 transition metals to group 7
Narasaka et al. found that the combination of NBu4ReO4 and p-TsOH.H2O catalyses the isomerisations with reasonable activity 〚15〛. However, in the presence of this catalytic system, a dehydration of the allylic alcohols proceeds gradually to give the conjugated dienes. The mechanism of the reaction has been investigated using substrate trans-6-benzyl-2-cyclohexen-1-ol (3) (Fig. 9). The treatment of 3 with 10% of NBu4ReO4 and 5% of p-TsOH·H2O afforded the rearranged products and the starting material as diastereomer mixtures after 5 min. These observations indicate that the allylic alcohol does not rearrange via the Chabardes mechanism. The reaction probably occurs (in part or completely) through the free allylic cation as an intermediate.
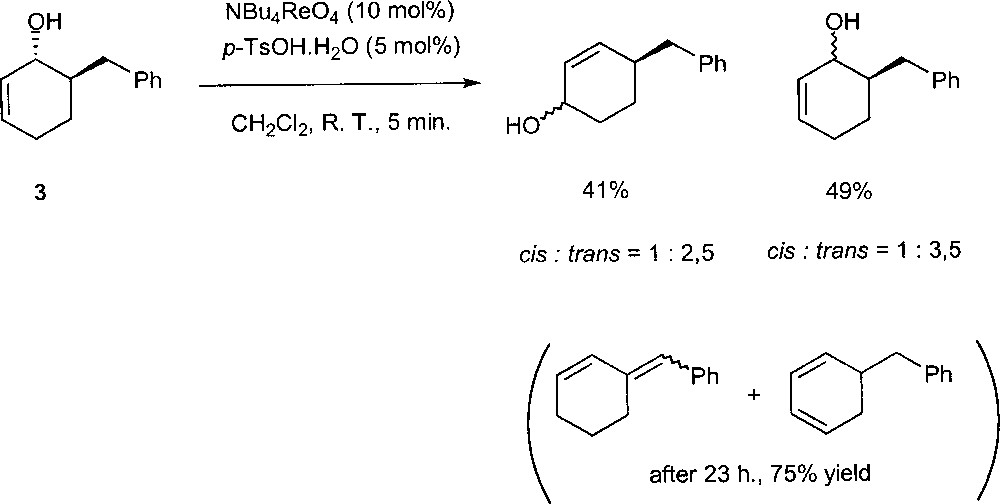
Rearrangement of trans-6-benzyl-2-cyclohexen-1-ol (3) in the presence of NBu4ReO4 and p-TsOH·H2O.
5 Well-defined oxo rhenium catalysts
An increase in the number of oxo ligands around the metal may give a greater stabilisation of the transition state and these oxo spectator ligands help to stabilise the negative charge developed on the metal during the rearrangement process (see TS1 in Fig. 7). Based on this idea, Osborn and co-workers found that the trioxorhenium complexes ReO3(OSiR3) (R = Me, Ph) are the most efficient catalysts yet known for the isomerisation process 〚16〛. In contrast with molybdenum (VI) complexes, these rhenium complexes are stable towards reduction, giving long-lived catalysts. For example, using hex-1-en-3-ol with 2 mol% of ReO3(OSiMe3) in acetonitrile at room temperature, the equilibrium with hex-2-en-1-ol is reached in less than 10 min (Fig. 10). In contrast, the catalyst MoO2(OtBu)2 requires about one day to reach this equilibrium under similar conditions.
Rearrangement of hexenol in the presence of ReO3(OSiMe3).
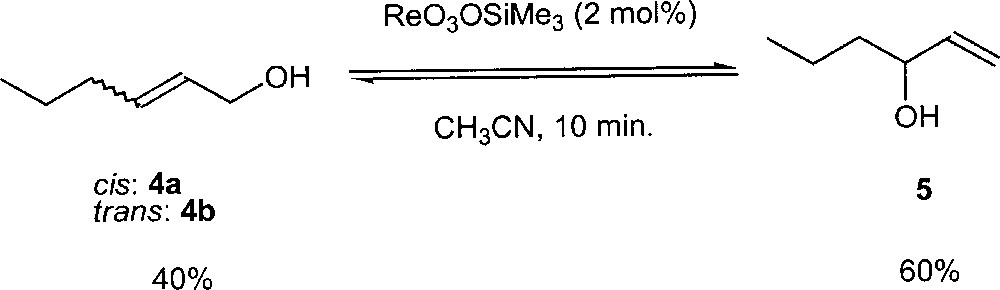
Kinetic studies have been carried out using ReO3(OSiPh3) as catalyst and the three allylic hexenols 4a, 4b and 5 separately as substrates. Interestingly, the kinetic law was found to be:
The kinetic data are consistent with the intramolecular rearrangement as the rate-determining step and not the alcohol-alkoxo exchange step (see step II and III in Fig. 3).
Very recently, Osborn’s rhenium catalyst has been used in the enantioselective total synthesis of (–) galanthamine, a molecule that has potentially clinical application for the treatment of Alzheimer’s disease 〚17〛. Treatment of (6) with ReO3(OSiPh3) gave the desired product with 50% yield (Fig. 11). Note that the hydroxy group of the substrate resides on the same face of the olefinic double bond.
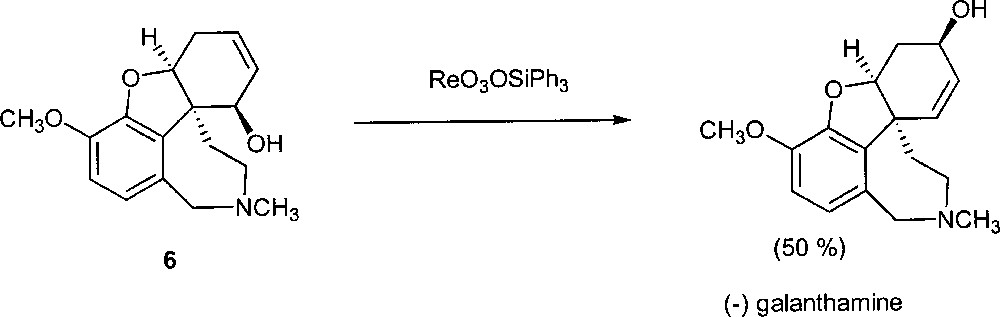
The synthesis of (-) galanthamine.
Wilkinson et al. reported that ReO3(OSiMe3) can react with the protected alcohol ROSiMe3 to give the corresponding complex ReO3(OR) in good yield 〚18〛. Based on that result, Osborn et al. found that this reaction can be applied to the catalytic isomerisation of trialkylsilyl protected allylic alcohols R’–CH=CH–CH2–OSiR3 〚19〛. Reaction rates for the isomerisation were slower than the corresponding unprotected alcohols and the ratio between the primary and the secondary protected alcohol was modified. When the alcohol was replaced by the trimethylsiloxy group, the proportion of primary allylic ether increased by about 15% at the reaction equilibrium.
Methyl trioxorhenium (MTO) MeReO3 has also been reported as a catalyst for the reaction. According to a patent by BASF, MeReO3 can catalyse the isomerisation of allylic alcohols at 60 °C, with a low catalyst loading (150:1) 〚20, 21〛. Espenson et al. also studied the use of MTO for this kind of transformation 〚22〛. In the presence of 5 mol% of methyltrioxorhenium in benzene at room temperature and oct-2-en-1-ol as substrate, the equilibrium with the octen-3-ol isomer is reached in three days (Fig. 12). The reaction was found to be very sensitive to the presence of water. Addition of water showed that the isomerisation was greatly inhibited. They speculate that MTO with H2O produces a dead-end species MeReO2(OH)2. An 18O-labelling experiment has been conducted to gain more insight into the mechanism. A stoichiometric reaction between 3-methyl-2-buten-1-ol and MeRe18O3 was run and no 18O incorporation into the rearranged allyl alcohol was observed. Because of this observation, they suggested a mechanism that does not involve an oxo ligand in the rearrangement (Fig. 13).
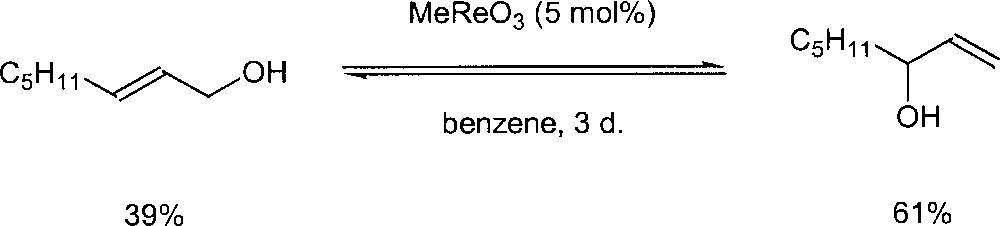
Rearrangement of octenol in the presence of methyl trioxorhenium.

Suggested mechanism of the isomerisation of allylic alcohol in the presence of MTO.
Mayer studied the chemistry of rhenium (III) oxo-bis(acetylene) compounds. No allyl transfer in the rhenium-oxo-bis(acetylene) complex depicted in Fig. 14 was detected 〚23〛. This system does not interconvert over a week at 70 °C, prior to decomposition. These results seem to indicate that a high oxidation state of the transition metal is required to get a catalytic activity for the isomerisation of allylic alcohol.
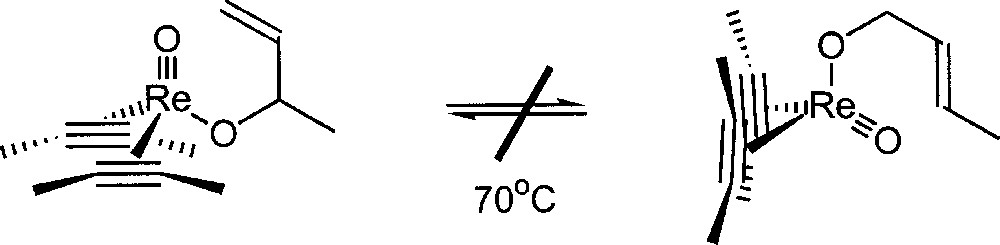
Reactivity of a rhenium oxo allyloxo bis(acetylene) complex.
6 Theoretical investigations
The mechanism of the reaction involving ReO3OR as catalyst has been investigated by ab initio calculations at the HF–SCF and MP2 levels 〚24〛. These calculations confirm the original proposal of the involvement of a cyclic transition state. The calculations point to a cyclic transition state that consists on a perrhenate anionic moiety and an allylic cationic group. The geometry of the transition state is chair-like (Fig. 15). The structure is very similar to that of the transition state found in 1,3-acyloxy shifts in allyl esters 〚25〛. However, the corresponding energy barrier is much lower as a result of a smaller repulsion between the oxo lone pairs and the π-allyl orbitals. The authors also found that this repulsion is highly dependent on the folding angles in the chair structure of the ReO2C3 skeleton. These angles can be strongly affected by microsolvation effects of the alcohols present in the reaction medium.
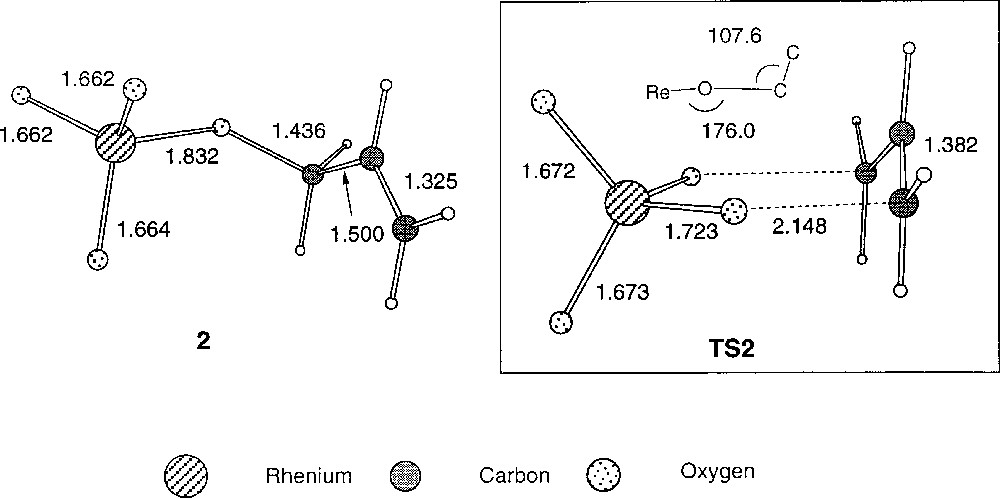
Optimised geometries of the reactant and the corresponding transition state for the allylic rearrangement of the system ReO3(OCH2CH=CH2) together with selected bond lengths 〚Å〛 and angles 〚°〛 (HF results).
7 Conclusion
Trialkyl oxovanadates (V) catalyse the isomerisation of allylic alcohols. These catalysts require a temperature higher than 150 °C. Since this first discovery, it has been shown that many other high oxidation state oxo complexes are efficient catalysts for the isomerisation process. New vanadium (V), molybdenum (VI) or rhenium (VII) oxo catalysts have been reported with usually good activities and selectivities even at room temperature. These catalysts have provided a useful tool for organic synthesis. A mechanism that involves a cyclic transition state and which incorporates a metal oxo unit has been proposed, which was subsequently confirmed by numerous investigations including theoretical calculations. However, in the case of methyltrioxorhenium as catalyst, a different mechanism has been suggested.